Insights into the Mechanism of Ethionamide Resistance in Mycobacterium tuberculosis through an in silico Structural Evaluation of EthA and Mutants Identified in Clinical Isolates

 , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
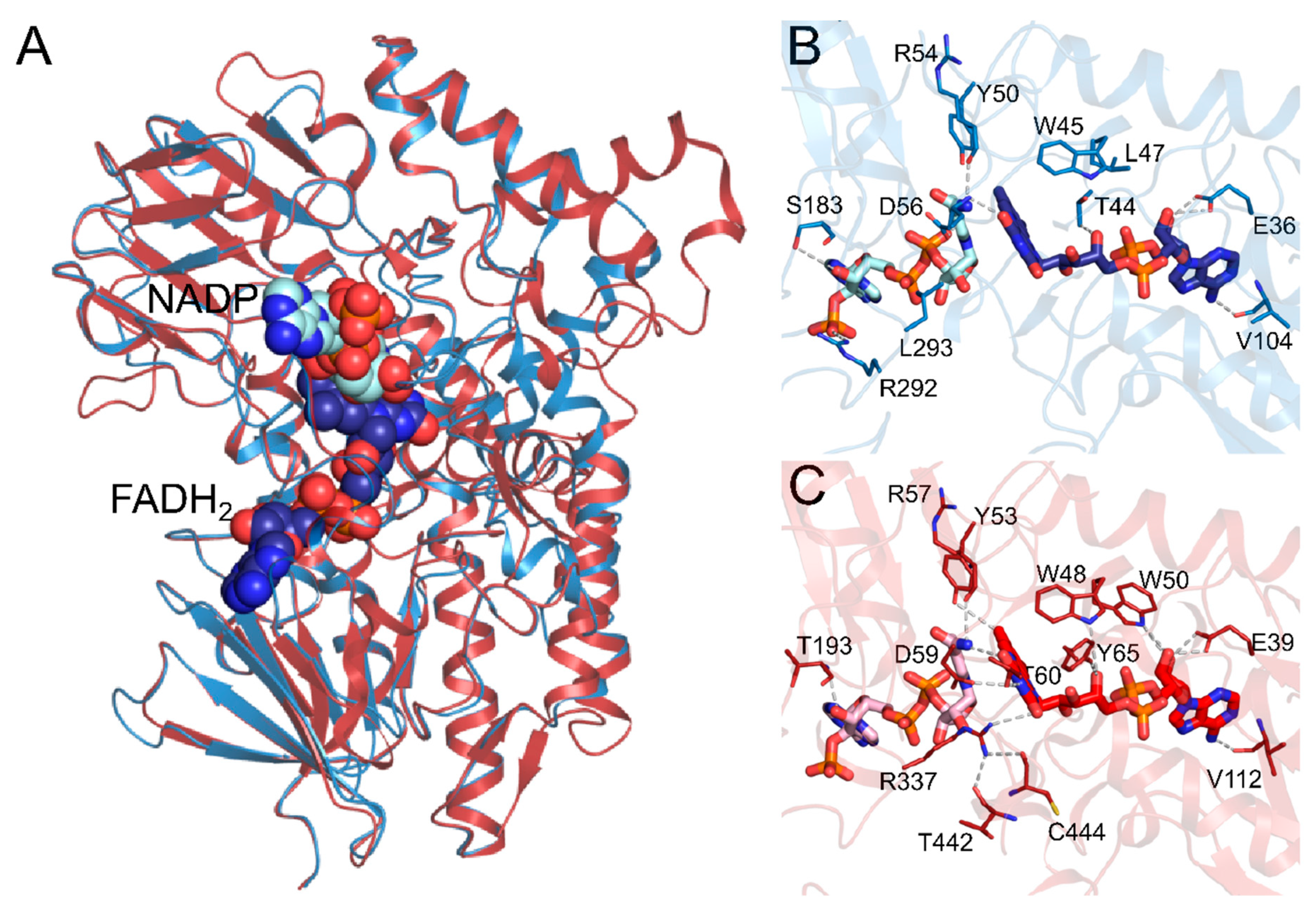
2.1. Comparative Modeling
2.2. Clustering and Molecular Docking
2.3. Assessing the Dynamic Properties of the EthA in Complex with ETH
2.4. Free Energy Changes for Y50C and T453I EthA Mutants
3. Materials and Methods
3.1. Comparative Modeling
3.2. Molecular Dynamics (MD)
3.3. Clustering and Free Energy Landscape
3.4. Docking
3.5. Thermodynamic Integration (TI)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Gilpin, C.; Korobitsyn, A.; Migliori, G.B.; Raviglione, M.C.; Weyer, K. The World Health Organization standards for tuberculosis care and management. Eur. Respir. J. 2018, 51, 1800098. [Google Scholar] [CrossRef]
- API Consensus Expert Committee. API TB Consensus Guidelines 2006: Management of pulmonary tuberculosis, extra-pulmonary tuberculosis and tuberculosis in special situations. J. Assoc. Physicians India 2006, 54, 219–234. [Google Scholar]
- Ramachandran, G.; Swaminathan, S. Safety and tolerability profile of second-line anti-tuberculosis medications. Drug Saf. 2015, 38, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Baulard, A.R.; Betts, J.C.; Engohang-Ndong, J.; Quan, S.; McAdam, R.A.; Brennan, P.J.; Locht, C.; Besra, G.S. Activation of the pro-drug ethionamide is regulated in mycobacteria. J. Biol. Chem. 2000, 275, 28326–28331. [Google Scholar] [CrossRef] [PubMed]
- DeBarber, A.E.; Mdluli, K.; Bosman, M.; Bekker, L.G.; Barry, C.E., 3rd. Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9677–9682. [Google Scholar] [CrossRef] [PubMed]
- Vannelli, T.A.; Dykman, A.; Ortiz de Montellano, P.R. The antituberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J. Biol. Chem. 2002, 277, 12824–12829. [Google Scholar] [CrossRef]
- Grant, S.S.; Wellington, S.; Kawate, T.; Desjardins, C.A.; Silvis, M.R.; Wivagg, C.; Thompson, M.; Gordon, K.; Kazyanskaya, E.; Nietupski, R.; et al. Baeyer-Villiger monooxygenases EthA and MymA are required for activation of replicating and non-replicating Mycobacterium tuberculosis inhibitors. Cell Chem. Biol. 2016, 23, 666–677. [Google Scholar] [CrossRef]
- Blondiaux, N.; Moune, M.; Desroses, M.; Frita, R.; Flipo, M.; Mathys, V.; Soetaert, K.; Kiass, M.; Delorme, V.; Djaout, K.; et al. Reversion of antibiotic resistance in Mycobacterium tuberculosis by spiroisoxazoline SMARt-420. Science 2017, 17, 1206–1211. [Google Scholar] [CrossRef]
- Hicks, N.D.; Carey, A.F.; Yang, J.; Zhao, Y.; Fortune, S.M. Bacterial genome-wide association identifies novel factors that contribute to ethionamide and prothionamide susceptibility in Mycobacterium tuberculosis. mBio 2019, 10, e00616. [Google Scholar] [CrossRef]
