1. Introduction
Baeyer-Villiger monooxygenases (BVMOs) are flavin-dependent enzymes that catalyze the conversion of ketones to esters [
1,
2]. Naturally, they enable the use of alternative carbon sources or the synthesis of various secondary metabolites by microorganisms [
3]. However, their use as alternatives to chemical Baeyer-Villiger catalysts, and their ability to also catalyze heteroatom oxidation, has driven much of the research over the past two decades. This has led the search for new and improved BVMOs to expand on the current substrate-scope, enhanced regio-, enantio- and stereospecificity/selectivity, as well as stability of these promising biocatalysts [
1,
4,
5,
6].
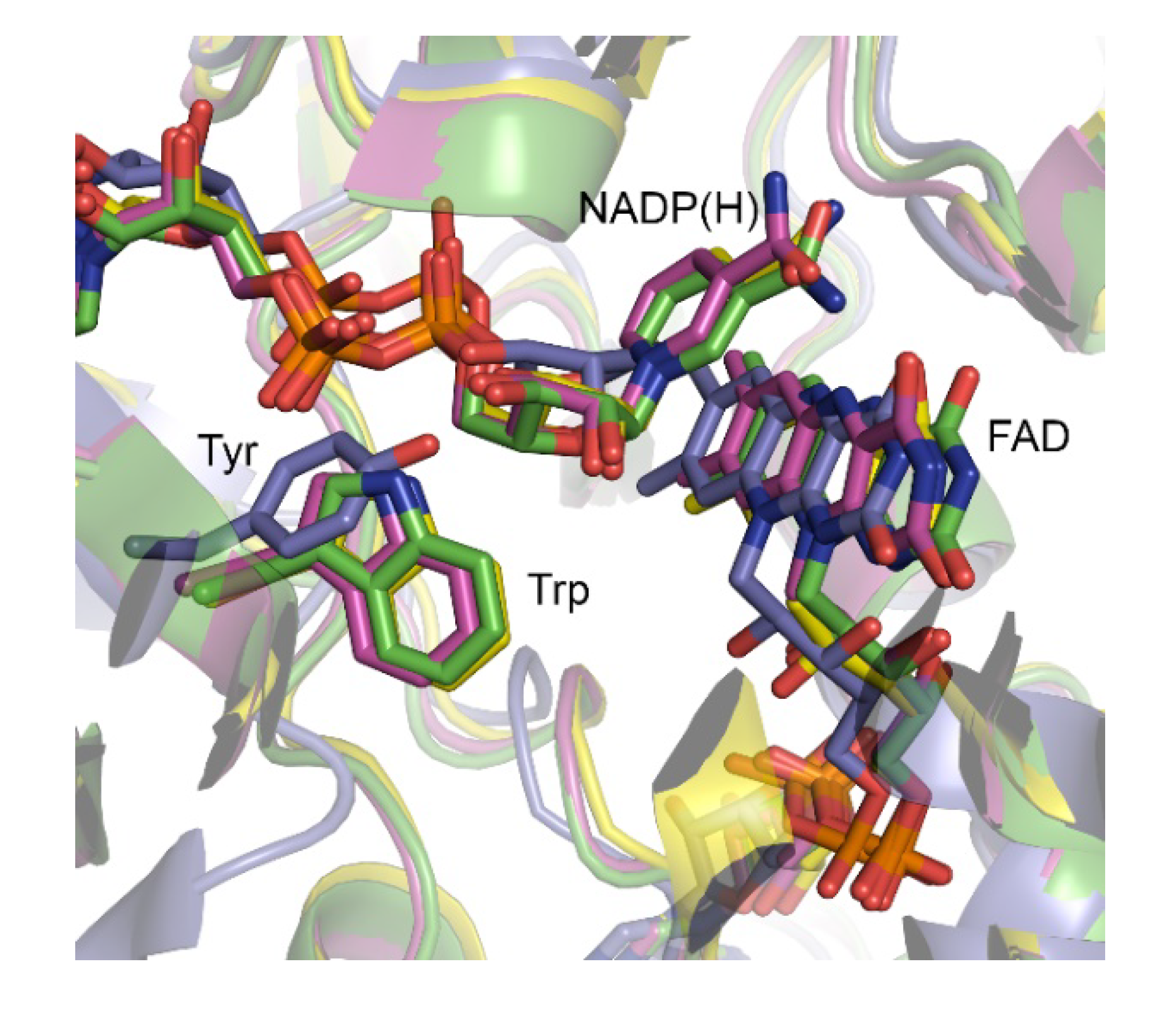
In general, BVMOs collectively refer to a group of structurally unrelated flavin-containing monooxygenases, typically identified either as Type I, II, and O (atypical) BVMOs [
2]. Type I BVMOs belong to group B of the flavin-dependent monooxygenases that bind flavin adenine dinucleotide (FAD) as co-factor. Mechanistically, enzyme-bound FAD is reduced by NAD(P)H, and after molecular oxygen is activated through the formation of a peroxyflavin, which then nucleophilically adds to the carbonyl group of the substrate. The reaction mechanism and structure of type I BVMOs have been extensively studied, with cyclohexanone monooxygenase (CHMO) [
7,
8,
9,
10] and phenylacetone monooxygenase (PAMO) [
11,
12,
13] typically serving as the prototypes. Co-crystallization studies have revealed BVMOs to be highly flexible enzymes, able to undergo large domain movements during catalysis. In the absence of NADPH and substrate, the enzyme is in an ‘open’ conformation. Upon the binding of NADPH, several linker regions are flexed to position the nicotinamide moiety of NADPH in proximity to the isoalloxazine ring for reduction of FAD, which is stabilized pre- and post-oxygen activation by the oxidized NADP
+ and the active site arginine. The NADP
+ molecule is positioned further away from the FAD and the substrate is guided towards the active site by a ‘control’ loop that becomes structured, and the substrate adopts the Criegee conformation. Post BV oxidation, the loop relaxes, allowing the product to diffuse out of the active site. As NADP(H) remains bound during the entire catalytic cycle, the conformation of the ’control loop‘ is crucial in understanding its role during catalysis [
10,
14]. Central to the ’control loop’ is a conserved tryptophan (W492 in CHMO) which interacts with NADP(H). It has been postulated that it acts as a gatekeeper, preventing dissociation of the cofactor or substrate during catalysis. Indeed, mutation of the W492 to alanine (A) in CHMO resulted in reduced activity (14%) with cyclohexanone [
7] and increased uncoupling [
9]. Surprisingly, an altered selectivity was observed with the four substrates tested [
9]. This, together with small-angle X-ray scattering (SAXS) and targeted mutation studies, suggests that the control loop has a more complex role, affecting not only overall structure, but also the active site environment [
10].
