Lipase-Catalyzed Kinetic Resolution of Alcohols as Intermediates for the Synthesis of Heart Rate Reducing Agent Ivabradine




Abstract
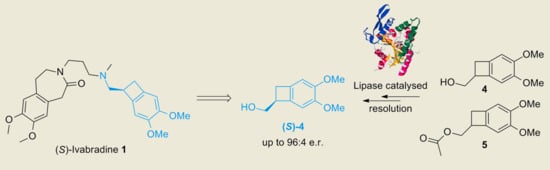
1. Introduction
2. Results and Discussion
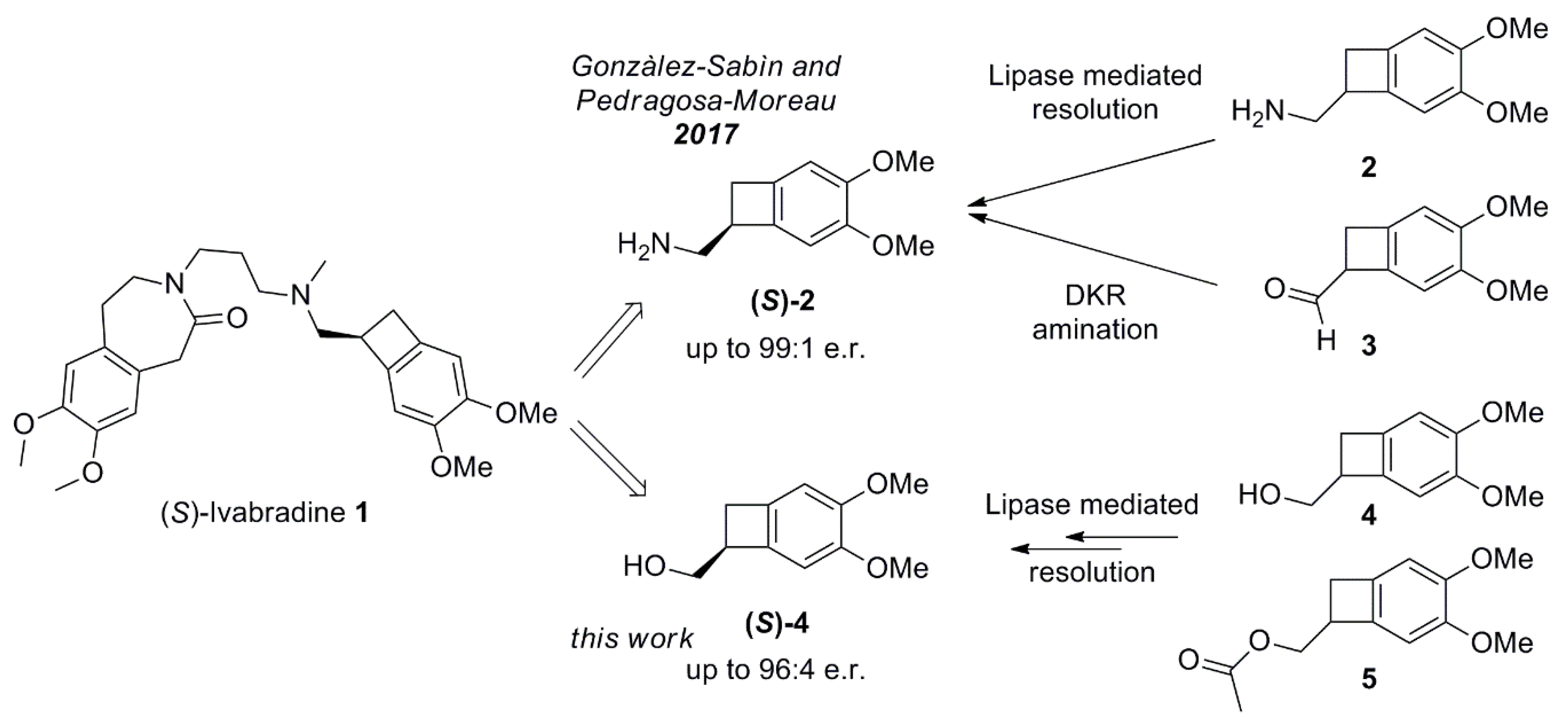
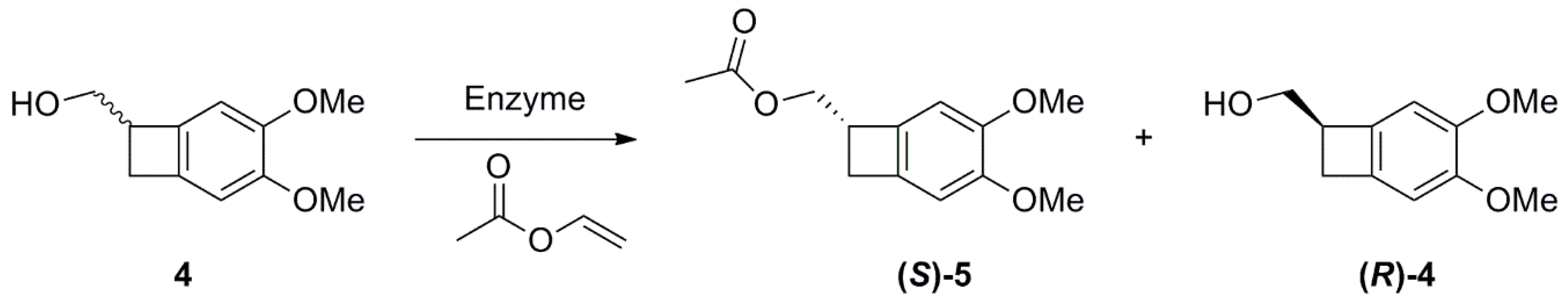
2.1. Studies on the Enzymes-Catalyzed Acetylation of Racemic 4
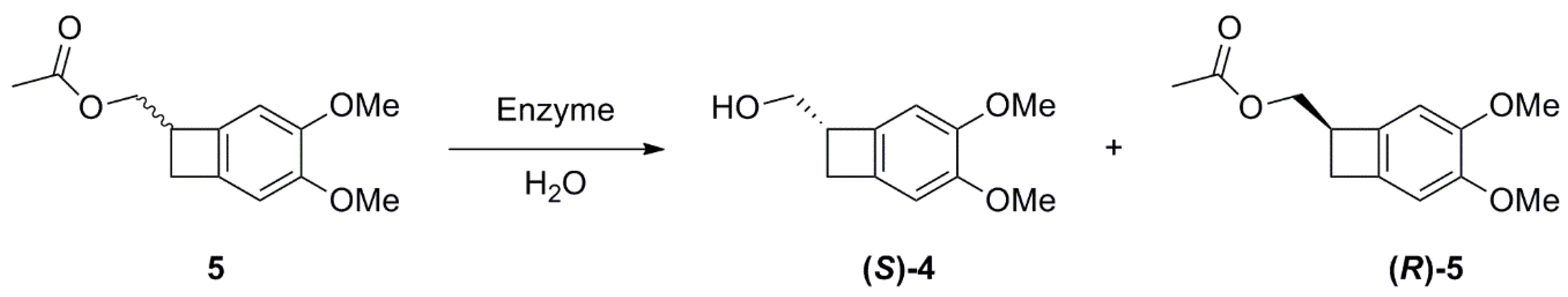
2.2. Studies on the Enzymes-Catalyzed Hydrolysis of Racemic 5
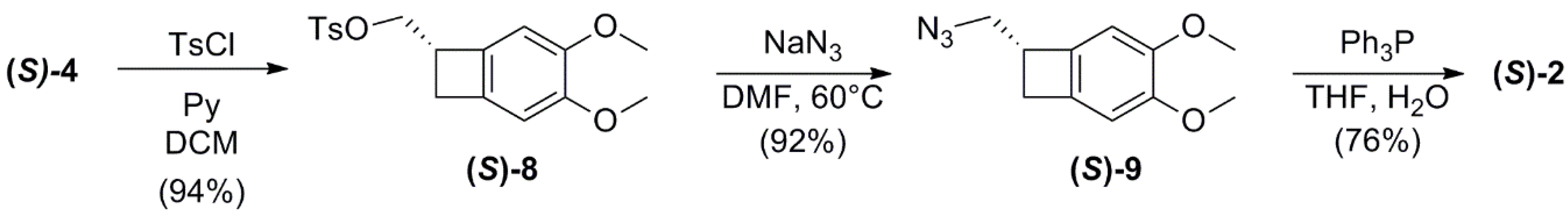
2.3. Conversion of Alcohol (S)-4 into Amine (S)-2
3. Materials and Methods
3.1. General Remarks
3.2. Synthetic Procedures
Methyl 3,4-dimethoxybicyclo[4.2.0]octa-1,3,5-triene-7-carboxylate 7
(3,4-dimethoxybicyclo[4.2.0]octa-1,3,5-trien-7-yl)methanol 4
(3,4-dimethoxybicyclo[4.2.0]octa-1(6),2,4-trien-7-yl)methyl acetate 5
General Procedure for enzyme-mediated acetylation of (3,4-dimethoxybicyclo[4.2.0]octa-1,3,5-trien-7-yl)methanol 4
General Procedure for enzyme-mediated hydrolysis of (3,4-dimethoxybicyclo[4.2.0]octa-1,3,5-trien-7-yl)methyl acetate 5
Preparative Synthesis of (S)-4
(S)-(3,4-dimethoxybicyclo[4.2.0]octa-1,3,5-trien-7-yl)methyl 4-methylbenzenesulfonate (S)-8
(S)-7-(azidomethyl)-bicyclo-[4.2.0]-octa-1,3,5-triene (S)-9
(S)-(3,4-dimethoxybicyclo[4.2.0]octa-1,3,5-trien-7-yl)-methanamine (S)-2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Parker, J.D.; Parker, J.O. Stable Angina Pectoris: The Medical Management of Symptomatic Myocardial Ischemia. Can. J. Cardiol. 2012, 28, S70–S80. [Google Scholar] [CrossRef]
- Hori, M.; Okamoto, H. Heart rate as a target of treatment of chronic heart failure. J. Cardiol. 2012, 60, 86–90. [Google Scholar] [CrossRef]
- Pedragosa-Moreau, S.; Le Flohic, A.; Thienpondt, V.; Lefoulon, F.; Petit, A.M.; Ríos-Lombardía, N.; Morís, F.; González-Sabín, J. Exploiting the Biocatalytic Toolbox for the Asymmetric Synthesis of the Heart-Rate Reducing Agent Ivabradine. Adv. Synth. Catal. 2017, 359, 485–493. [Google Scholar] [CrossRef]