- Vilchèze, C.; Av-Gay, Y.; Attarian, R.; Liu, Z.; Hazbón, M.H.; Colangeli, R.; Chen, B.; Liu, W.; Alland, D.; Sacchettini, J.C.; et al. Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol. Microbiol. 2008, 69, 1316–1329. [Google Scholar] [CrossRef]
- Laborde, J.; Deraeve, C.; Bernardes-Génisson, V. Update of antitubercular prodrugs from a molecular perspective: Mechanisms of action, bioactivation pathways, and associated resistance. Chem. Med. Chem. 2017, 12, 1657–1676. [Google Scholar] [CrossRef] [PubMed]
- Ang, M.L.; Siti, Z.Z.; Shui, G.; Dianišková, P.; Madacki, J.; Lin, W.; Koh, V.H.; Gomez, J.M.; Sudarkodi, S.; Bendt, A.; et al. An ethA-ethR-deficient Mycobacterium bovis BCG mutant displays increased adherence to mammalian cells and greater persistence in vivo, which correlate with altered mycolic acid composition. Infect. Immun. 2014, 82, 1850–1859. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Toriyama, S.; Imaizumi, S.; Tomiyasu, I.; Masui, M.; Yano, I. Incorporation of 18O into long-chain, secondary alcohols derived from ester mycolic acids in Mycobacterium phlei. BBA Lipid Lipid Met. 1982, 712, 427–429. [Google Scholar] [CrossRef]
- Kamerbeek, N.M.; Janssen, D.B.; van Berkel, W.J.H.; Fraaije, M.W. Baeyer–Villiger monooxygenases, an emerging family of flavin-dependent biocatalysts. Adv. Synth. Catal. 2003, 345, 667–678. [Google Scholar] [CrossRef]
- Torres Pazmiño, D.E.; Baas, B.-J.; Janssen, D.B.; Fraaije, M.W. Kinetic mechanism of phenylacetone monooxygenase from Thermobifida fusca. Biochemistry 2008, 47, 4082–4093. [Google Scholar] [CrossRef]
- Leisch, H.; Morley, K.; Lau, P.C.K. Baeyer−Villiger monooxygenases: More than just green chemistry. Chem. Rev. 2011, 11, 4165–4222. [Google Scholar] [CrossRef]
- da Silva, D.A.; Ferreira, N.V.; Rego, A.M.; Barbosa, P.C.; Machado, R.F.; Pimentel, A.; dos Reis, L.M.; de Pina, L.C.; Redner, P.; de Souza Caldas, P.C.; et al. Integrated analysis of ethionamide resistance loci in Mycobacterium tuberculosis clinical isolates. Tuberculosis 2018, 113, 163–174. [Google Scholar] [CrossRef]
- Fraaije, M.W.; Kamerbeek, N.M.; Heidekamp, A.J.; Fortin, R.; Janssen, D.B. The prodrug activator EtaA from Mycobacterium tuberculosis is a Baeyer-Villiger monooxygenase. J. Biol. Chem. 2004, 279, 3354–3360. [Google Scholar] [CrossRef]