Our research group has explored the structure-function relationships of paralogous BVMOs from
Aspergillus flavus [
15,
16,
17]. One of the
A. flavus BVMOs, BVMO
AFL210, does not contain this conserved W in its control loop, but rather a threonine (T) in the equivalent position. Despite this natural variation, BVMO
AFL210 displays high activity towards a variety of substrates [
17]. Here, we describe the structural and catalytic characterization of this BVMO and the role of T513 on its activity and selectivity.
2. Results
BVMO
AFL210 is a versatile BVMO, able to convert 24 of the 26 substrates previously tested, with the exceptions being cylcooctanone and cyclododecanone [
17]. Multiple sequence alignments revealed the two conserved Rossmann fold motifs (GxGxxG) as well the conserved type I BVMO fingerprint 1 [
18] and 2 motifs [
19]. BVMO
AFL210, however, contains a Thr rather than the conserved Trp usually observed in the ‘control loop’. Trp typically serves in contacting and positioning the NADP(H) during catalysis via its nicotinamide-linked ribose unit (
Figure 1). More extensive comparison with the 120 characterized type I BVMOs showed that although this position is decidedly conserved in a large group of BVMOs and typically occupied by a tryptophan, variations do occur. The lack of structural data, and poor alignments due to variation in the sequence length of the ‘control loop’ in these BVMOs, does however leave uncertainty as to the correlations between sequence and structure. Structural studies with polycyclic ketone monooxygenase (PockeMO) revealed a Tyr to occupy the equivalent position in structure space [
20]. In an attempt to elucidate the role of Thr513 in the ‘control loop’ of BVMO
AFL210, we therefore turned to determining the structure using X-ray crystallography.
2.1. Overall Structure and Catalytic site of BVMOAFL210
BVMO
AFL210 was expressed in
E. coli as monomeric C-terminally hexa-histidine tagged protein and purified to near homogeneity using immobilized metal affinity and size-exclusion chromatography. The purified protein was co-crystallized with an excess of NADP
+, with yellow crystals which diffracted to 2.09 Å forming within 1 week (
Supplementary Table S1). The protein crystallized in spacegroup
P2
1 and contained 4 copies in the asymmetric unit (ASU). The structure was solved by molecular replacement using BVMO
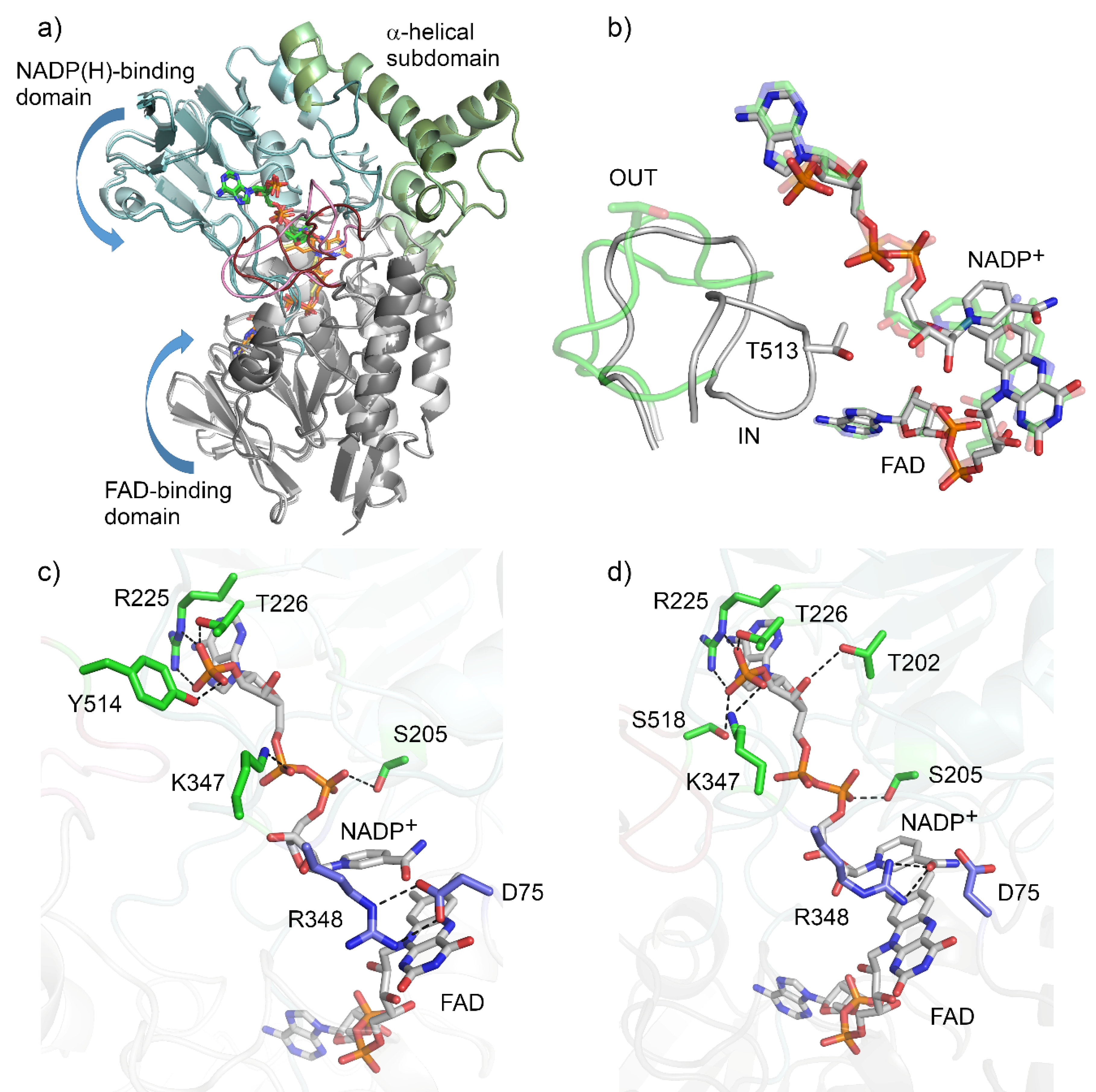
AFL838 (48% sequence identity) as the search model. Conformational heterogeneity was observed as two distinct conformations were adopted, with chains A and C in an ‘open’ conformation, and chains B and D in a ‘closed’ or ‘tight’ conformation (
Figure 2a). Of the four copies in the ASU, one (chain D) had poor density, especially for the ‘control loop’, confirming the general mobility of this loop. Much of chain D was therefore inferred from non-crystallographic symmetry based on chain B.
The structure of BVMO
AFL210 shares an overall fold with the type I BVMOs from bacteria and fungi, which is a Rossmann or GR2 fold that consists of an FAD-binding domain and an NADPH-binding domain (
