New Trends in Enantioselective Cross-Dehydrogenative Coupling




Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
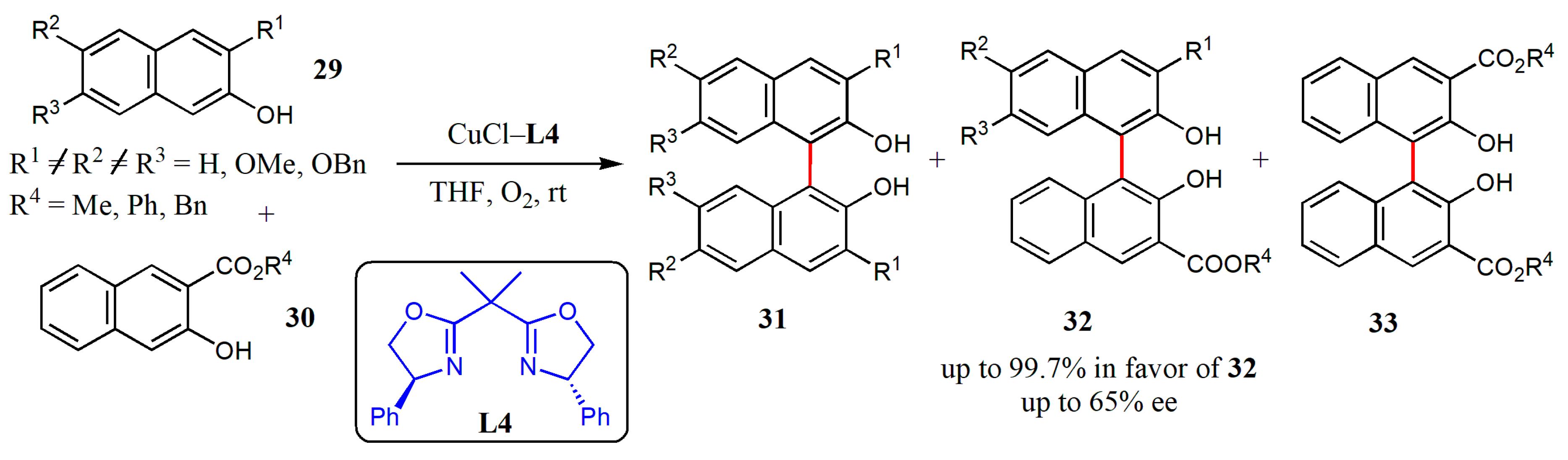{kind=link}
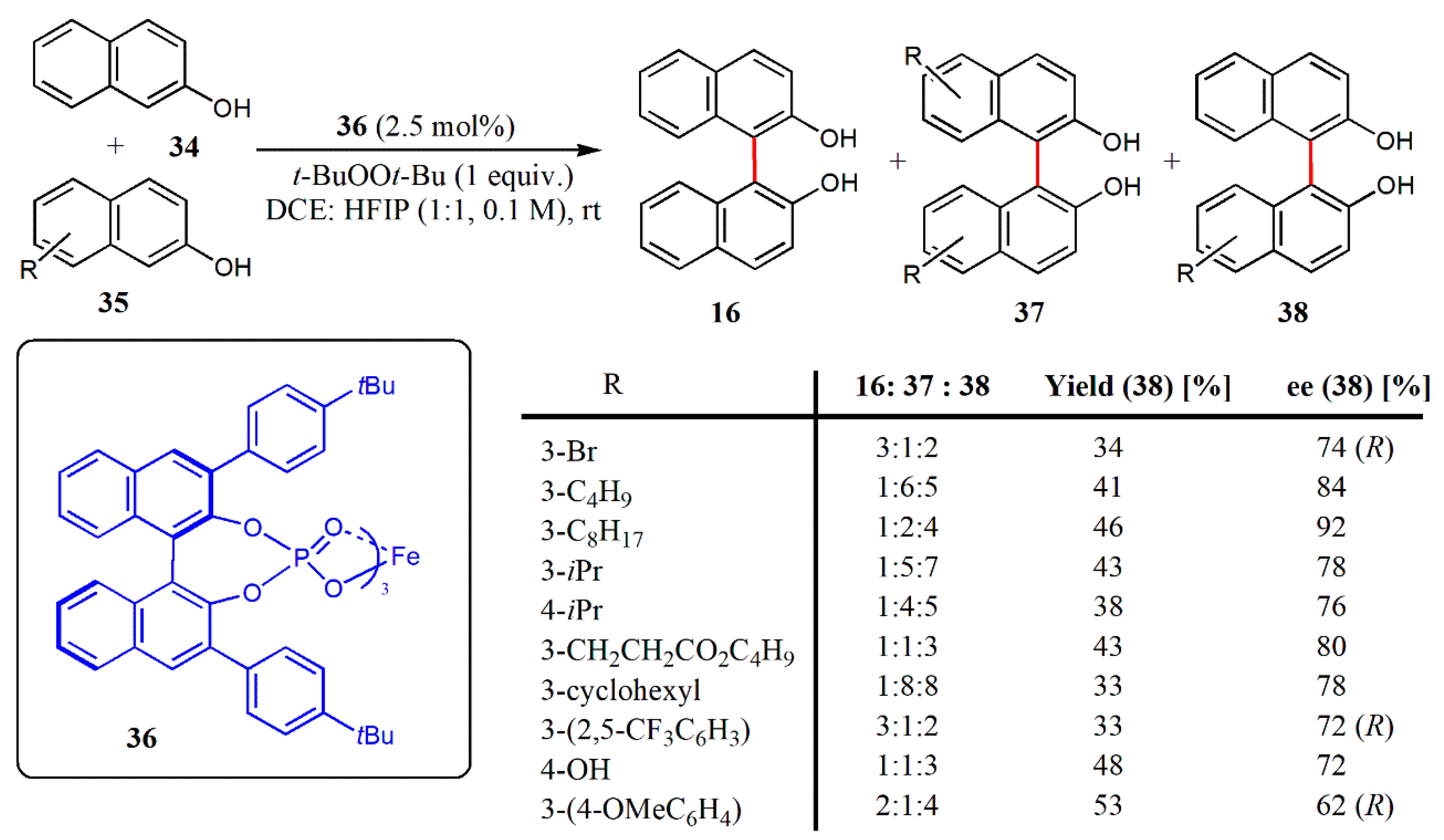{kind=link}
{kind=link}