- Romanelli, M.N.; Sartiani, L.; Masi, A.; Mannaioni, G.; Manetti, D.; Mugelli, A.; Cerbai, E. HCN Channels Modulators: The Need for Selectivity. Curr. Top. Med. Chem. 2016, 16, 1764–1791. [Google Scholar] [CrossRef]
- Sartiani, L.; Mannaioni, G.; Masi, A.; Romanelli, M.N.; Cerbai, E. The hyperpolarization-activated cyclic nucleotide-gated channels: From biophysics to pharmacology of a unique family of ion channels. Pharmacol. Rev. 2017, 69, 354–395. [Google Scholar] [CrossRef]
- Badu-Boateng, C.; Jennings, R.; Hammersley, D. The therapeutic role of ivabradine in heart failure. Ther. Adv. Chronic Dis. 2018, 9, 199–207. [Google Scholar] [CrossRef]
- Pascual Izco, M.; Alonso Salinas, G.L.; Sanmartín Fernández, M.; Del Castillo Carnevalli, H.; Jiménez Mena, M.; Camino López, A.; Zamorano Gómez, J.L. Clinical Experience with Ivabradine in Acute Heart Failure. Cardiology 2016, 134, 372–374. [Google Scholar] [CrossRef]
- Hidalgo, F.J.; Anguita, M.; Castillo, J.C.; Rodríguez, S.; Pardo, L.; Durán, E.; Sánchez, J.J.; Ferreiro, C.; Pan, M.; Mesa, D.; et al. Effect of early treatment with ivabradine combined with beta-blockers versus beta-blockers alone in patients hospitalised with heart failure and reduced left ventricular ejection fraction (ETHIC-AHF): A randomised study. Int. J. Cardiol. 2016, 217, 7–11. [Google Scholar] [CrossRef]
- Camici, P.G.; Gloekler, S.; Levy, B.I.; Skalidis, E.; Tagliamonte, E.; Vardas, P.; Heusch, G. Ivabradine in chronic stable angina: Effects by and beyond heart rate reduction. Int. J. Cardiol. 2016, 215, 1–6. [Google Scholar] [CrossRef]
- Weeda, E.R.; Nguyen, E.; White, C.M. Role of Ivabradine in the Treatment of Patients With Cardiovascular Disease. Ann. Pharmacother. 2016, 50, 475–485. [Google Scholar] [CrossRef]
- Malinowski, J.T.; St. Jean, D.J. Next-generation small molecule therapies for heart failure: 2015 and beyond. Bioorganic Med. Chem. Lett. 2018, 28, 1429–1435. [Google Scholar] [CrossRef]