Figure 2). The first 17 residues, as well as the C-terminal hexa-histidine, were not modelled due to a lack of electron density. The NADP(H)-binding domain consisted of residues 170–224 and 355–397, and contained the type I BVMO fingerprint motif (FXGXXXHXXXWP, residues 175–186), while the α-helical subdomain spanned residues 240–335. Residues 17–156 and 405–555 formed the FAD binding domain. The domains are connected with four linker regions composed of residues 157–169, 225–239, 336–354 and 398–404. Good electron density was observed for the bound FAD and NADP
+ (
Figure S1). The adenine portion of the FAD is anchored within the FAD binding domain by hydrogen bonds to side chains of conserved residues (E55, W66 and Y81) and to the backbone of the main chain. While the
si face of the isoalloxazine ring points toward the FAD binding domain, the
re face is positioned towards the active site. NADP
+ is bound to the NADP-binding domain by hydrogen bonds with the main-chain and side-chains of the flexible linkers (R225, T226, K347), the NADP-binding domain (S205) and the control loop in both conformations (
Figure 2c,d). In the open conformation, NADP
+ is bound by additional hydrogen bonds with adenosine phosphate and the side-chain of Y514 from the ‘control loop’. However, in the closed conformation, these hydrogen bonds are formed by S518 and the rotated side chain of K347. The nicotinamide moiety of the NADP
+ is now positioned deeper into the active site, interacting with R348 (
Figure 2b).
The second type I BVMO signature motif (GGXWXXXXYPGXXXD) spans residues 61–75 and shapes the one side of the FAD binding site, also contributing D75 which is located above the re face of the isoalloxazine ring of FAD and is critical for catalysis. In BVMOAFL210, R348 is responsible for stabilizing the peroxyanion and the Criegee intermediate, and forms two hydrogen-bonded salt bridges with D75 when located in the OUT position (chain A). Both D75 and R348 adopt alternative conformations in the IN position (chain B), with D75 rotating away from the FAD and R348 adopting a conformation which brings it closer to C4a of the FAD.
The active site of BVMO
AFL210 contains a bulge at residues G293–F294 that is present in PAMO [
13] and BVMO
AFL838, but absent in CHMO, and has been implicated in the narrowed substrate scope of PAMO [
21]. BVMO
AFL210, in contrast to BVMO
AFL838, does convert small cyclic and substituted cyclic ketones; in addition to ketones converted by both BVMOs, including linear, aromatic, and the bicyclic ketone,
rac-
cis-bicyclo[3.2.0]hept-2-en-6-one.
The ‘control loop’ (residues 508–525) is often disordered in the absence of NADP(H). Density for the ‘control loop’ (residues 508–525) could be resolved for both conformations of BVMO
AFL210. In the open conformation (chain A), the loop adopts an extended, solvent-exposed unstructured loop (
Figure 2b), similar to that seen with PAMO in the absence of NADP(H) [
13]. In this conformation, T513 is also solvent exposed and not located close to the active or NADPH binding sites. In the closed conformation, the ‘control loop’ is folded inwards, with Thr513 assuming a position structurally equivalent to that of W492 in CHMO. However, unlike W492 in CHMO, T513 of BVMO
AFL210 is not within hydrogen bonding distance of the NADP
+.