{kind=link}
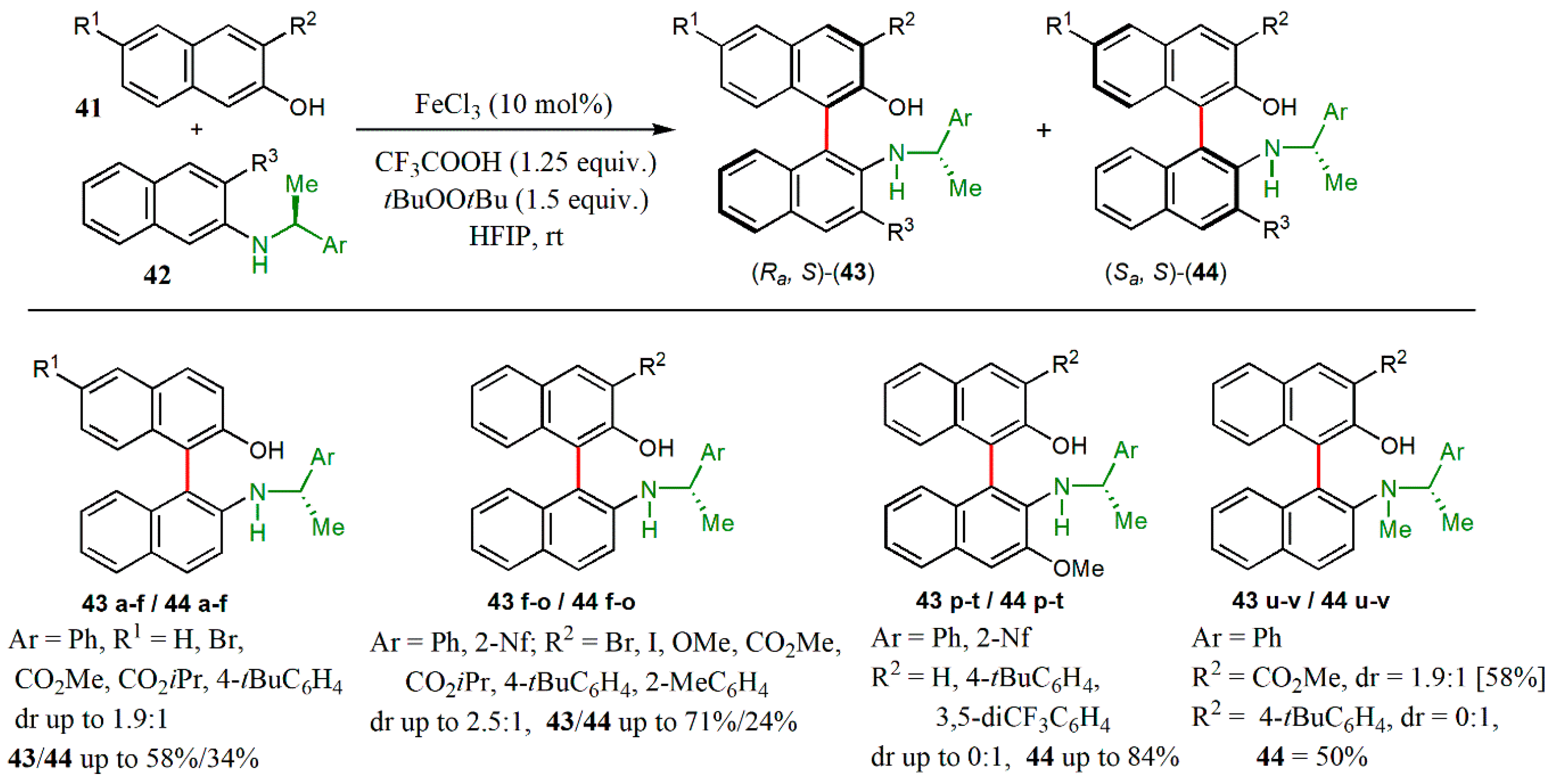{kind=link}
{kind=link}
{kind=link}
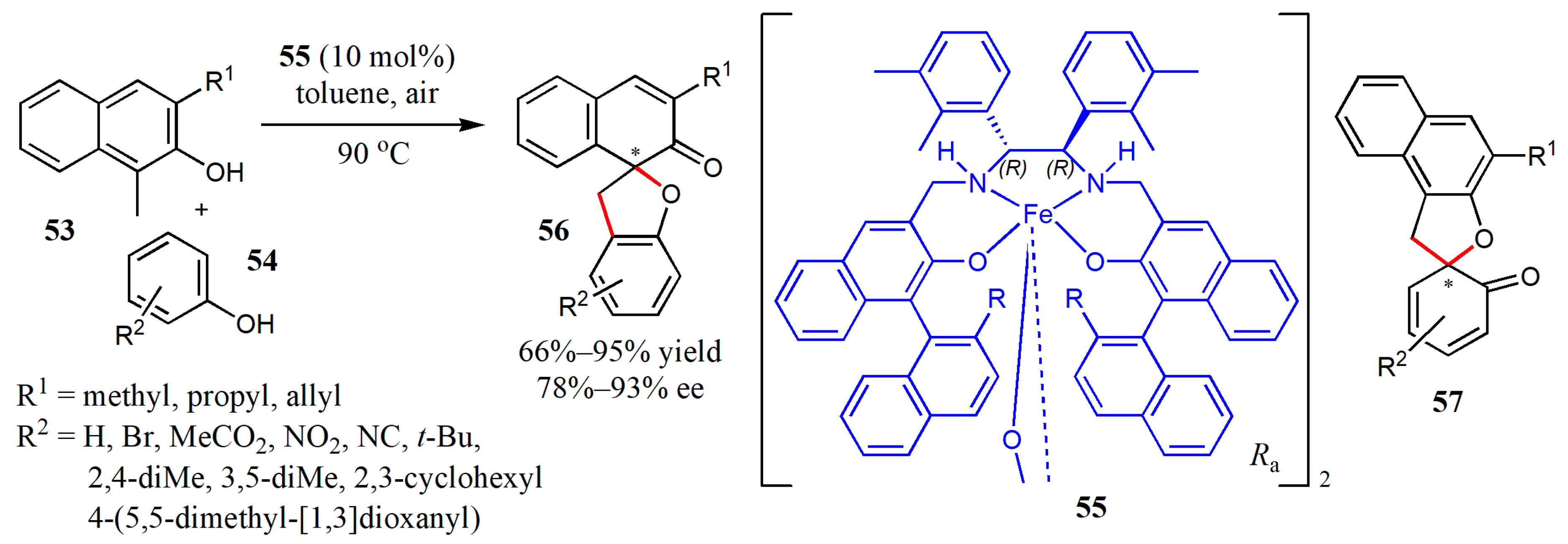