- Leisch, H.; Shi, R.; Grosse, S.; Morley, K.; Bergeron, H.; Cygler, M.; Iwaki, H.; Hasegawa, Y.; Lau, P.C. Cloning, baeyer-villiger biooxidations, and structures of the camphor pathway 2-oxo-δ3-4, 5, 5-trimethylcyclopentenylacetyl-coenzyme a monooxygenase of Pseudomonas putida ATCC 17453. Appl. Environ. Microbiol. 2012, 78, 2200–2212. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Mirza, I.A.; Yachnin, B.J.; Wang, S.; Grosse, S.; Bergeron, H.; Imura, A.; Iwaki, H.; Hasegawa, Y.; Lau, P.C.; Berghuis, A.M. Crystal structures of cyclohexanone monooxygenase reveal complex domain movements and a sliding cofactor. J. Am. Chem. Soc. 2009, 131, 8848–8854. [Google Scholar] [CrossRef] [PubMed]
- Fürst, M.J.; Fiorentini, F.; Fraaije, M.W. Beyond active site residues: Overall structural dynamics control catalysis in flavin-containing and heme-containing monooxygenases. Curr. Opin. Struct. Biol. 2019, 59, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Minerdi, D.; Zgrablic, I.; Sadeghi, S.J.; Gilardi, G. Identification of a novel Baeyer-Villiger monooxygenase from Acinetobacter radioresistens: Close relationship to the Mycobacterium tuberculosis prodrug activator EtaA. Microb. Biotech. 2012, 5, 700–716. [Google Scholar] [CrossRef]
- Heinig, M.; Frishman, D. STRIDE: A Web server for secondary structure assignment from known atomic coordinates of proteins. Nucl. Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef]
- Malito, E.; Alfieri, A.; Fraaije, M.W.; Mattevi, A. Crystal structure of a Baeyer–Villiger monooxygenase. Proc. Natl. Acad. Sci. USA 2004, 101, 13157–13162. [Google Scholar] [CrossRef]
- Leung, K.L.; Yip, C.W.; Yeung, Y.L.; Wong, K.L.; Chan, W.Y.; Chan, M.Y.; Kam, K.M. Usefulness of resistant gene markers for predicting treatment outcome on second-line antituberculosis drugs. J. Appl. Microbiol. 2010, 109, 2087–2094. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comp. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Lee, T.-S.; Hu, Y.; Sherborne, B.; Guo, Z.; York, D.M. Toward fast and accurate binding affinity prediction with pmemdGTI: An efficient implementation of GPU-accelerated thermodynamic integration. J. Chem. Theory Comput. 2017, 13, 3077–3084. [Google Scholar] [CrossRef]
- Kaus, J.W.; Pierce, L.T.; Walker, R.C.; McCammon, J.A. Improving the efficiency of free energy calculations in the amber molecular dynamics package. J. Chem. Theory Comput. 2013, 9, 4131–4139. [Google Scholar] [CrossRef]
- Mermelstein, D.J.; Lin, C.; Nelson, G.; Kretsch, R.; McCammon, J.A.; Walker, R.C. Fast and flexible gpu accelerated binding free energy calculations within the amber molecular dynamics package. J. Comput. Chem. 2018, 39, 1354–1358. [Google Scholar] [CrossRef] [PubMed]
- Klimovich, P.V.; Shirts, M.R.; Mobley, D.L. Guidelines for the analysis of free energy calculations. J. Comput. Aided Mol. Des. 2015, 29, 397–411. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EthA Conformation | ETH Interaction | Interaction Energy (Kcal/mol) |
|---|---|---|
| 01346 | NADP | −4.9 |
| 13841 | R456 | −5.6 |