Despite various attempts to obtain crystal structures of BVMOAFL210 with NADP+ and different substrates, no electron density for substrates could be observed in any of the structures that were co-crystallized with these compounds. We therefore decided to probe the role of Thr513 using site-directed mutagenesis.
2.2. Mutation of T513 and the Effect on Activity
T513 was mutated to glycine (T513G), tyrosine (T513Y) and tryptophan (T513W). All the mutants expressed as soluble enzymes (
Figure S2) and were evaluated using whole-cell biotransformations to determine whether the mutations altered the substrate scope. The substrate acceptance profiles of the mutants were nearly identical to that of the wild-type (WT) BVMO
AFL210 (
Supplementary Table S2). However, while acetophenone was converted by the wild-type and T513G (10%–20% conversion) only trace amounts of conversion (<1%) could be detected for both T513Y and T513W. A significant reduction of conversion was also observed for 4-(4-hydroxyphenyl)-2-butanone by all the mutants. Contrary to expected, the mutation to T513G did not expand the active site to accommodate larger cyclic ketones, such as cyclooctanone and cyclododecanone, which are not accepted by the wild-type. As the expression levels between the WT and the mutants differed significantly (
Supplementary Figure S1), the WT and the three mutants were purified, and their specific activity determined. As mutations of W492 in CHMO have been shown to significantly affect ‘uncoupling’, we first evaluated the mutants’ NADPH consumption rates in the absence of substrate. Lower stability of the peroxyflavin was observed for the bulkier substitutions (Trp and Tyr), with NADPH reduction rates in the absence of substrate increasing to 0.27 s
−1 and 0.35 s
−1 respectively, compared to the WT (0.07 s
−1). Mutagenesis to a glycine had the lowest effect (0.10 s
−1).
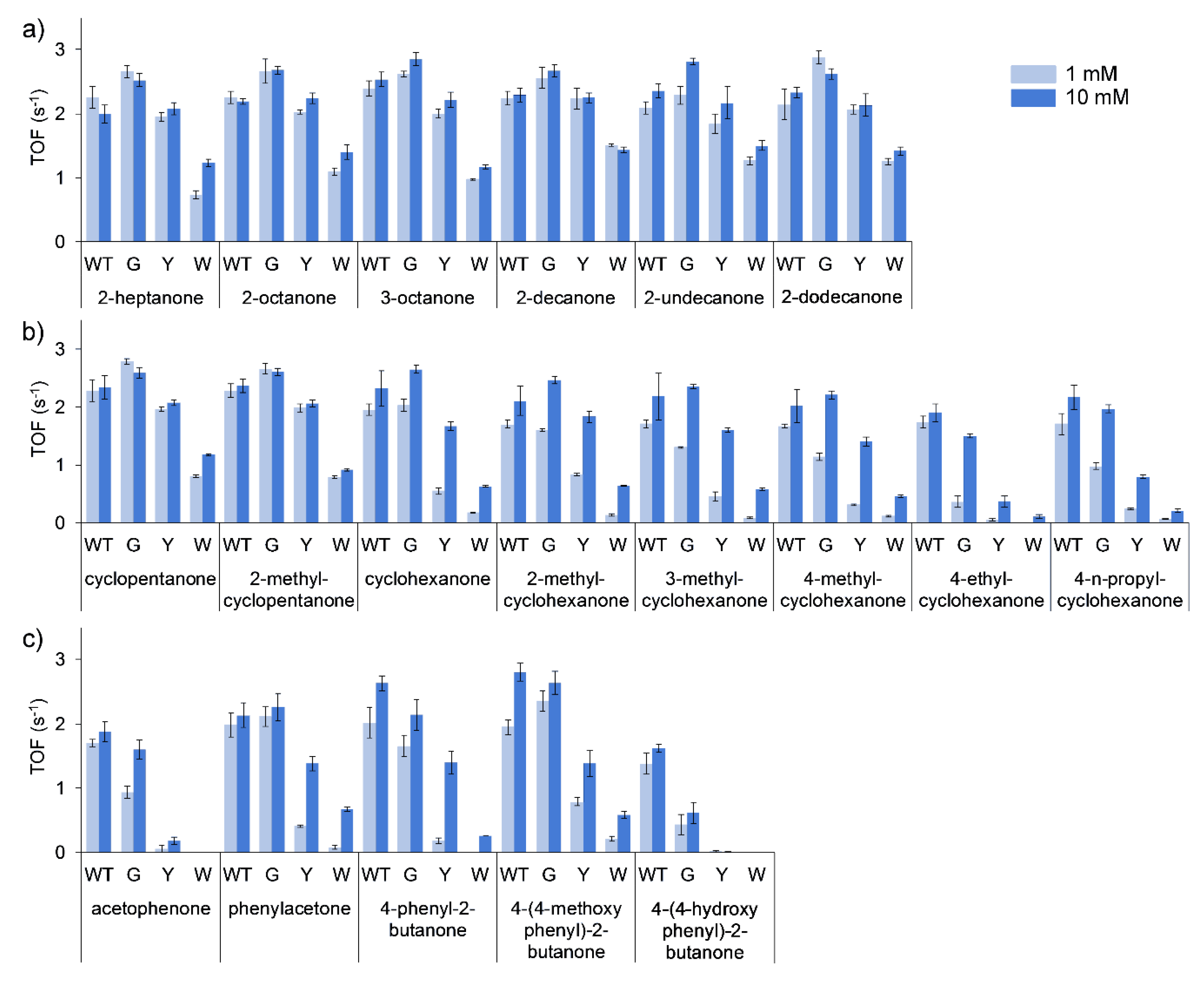
Turnover frequencies (TOFs) of all the purified enzymes were performed using 1 and 10 mM of substrate. The general trend for all of the substrates evaluated was a decrease in specific activity (TOF) by the purified mutants in the order of G > Y > W. This was the least pronounced in the conversion of linear ketones (
Figure 3a), with TOFs of T513G equal to marginally higher than that of the WT, and T513Y equal or marginally lower than that of the WT. T513W, however, only retained approximately half of the catalytic activity observed for the WT. More marked differences in the conversion of cyclic (
Figure 3b) and aromatic ketones (
Figure 3c) were observed, with considerably decreased activity seen for T513Y and T513W for certain substrates such as acetophenone, 4-phenyl-2-butanone and 4-ethylcyclohexanone. In mutation studies with CHMO, W492A mutants only retained 14% of activity towards cyclohexanone, however, mutation of residue 513 to glycine in BVMO
AFL210 showed that the mutant largely retained activity in most of the cases, especially in reactions with 10 mM of substrate. Additionally, large increases in the observed
KM for certain substrates can be assumed for the mutants, such as cyclohexanone and the substituted cyclohexanones, phenylacetone and 4-phenyl-2-butanone.