{kind=link}
{kind=link}
{kind=link}
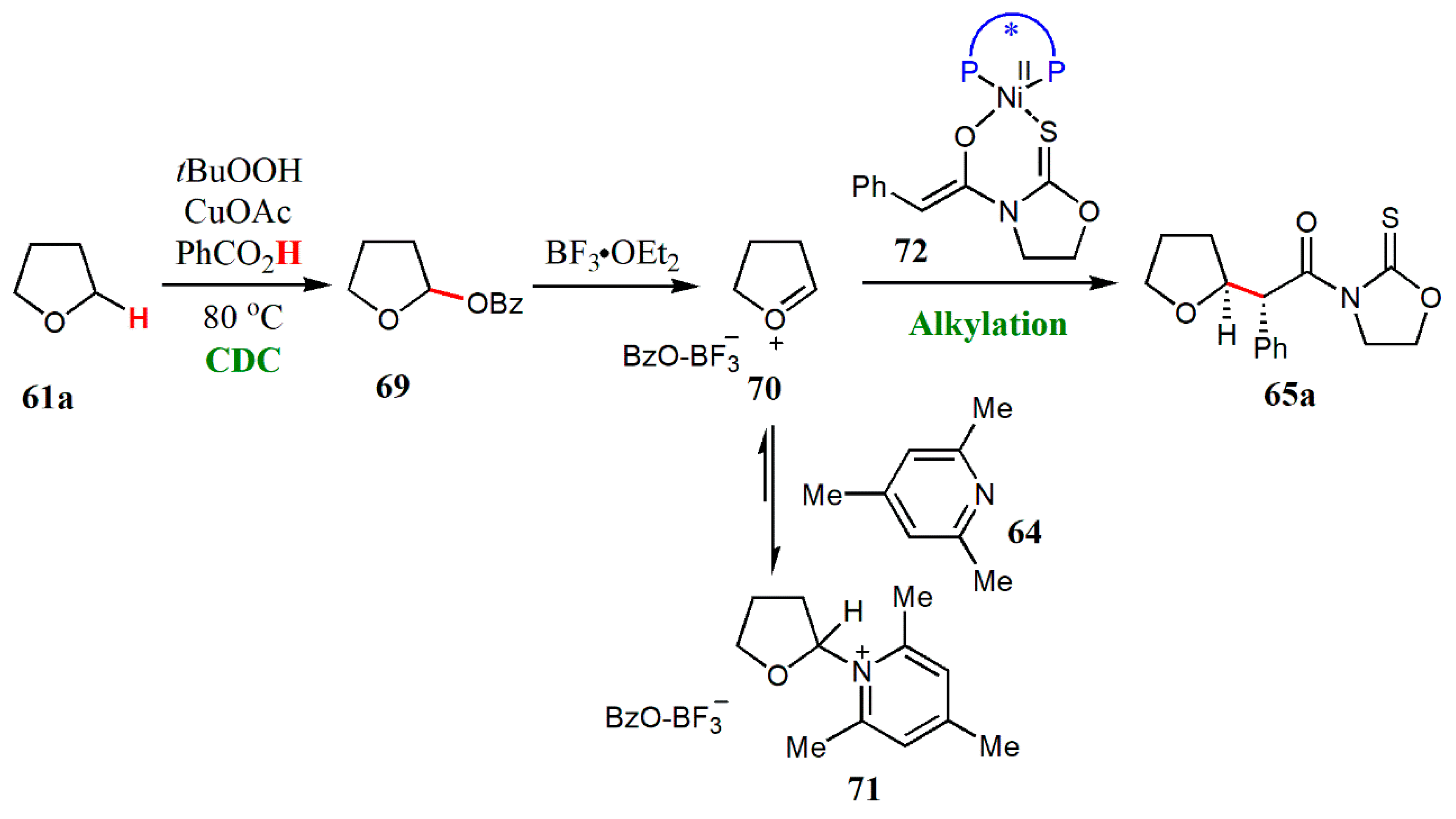{kind=link}
{kind=link}