| 14053 | D290, Q291, L293, R456, NADP | −5.5 |
| Molecules | 01346 (Å) | 13841 (Å) | 14053 (Å) |
|---|---|---|---|
| EthA | 2.3 ± 0.3 | 2.1 ± 0.4 | 2.0 ± 0.4 |
| ETH | 7.0 ± 1.2 | 4.9 ± 3.0 | 2.9 ± 1.1 |
| NADP | 1.6 ± 0.3 | 1.2 ± 0.3 | 1.2 ± 0.2 |
| FADH2 | 1.7 ± 0.8 | 1.4 ± 0.3 | 2.9 ± 0.7 |
| Residue Pairs | Occupancy (%) | |||
|---|---|---|---|---|
| Donor | Acceptor | 01346 | 13841 | 14053 |
| R456-Side | ETH | - | 27.55 | 64.46 |
| ETH | Q291-Main | 0.11 | 13.51 | 62.47 |
| ETH | T453-Side | - | 11.79 | 26.12 |
| ETH | C294-Main | 12.70 | - | - |
| ETH | A237-Main | 19.24 | - | - |
| W240-Main | ETH | 10.59 | - | - |
| Y50 ↔ C50 | T453 ↔ I453 | ||||
|---|---|---|---|---|---|
| APO | HOLO | APO | HOLO | ||
| Forward | ΔGrecharge | −17.92 ± 0.09 | −17.45 ± 0.09 | −27.29 ± 0.08 | −29.32 ± 0.08 |
| ΔGvdw | 123.82 ± 0.27 | 127.45 ± 0.27 | 144.34 ± 0.22 | 155.08 ± 0.25 | |
| Backward* | ΔGrecharge | −19.40 ± 0.08 | −19.43 ± 0.08 | −27.52 ± 0.07 | −30.08 ± 0.07 |
| ΔGvdw | 124.93 ± 0.28 | 127.55 ± 0.23 | 144.20 ± 0.24 | 154.29 ± 0.27 | |
| Final | 105.72 ± 0.41 | 109.06± 0.38 | 116.87± 0.34 | 124.98 ± 0.38 | |
| ΔΔG (kcal/mol) | 3.34 ± 0.55 | 8.11 ± 0.51 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, V.C.; Antunes, D.; Santos, L.H.S.; Goliatt, P.V.Z.C.; Caffarena, E.R.; Guimarães, A.C.R.; Galvão, T.C. Insights into the Mechanism of Ethionamide Resistance in Mycobacterium tuberculosis through an in silico Structural Evaluation of EthA and Mutants Identified in Clinical Isolates. Catalysts 2020, 10, 543. https://doi.org/10.3390/catal10050543
de Souza VC, Antunes D, Santos LHS, Goliatt PVZC, Caffarena ER, Guimarães ACR, Galvão TC. Insights into the Mechanism of Ethionamide Resistance in Mycobacterium tuberculosis through an in silico Structural Evaluation of EthA and Mutants Identified in Clinical Isolates. Catalysts. 2020; 10(5):543. https://doi.org/10.3390/catal10050543
Chicago/Turabian Stylede Souza, Vinicius Carius, Deborah Antunes, Lucianna H.S. Santos, Priscila Vanessa Zabala Capriles Goliatt, Ernesto Raul Caffarena, Ana Carolina Ramos Guimarães, and Teca Calcagno Galvão. 2020. "Insights into the Mechanism of Ethionamide Resistance in Mycobacterium tuberculosis through an in silico Structural Evaluation of EthA and Mutants Identified in Clinical Isolates" Catalysts 10, no. 5: 543. https://doi.org/10.3390/catal10050543
APA Stylede Souza, V. C., Antunes, D., Santos, L. H. S., Goliatt, P. V. Z. C., Caffarena, E. R., Guimarães, A. C. R., & Galvão, T. C. (2020). Insights into the Mechanism of Ethionamide Resistance in Mycobacterium tuberculosis through an in silico Structural Evaluation of EthA and Mutants Identified in Clinical Isolates. Catalysts, 10(5), 543. https://doi.org/10.3390/catal10050543