2.3. Mutation of T513 and the Effect on Selectivity
The absolute regioselectivity of wild-type BVMO
AFL210 towards linear and aromatic ketones were retained by the mutants. The regioselectivity of the variants towards racemic mixtures of 2-methylcyclopentanone, 2-methylcyclohexanone and
cis-bicyclo[3.2.0]hept-2-en-6-one was, however, altered (
Figure 4a–c). In general, the T513G mutant of BVMO
AFL210 did not change the regioselectivity considerably, whereas the mutants with the bulkier side chains were more significant, and also comparable, in effect. In CHMO, a W492A mutant was shown as more selective for all substrates evaluated [
9], however, the change in regioselectivity of the BVMO
AFL210 mutants could not be extended from one substrate to the other, with divergent profiles observed for each substrate. T513Y and T51W showed increased abnormal (distal) lactone formation with 2-methylcyclopentanone and
cis-bicyclo[3.2.0]hept-2-en-6-one but higher regioselectivity towards 2-methylcyclohexanone in the production of the normal (proximal) lactone. To further explore the change in regioselectivity, chiral analysis of the conversion of
rac-2-methylcyclohexanone was performed. Similar to conversions with
rac-
cis-bicyclo[3.2.0]hept-2-en-6-one, BVMO
AFL210 WT showed no kinetic resolution with 2-methylcyclohexanone. Both T513Y and T513W, however, displayed marginal
R-selectivity. In addition, whereas the WT converts
R-2-methylcyclohexanone almost exclusively to the normal lactone, and
S-2-methylcyclohexanone to near equal amounts of the normal and the abnormal lactone, all three mutants displayed increased regioselectivity for the normal lactone from the
S-enantiomer, especially with the bulkier Y and W substitutions (
Supplementary Figure S3).
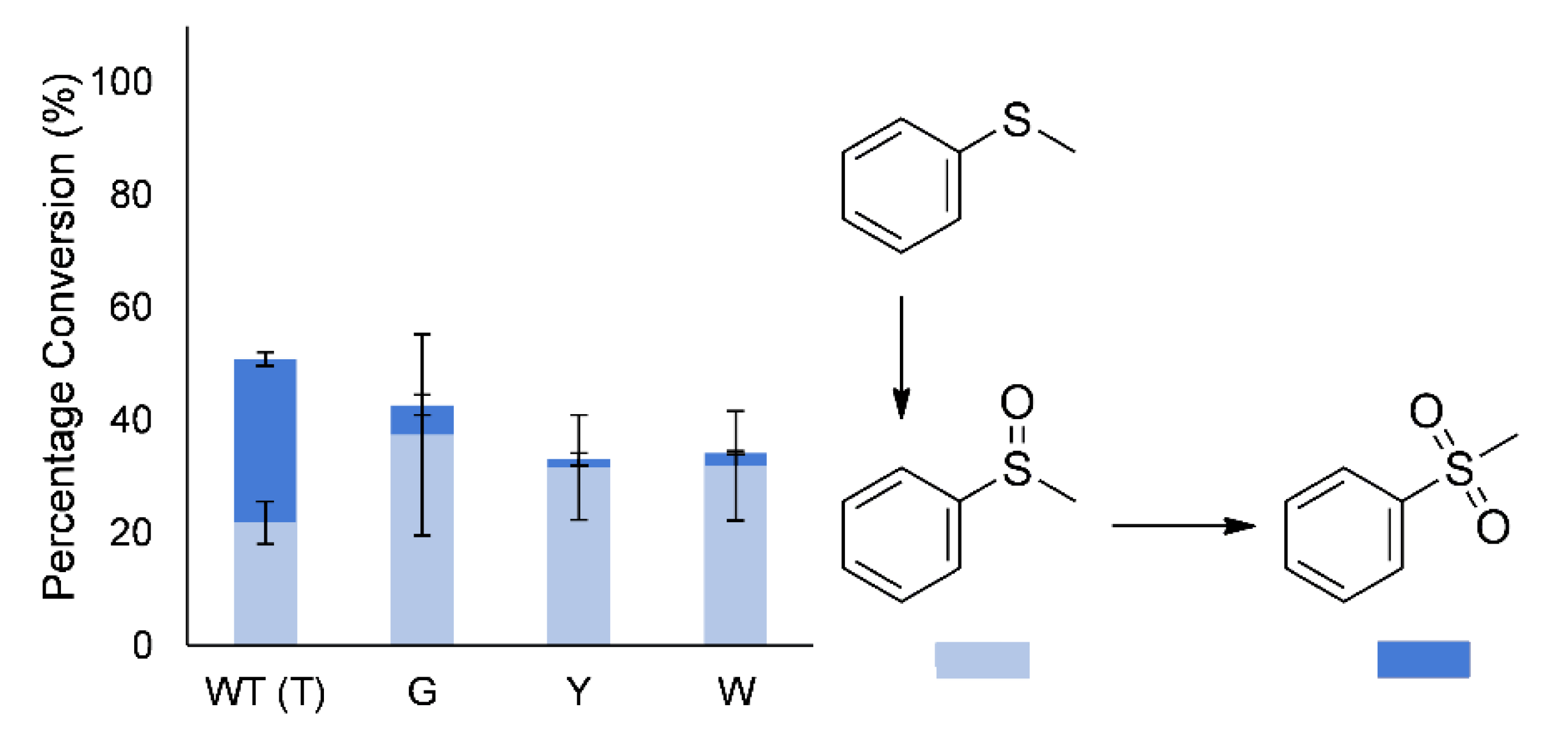
In addition to regio- and enantioselectivity, the sulfoxidation of thioanisole (methyl phenyl sulfide) to the sulfoxide and sulfone had been altered. The wild-type enzyme produces a large amount of the overoxidized sulfone during early conversion, whereas the mutants largely produced mostly the sulfoxide (86%–95% of products;
Figure 5). However, after complete conversion of the thioanisole, the sulfoxide is again further oxidized to the sulfone by the mutants, indicating only a change in selectivity.
3. Discussion
With the exception of directed evolution studies influencing the substrate scope and selectivity/specificity through allosteric effects [
22,
23], protein engineering of BVMOs to improve substrate acceptance or regio-, enantio-, and stereospecificity/selectivity has mostly focused on “hot spot” residues lining the active site [
4,
5]. NADP(H), however, remains bound during the entire catalytic cycle of BVMOs, and early kinetic [
24] and structural studies [
25] on FMO have already revealed its dual function in this class of enzymes. NADP(H) is not only responsible for flavin reduction, but also stabilization of the (hydro)peroxyflavin reaction intermediate [