{kind=link}
{kind=link}
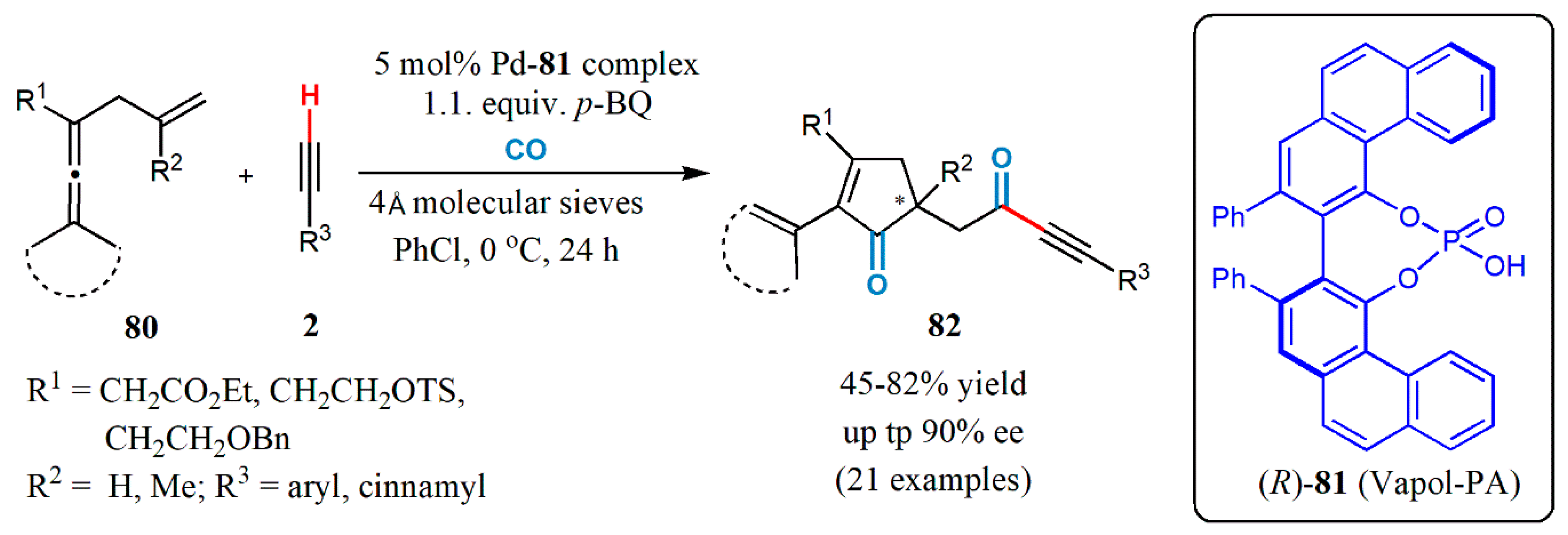{kind=link}
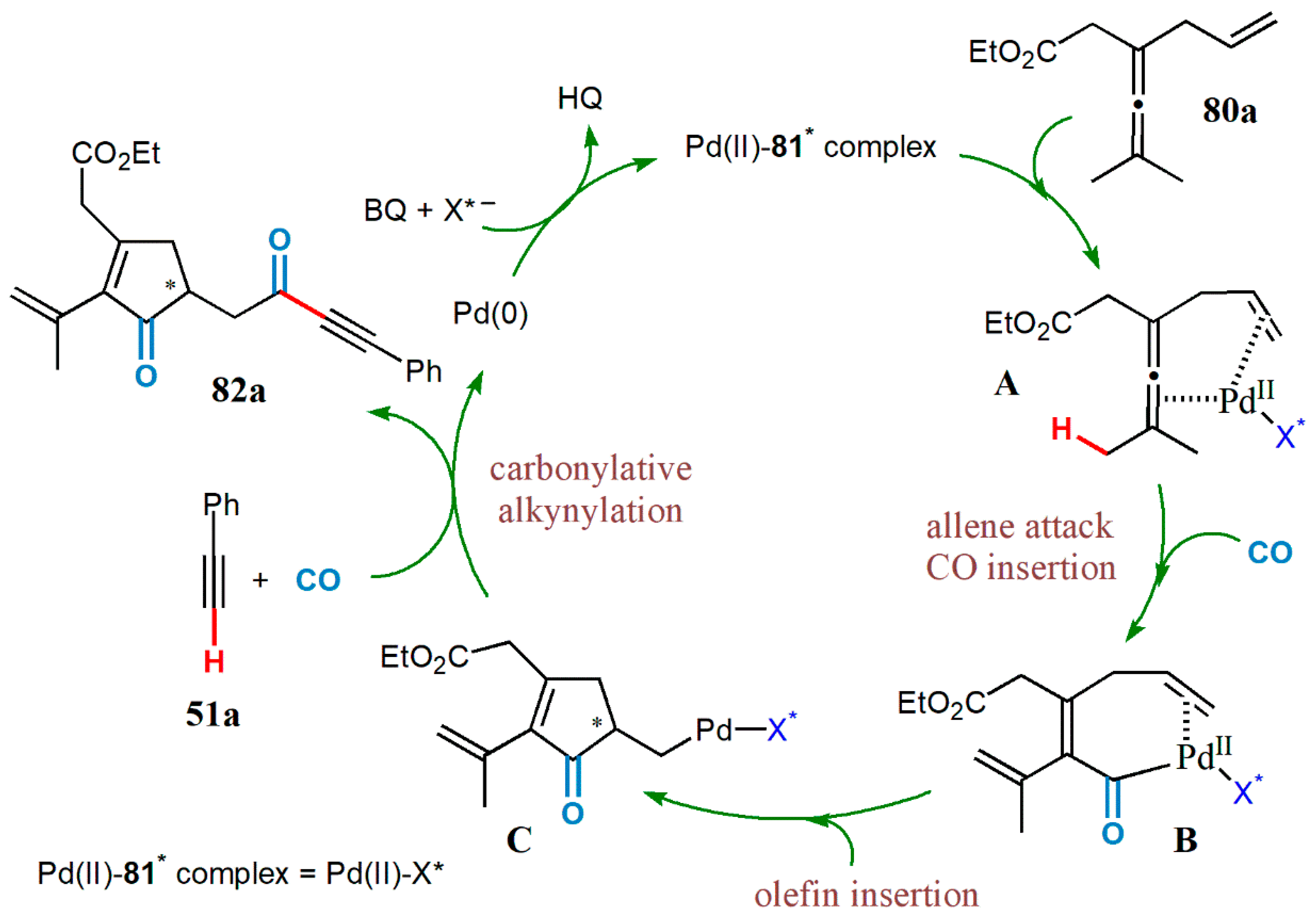{