- Sharma, V.; Dewangan, H.K.; Maurya, L.; Vats, K.; Verma, H.; Singh, S. Rational design and in-vivo estimation of Ivabradine Hydrochloride loaded nanoparticles for management of stable angina. J. Drug Deliv. Sci. Technol. 2019, 54, 101337. [Google Scholar] [CrossRef]
- Peglion, J.L.; Vian, J.; Vilaine, J.P.; Villeneuve, N.; Janiak, P.; Bidouard, J.P. Benzocyclobutyl- or indanyl-alkyl-amino-alkyl substituted 3-benzazepin-2-ones Useful in the Treatment of Cardiovascular Diseases. EP Patent 0534859A1, 25 September 1992. [Google Scholar]
- Lerestif, M.J.; Gonzales, I.; Lecouve, J.P.; Brigot, D. Method of Synthesising (1s)-4,5-dimethoxy-1-(methylaminomethyl)-benzocyclobutane and the Addition Salts Thereof, and Use of Same for the Synthesis of Ivabradine and the Pharmaceutically-Acceptable Addition Salts Thereof. WO Patent 2005123659A1, 29 December 2005. [Google Scholar]
- Ujvári, V.; Bódi, J.; Faragó, J.; Szöké, K.; Faigl, F.; Német, Z.; Temesvári, K.; Kiss, R.; Mátravoelgyi, B.; Kassai, F.; et al. Industrial Process for the Synthesis of Ivabradine Salts. WO Patent 2011138625A1, 10 November 2011. [Google Scholar]
- Liu, X.; Liu, Y.; He, H.; Cai, Z.; Yang, Y. New synthetic route to (1s)-4,5-dimethoxy-1-[(methylamino)methyl] benzocyclobutane, a key intermediate of ivabradine. Synth. Commun. 2014, 44, 451–456. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Arends, I.W.C.E.; Hanefeld, U. Introduction: Green Chemistry and Catalysis. In Green Chemistry and Catalysis; Wiley Online Books; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; pp. 1–47. ISBN 9783527611003. [Google Scholar]
- Sheldon, R.A.; Woodley, J.M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef]
- Woodley, J.M. New opportunities for biocatalysis: Making pharmaceutical processes greener. Trends Biotechnol. 2008, 26, 321–327. [Google Scholar] [CrossRef]
- Pollard, D.J.; Woodley, J.M. Biocatalysis for pharmaceutical intermediates: The future is now. Trends Biotechnol. 2007, 25, 66–73. [Google Scholar] [CrossRef]