7,
8,
26,
27], as the nicotinamide ring and adjacent ribose of NADP
+ forms an integral part of the active site. The first crystal structures of CHMO revealed a sliding co-factor, whereby domain rotation and the ordering of a disordered ‘control loop’ slides NADP
+ further into the active site via interaction with W492 [
7]. The complexity of domain movement and NADP(H) position was further uncovered in structural studies with bound substrate [
8] and product [
9]. The ‘control loop’ is postulated to not only have a direct impact on the overall structure but also the active site environment [
10]. Mutation of the ‘control loop’ Trp (W) to Ala (A) in CHMO resulted in reduced activity, underscoring its importance [
7]. The same conserved Trp (W501) of PAMO was also later mutated to Ala, resulting in reduced activity with phenylacetone and a high uncoupling rate (0.4 s
−1), whereafter the authors concluded that W501 should not be targeted in mutant libraries [
28].
BVMO
AFL210 has a ‘control loop’ that is conserved in length with respect to CHMO and PAMO. A Thr (T513), however, occupies the equivalent position to W492 and W501 in CHMO and PAMO, respectively. Crystallization of BVMO
AFL210 revealed two distinct conformations of the ‘control loop’ in the presence of NADP
+. In the ‘open’ conformation, the ‘control loop’ adopts an unstructured loop, with T513 solvent exposed. In other BVMOs in the ‘open’ conformation, the loop is often completely disordered, or can adopt secondary structure such as a partial α-helix [
16] or β-hairpin [
29]. In the ‘closed’ conformation, the loop rearranges as has been observed with other BVMOs [
7,
12,
29,
30], with T513 occupying the same position as the conserved Trp in related BVMOs.
Despite this natural distinction, BVMOAFL210 has a broad substrate scope. Contrary to expectation, mutagenesis of T513 to the typically observed Trp decreased the peroxyflavin stability and reduced the overall specific activity. Surprisingly, the T513G mutant, which would give the most conformational freedom with no predicted contacts with the NADP(H), showed minimal decreased peroxyflavin stability and the lowest effect on specific activity for most substrates. The effect of the T513 mutations were also substrate specific, with T513G retaining nearly the same TOF as the WT with linear ketones. The effect of the glycine mutation was, however, more pronounced in 3- and 4-substituted cyclohexanones and some aryl ketones such as acetophenone. Moreover, the reduced activity was higher at lower substrate concentrations, suggesting a decreased affinity (KM) for these substrates. These substrate and concentration-specific effects were even more evident with the bulkier (T513Y/W) mutations, where the TOF decreased to only approximately 50% for T513W with linear ketones, but substantially with most of the cyclohexanone derivatives and aryl ketones.