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
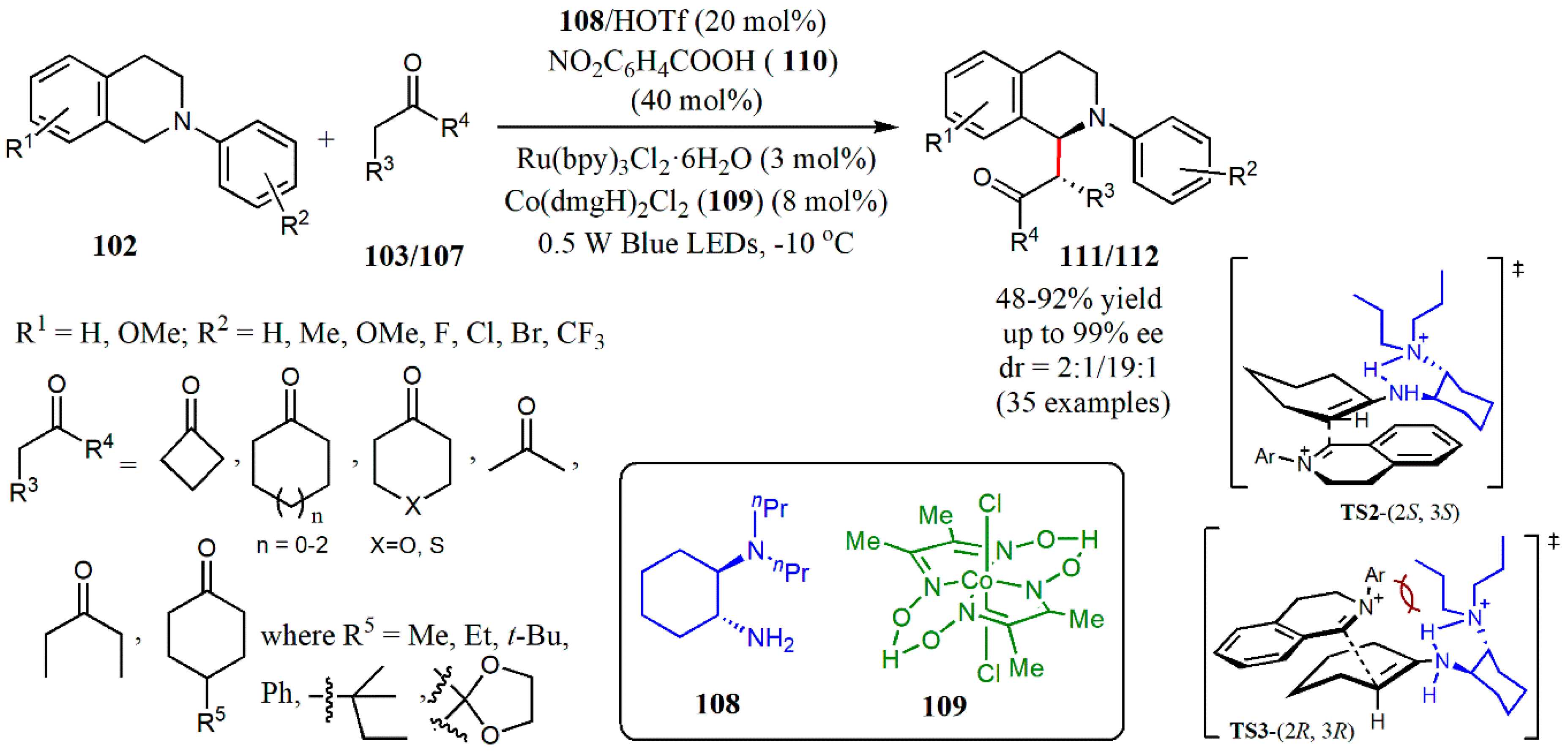{