- Patel, R.N. Biocatalysis for synthesis of pharmaceuticals. Bioorg. Med. Chem. 2018, 26, 1252–1274. [Google Scholar] [CrossRef]
- Hansen, K.B.; Hsiao, Y.; Xu, F.; Rivera, N.; Clausen, A.; Kubryk, M.; Krska, S.; Rosner, T.; Simmons, B.; Balsells, J.; et al. Highly Efficient Asymmetric Synthesis of Sitagliptin. J. Am. Chem. Soc. 2009, 131, 8798–8804. [Google Scholar] [CrossRef]
- Desai, A.A. Sitagliptin Manufacture: A Compelling Tale of Green Chemistry, Process Intensification, and Industrial Asymmetric Catalysis. Angew. Chemie Int. Ed. 2011, 50, 1974–1976. [Google Scholar] [CrossRef]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef]
- Ran, N.; Zhao, L.; Chen, Z.; Tao, J. Recent applications of biocatalysis in developing green chemistry for chemical synthesis at the industrial scale. Green Chem. 2008, 10, 361–372. [Google Scholar] [CrossRef]
- Kazlauskas, R.J.; Kim, B.-G. Biotechnology Tools for Green Synthesis: Enzymes, Metabolic Pathways, and their Improvement by Engineering. Biocatal. Green Chem. Chem. Process Dev. 2011, 1–22. [Google Scholar] [CrossRef]
- Sheldon, R.A. Enzyme Immobilization: The Quest for Optimum Performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Fiorati, A.; Berglund, P.; Humble, M.S.; Tessaro, D. Application of Transaminases in a Disperse System for the Bioamination of Hydrophobic Substrates. Adv. Synth. Catal. 2020, 362, 1156–1166. [Google Scholar] [CrossRef]
- Turner, N.J.; Truppo, M.D. Biocatalysis enters a new era. Curr. Opin. Chem. Biol. 2013, 17, 212–214. [Google Scholar] [CrossRef]
- Adrio, J.L.; Demain, A.L. Microbial enzymes: Tools for biotechnological processes. Biomolecules 2014, 4, 117–139. [Google Scholar] [CrossRef]
- García-Urdiales, E.; Alfonso, I.; Gotor, V. Enantioselective Enzymatic Desymmetrizations in Organic Synthesis. Chem. Rev. 2005, 105, 313–354. [Google Scholar] [CrossRef]
- Adlercreutz, P. Immobilisation and application of lipases in organic media. Chem. Soc. Rev. 2013, 42, 6406–6436. [Google Scholar] [CrossRef]