The T513 mutations also greatly influenced the regioselectivity with 2-methyl substituted cyclic ketones, as well as cis-bicyclo[3.2.0]hept-2-en-6-one. Whereas substitution of T513 with a bulkier amino acid (W/Y) resulted in lower regioselectivity (increased distal regioisomer) with 2-methylcyclopentanone, improved regioselectivity for the formation of the normal lactone (proximal regioisomer) was observed with the same mutations and 2-methylcyclohexanone. Regioselectivity with rac-cis-bicyclo[3.2.0]hept-2-en-6-one was also affected by the T513 mutations. During sufloxidation of thioanisole, decreased overoxidation (sulfoxide to sulfone) during the early stages of the reaction was observed with all of the T513 mutants.
In co-crystallization studies with CHMO, W492, the corresponding residue of CHMO, does not directly bind cyclohexanone. However, mutation studies with the corresponding residues in CHMO, PAMO (W501) and 2-oxo-Δ
3-4,5,5-trimethylcyclopentenylacetyl-coenzyme A monooxygenase (OTEMO, W501) have revealed that it contributes to the regioselectivity of the enzymes. In OTEMO, W501A/V shifted the conversion of
(-)-cis-bicyclo[3.2.0]hept-2-en-6-one from equimolar products to heavily favour the distal lactone (95%) [
31]. In PAMO, an inverse in the ratio of normal (proximal) to abnormal (distal) lactone from
rac-
cis-bicyclo[3.2.0]hept-2-en-6-one was observed [
28]. Interestingly, directed evolution studies with
TmCHMO targeting the same amino acids in iterative saturation mutagenesis (ISM), found no hits with W492 in the reversal of enantioselectivity during the desymmetrization of the prochiral ketone 4-methylcyclohexanone [
32], but W492Y/F mutations did significantly change the regioselelctiviey with 1-phenyl-2-propanone (phenylacetone) and 1-phenyl-2-butanone [
33]. In silico analysis through substrate docking, molecular dynamics (MD) and quantum mechanical (QM/MM) simulations with density function theory (DFT) of the Criegee intermediate, however, do show the corresponding Trp in related BVMOs to contact the substrate or intermediate, rationalizing its involvement in changes in selectivity/specificity [
30,
33,
34,
35]. In PAMO, the same changes in regioselectivity observed with W501A was also observed in Y502A, of which the side chain would not directly contact the substrate or reaction intermediates [
28]. Changes to the overall ‘control loop’ structure could thus potentially be induced by these mutations, with conformations not yet observed in crystallographic studies actually responsible for the changes observed in selectivity and specificity.
4. Materials and Methods
4.1. Expression and Purification of BVMOAFL210
The cloning of BVMO
AFL210 was described previously [
17]. The C-terminally His-tagged BVMO
AFL210 was expressed in
E. coli BL21(DE3) Gold (Agilent Technologies, Santa Clara, CA, USA) from the pET-22b(+) vector (Novagen/Merck, Darmstadt, Germany) in ZYP5052 auto-induction media [
36] supplemented with 100 μg.mL
−1 ampicillin, for 36 h at 20 °C in an orbital shaker (200 rpm). The cells were harvested and resuspended in immobilized-metal affinity chromatography (IMAC) binding buffer, comprising 50 mM Tris pH 7.4, 500 mM NaCl and 30 mM imidazole. The cells were lysed by single passage through a continuous flow cell disruptor at 4 °C and 30 kPsi (Constant Systems Ltd, Northants, UK). The lysate was centrifuged at 20,000 ×g (4 °C, 30 min) to remove insoluble cell debris, and ultracentrifuged at 90,000 ×g (4 °C, 90 min) to remove the membrane fraction.
The clear lysate was loaded onto a HisTrap FastFlow (FF) column (GE Healthcare, Chicago, IL, USA), equilibrated with IMAC binding buffer. The column was washed with 30 column volumes of binding buffer to remove unbound proteins. The bound proteins were eluted from the IMAC column by using buffer with a linear increasing gradient of imidazole (50 mM Tris pH 7.4, 500 mM NaCl, 30–500 mM imidazole). The purity of the fractions was evaluated using SDS-PAGE. PageRuler Prestained Protein Ladder (Thermo Fisher Scientific, Waltham, MA, USA) was used as molecular weight marker and the gels were stained with Coomassie Brilliant Blue R-250 (Sigma Aldrich, St. Louis, MO, USA) to visualize the proteins. Yellow fractions containing the purified BVMOAFL210 were pooled and concentrated to ~2.5 mL using an Amicon centrifugal ultrafiltration device (30 kDa NMWL, 3500 ×g, 4 °C, Merck-Millipore, Massachusetts, MA, USA). The concentrated fraction was incubated with an excess of FAD for 1 h at 4 °C, after which it was further purified using size-exclusion chromatography (SEC). The fraction was loaded onto a Sephacryl HR-100 size exclusion column (GE Healthcare, Chicago, IL, USA) equilibrated with 20 mM Tris pH 8, 100 mM NaCl, and eluted in the same buffer. Protein concentration was determined with the Pierce BCA kit (Thermo Fisher Scientific, Waltham, MA, USA) using bovine serum albumin (BSA) as standard, as per manufacturer’s instructions.