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
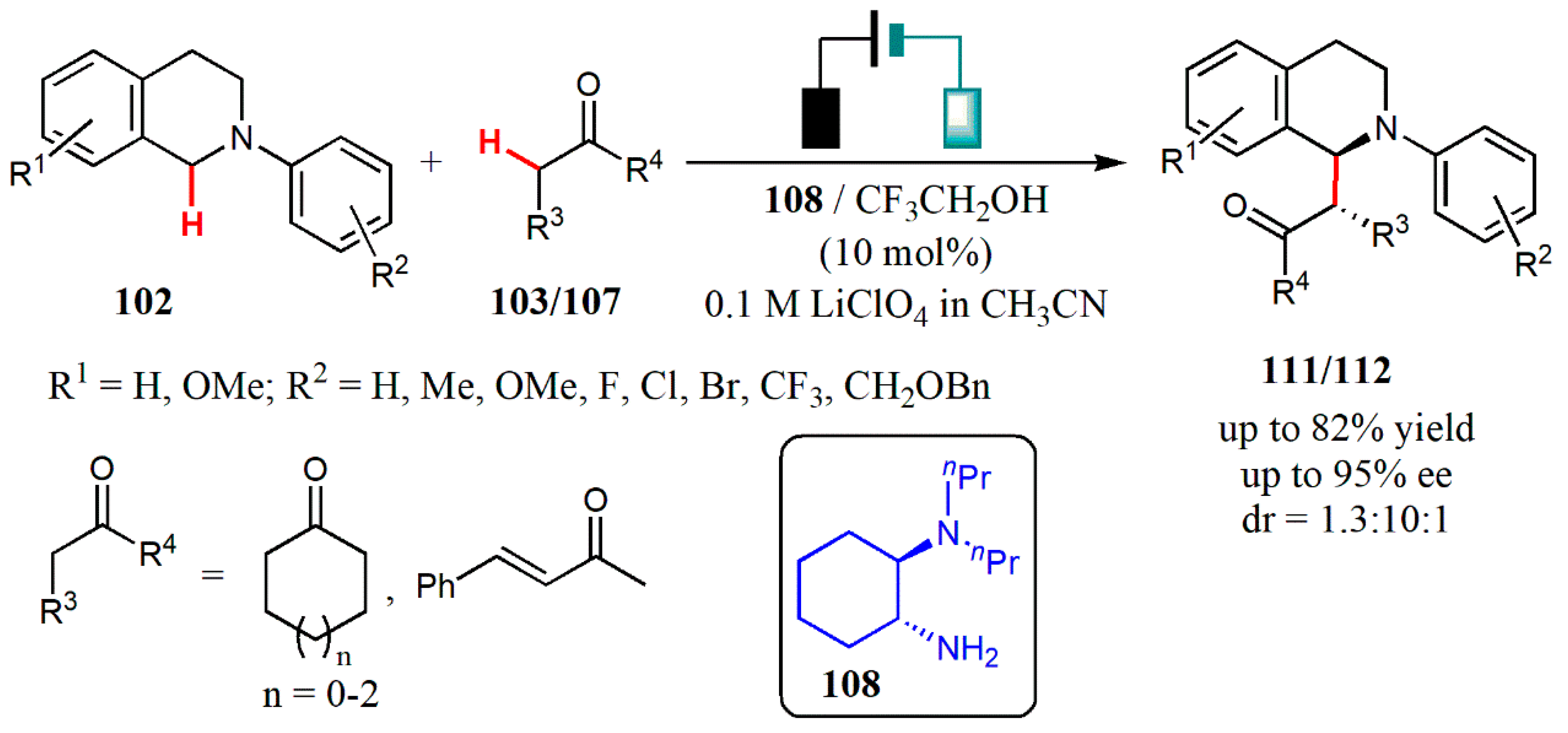{kind=link}
{kind=link}
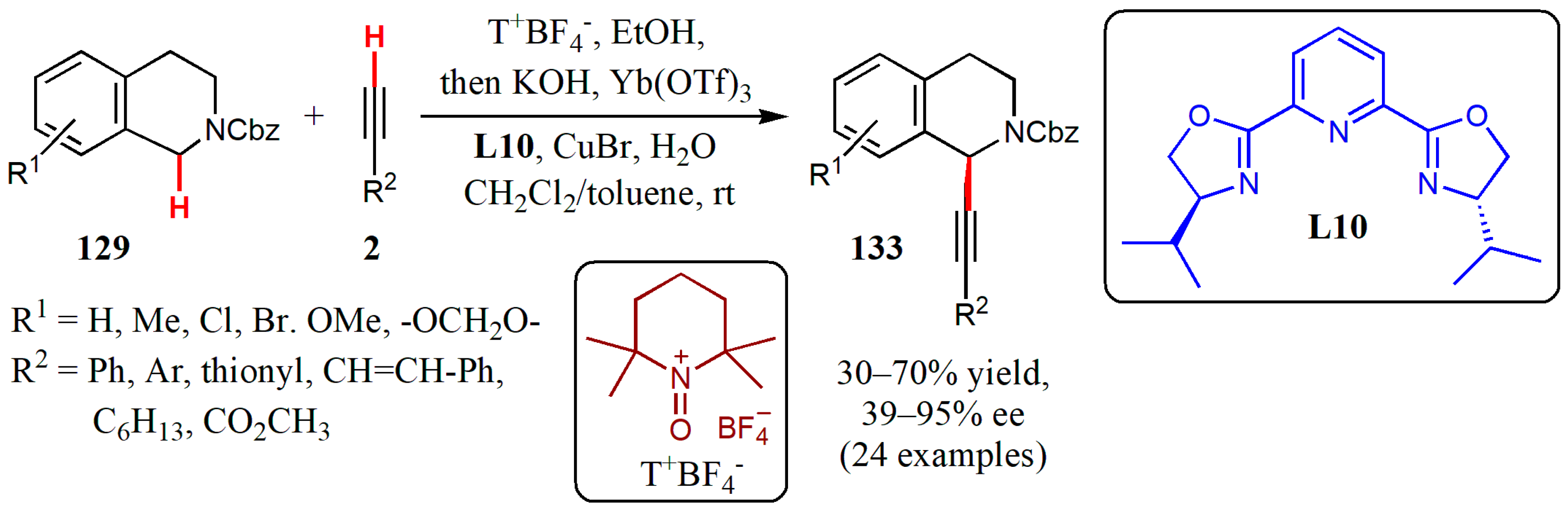