- Jaeger, K.-E.; Eggert, T. Lipases for biotechnology. Curr. Opin. Biotechnol. 2002, 13, 390–397. [Google Scholar] [CrossRef]
- Hasan, F.; Shah, A.A.; Hameed, A. Industrial applications of microbial lipases. Enzyme Microb. Technol. 2006, 39, 235–251. [Google Scholar] [CrossRef]
- Ollis, D.L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken, S.M.; Harel, M.; Remington, S.J.; Silman, I.; Schrag, J. The alpha/beta hydrolase fold. Protein Eng. 1992, 5, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Holmquist, M. Alpha/Beta-hydrolase fold enzymes: Structures, functions and mechanisms. Curr. Protein Pept. Sci. 2000, 1, 209–235. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, A.M. Improving enzymes by using them in organic solvents. Nature 2001, 409, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, P.; Muderhwa, J.M.; Graille, J.; Haas, M.J. Customizing lipases for biocatalysis: A survey of chemical, physical and molecular biological approaches. J. Mol. Catal. B Enzym. 2000, 9, 113–148. [Google Scholar] [CrossRef]
- Reetz, M.T. Lipases as practical biocatalysts. Curr. Opin. Chem. Biol. 2002, 6, 145–150. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Resch, V. The role of biocatalysis in the asymmetric synthesis of alkaloids. RSC Adv. 2013, 3, 17602–17632. [Google Scholar] [CrossRef]
- Simon, R.C.; Mutti, F.G.; Kroutil, W. Biocatalytic synthesis of enantiopure building blocks for pharmaceuticals. Drug Discov. Today Technol. 2013, 10, e37–e44. [Google Scholar] [CrossRef]
- Chen, C.S.; Fujimoto, Y.; Girdaukas, G.; Sih, C.J. Quantitative analyses of biochemical kinetic resolutions of enantiomers. J. Am. Chem. Soc. 1982, 104, 7294–7299. [Google Scholar] [CrossRef]
- Bose, P.; Kandadai, A.S.; Siripalli, U.B.R. Process for Preparation of Ivabradine. WO Patent 2010072409A1, 1 July 2010. [Google Scholar]