4.2. Crystallization and Structure Determination of BVMOAFL210
Purified BVMO
AFL210 was crystallized by hanging drop vapour diffusion with equal volumes (1 μL + 1 μL) of protein (16 mg.mL
−1) and precipitant (0.5 M ammonium citrate tribasic pH 9; 27.5% (v/v) PEG3350) at 16 °C, respectively. Crystals were cryocooled with the addition of 12% PEG400 in the mother liquor prior to X-ray diffraction, and data was collected at the Diamond Light Source (UK) at beamline i04-1. Data was indexed and integrated using the autoPROC STARANISO pipeline, using XDS [
37], scaled and merged using Aimless [
38] and STARANISO [
39] for anisotropic correction (ellipsoidal with resolution cutoff criteria - local (
I/σ
I) = 1.2). Molecular replacement was performed with Phaser [
40] using BVMO
AFL838 as search model (PDB ID 5J7X). The structure was refined by cycles of manual building in Coot [
41] and refinement in REFMAC5 [
42]. The structure was validated using Coot and other programs from the CCP4 suite [
43]. Figures were generated in Pymol. Coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under 6Y48.
4.3. Creation of T513 Mutants of BVMOAFL210
Mutants BVMOAFL210_T513Y, BVMOAFL210_T513G and BVMOAFL210_T513W were generated by inverse PCR using pET-22b(+):BVMOAFL210_CTH as template. The PCRs were performed with the KOD Hot Start DNA polymerase kit (Merck, Darmstadt, Germany), as per manufacturer’s instructions, with an annealing temperature of 62.7 °C. The reverse primer sequence was 5′-GGA CTT CGT GGT AGG GAA GAG TGT CTT GTC-3′ and the respective forward primers were 5′-TAC TAC ATG GGT GGT AGC ATG CCC GG-3′ for the T513Y mutant, and 5′-GGC TAC ATG GGT GGT AGC ATG CC-3′ for the T513G mutant, while the forward and reverse primers for the T513W mutant were 5′-GTG GTA GCA TGC CCG GAA AGG TC-3′ and 5′-CC ATG TAC CAG GAC TTC GTG GTA GGG-3′. The PCR products were treated with DpnI to remove template DNA and ligated to circularize the plasmids. E. coli TOP10 cells (Thermo Fisher Scientific, Waltham, MA, USA) were used for plasmid propagation and successful mutagenesis was verified by DNA sequencing of the plasmids.
4.4. Whole-Cell Biotransformations
Recombinant
E. coli BL21Gold(DE3) cells expressing the native or the respective mutant BVMOs were harvested by centrifugation and resuspended in buffer (200 mM Tris pH 8). Reactions were performed in 40 mL amber vials, with 1 mL reaction volumes containing 0.1 g cells, 200 mM Tris pH 8, 100 mM glucose, 100 mM glycerol, 10 mM substrate (
Scheme S1, SI) and 1% (v/v) methanol as co-solvent, in an orbital shaker (200 rpm) at 20 °C. After 8 h, the reactions were stopped and extracted with 1 mL ethyl acetate, containing 2 mM internal standard (1-undecanol or 3-octanol). The substrates and products were evaluated with GC-MS on a Finnigan TRACE GC Ultra-Trace DSQ (Thermo Fisher Scientific, Waltham, MA, USA) with a FactorFour™ VF-5ms column (60 m × 0.32 mm × 0.25 μm, Agilent Technologies, Santa Clara, CA, USA). Detailed GC-MS programs are given in
Table S3. SDS-PAGE analysis was used to evaluate the soluble expression levels of BVMO
AFL210 and the mutants after the cells were treated with lysozyme (1 g L
−1), followed by a freeze thaw cycle, and centrifugation at 20,000 ×g for 20 min, at 4 °C.
4.5. Kinetic Characterization
Turnover frequencies (TOF) were determined spectrophotometrically by monitoring the oxidation of NADPH at 340 nm (ε340nm = 6.22 mM−1 cm−1) in a DU800 UV-Vis spectrophotometer (Beckman Coulter, Brea, CA, USA) using purified enzyme. Reactions contained 0.26–0.395 μM purified enzyme as determined by the ε436nm of 12.65 mM−1 cm−1 for enzyme-bound FAD, 1 or 10 mM of substrate, 0.3 mM NADPH, 1% (v/v) methanol, in 100 mM Tris-HCl buffer (pH 9) containing 100 mM NaCl, at 25 °C.
5. Conclusions
Until recently, the ‘control loop’, and specifically its apparent conserved Trp, has been largely ignored in directed evolution studies due to the often-high uncoupling and reduced activity observed with mutations of Trp. Here, we report on a BVMO from
A. flavus that has a natural variation in the ‘control loop’, whereby a Thr occupy the equivalent position of Trp in similar BVMOs. As was observed with comparable BVMOs, mutation of this position brought about changes in the regioselectivity of the enzyme. A diverse panel of substrates tested revealed these changes to not only be dependent on the mutation (Gly vs. the bulkier amino acids Trp and Tyr) but also the substrate evaluated. Conserved amino acid changes had the smallest detrimental effect (peroxyflavin stability and reduced activity). Moreover, whereas the regioselectivity towards aliphatic and aryl ketones remained absolute between the different variants, changes were observed with 2-methyl substituted cyclic ketones. Mutations to the bulkier amino acids either significantly improved the regioselectivity for ‘normal’ lactone production from 2-methylcyclohexanone or allowed access to more of the ‘abnormal’ lactone from 2-methylcyclopentanone. This was similarly observed in directed evolution studies by Reetz and co-workers [
33], where W492Y in
TmCHMO did result in reduced activity, but at the gain of significantly improved regioselectivity for abnormal lactone products.
Further research is, however, still required to fully elucidate the role of the ‘control loop’ in the selectivity and specificity of BVMOs, as it not only contacts the substrate and intermediates during catalysis, but also positions the NADP+, which lines the active site with its nicotinamide and connected ribose unit.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