- Sun, P.Y.; Chen, Y.J.; Yu, G.L. Resolution of 4,5-dimethoxy-1-(methylaminomethyl)-benzocyclobutane. WO Patent 2009062377A1, 22 May 2009. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Time (h) | Conversion (%) a | e.r. (S)-5 b | E c |
|---|---|---|---|---|---|
| 1 | Esterase BS3 (CLEA) | 2 | 10 | 66:34 | 2 |
| 2 | Esterase BS2 (CLEA) | 48 | 21 | 57:43 | 1 |
| 3 | Acylase (Amano) | 3 | 6 | 59:41 | 1 |
| 4 | Candida Antarctica Lipase B (CAL-B) | 1 | 55 | 73:23 | 4 |
| 5 | Lipase PS (Amano) | 1 | 41 | 90:10 | 17 |
| 6 | Porcine Pancreas Lipase (PPL) (Type II) | 6 | 26 | 90:10 | 11 |
| Entry | Catalyst | Time (h) | Conversion (%) a | e.r. (S)-4 b | E c |
|---|---|---|---|---|---|
| 1 | Esterase BS3 (CLEA) | 20 | 35 | 79:21 | 5 |
| 2 | Esterase BS2 (CLEA) | 2 | 51 | 52:48 | 1 |
| 3 | Acylase (Amano) | 29 | 50 | 65:35 | 4 |
| 4 | Candida Antarctica Lipase B (CAL-B) | 0.5 | 44 | 82:18 | 7 |
| 5 | Lipase PS (Amano) | 0.5 | 32 | 96:4 | 37 |
| 6 | Porcine Pancreas Lipase (PPL) (Type II) | 5 | 30 | 87:13 | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morandini, A.; Rossetti, A.; Sacchetti, A. Lipase-Catalyzed Kinetic Resolution of Alcohols as Intermediates for the Synthesis of Heart Rate Reducing Agent Ivabradine. Catalysts 2021, 11, 53. https://doi.org/10.3390/catal11010053
Morandini A, Rossetti A, Sacchetti A. Lipase-Catalyzed Kinetic Resolution of Alcohols as Intermediates for the Synthesis of Heart Rate Reducing Agent Ivabradine. Catalysts. 2021; 11(1):53. https://doi.org/10.3390/catal11010053
Chicago/Turabian StyleMorandini, Anna, Arianna Rossetti, and Alessandro Sacchetti. 2021. "Lipase-Catalyzed Kinetic Resolution of Alcohols as Intermediates for the Synthesis of Heart Rate Reducing Agent Ivabradine" Catalysts 11, no. 1: 53. https://doi.org/10.3390/catal11010053
APA StyleMorandini, A., Rossetti, A., & Sacchetti, A. (2021). Lipase-Catalyzed Kinetic Resolution of Alcohols as Intermediates for the Synthesis of Heart Rate Reducing Agent Ivabradine. Catalysts, 11(1), 53. https://doi.org/10.3390/catal11010053