{kind=link}
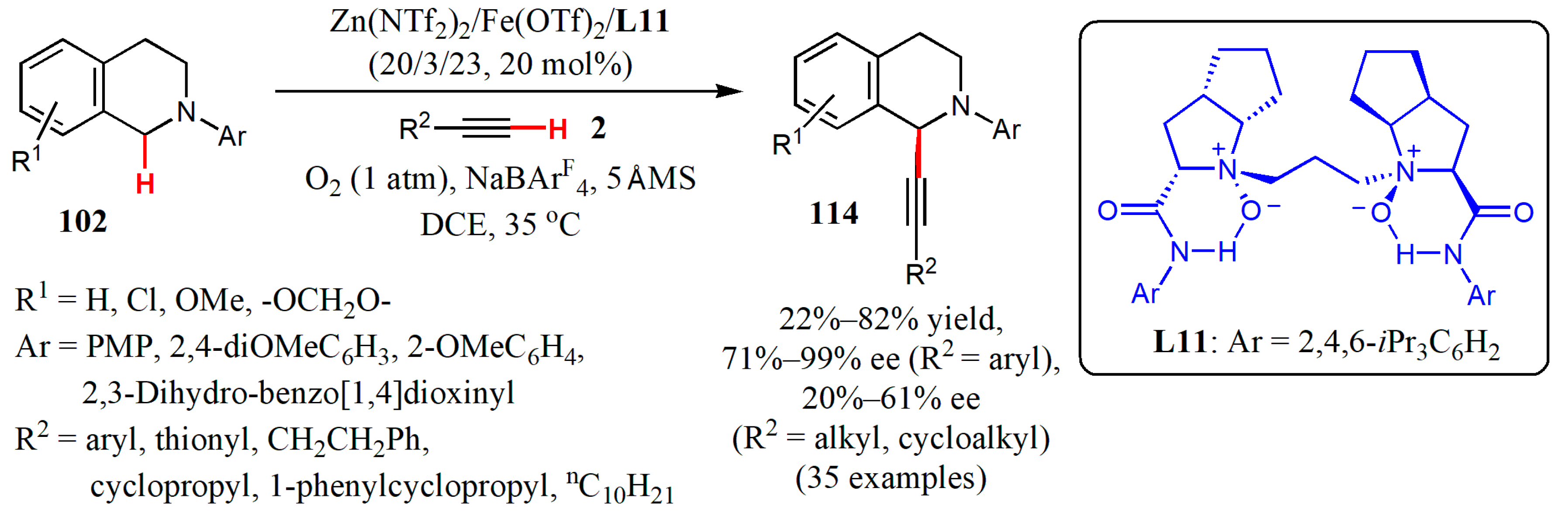{kind=link}
{kind=link}
{kind=link}
{kind=link}
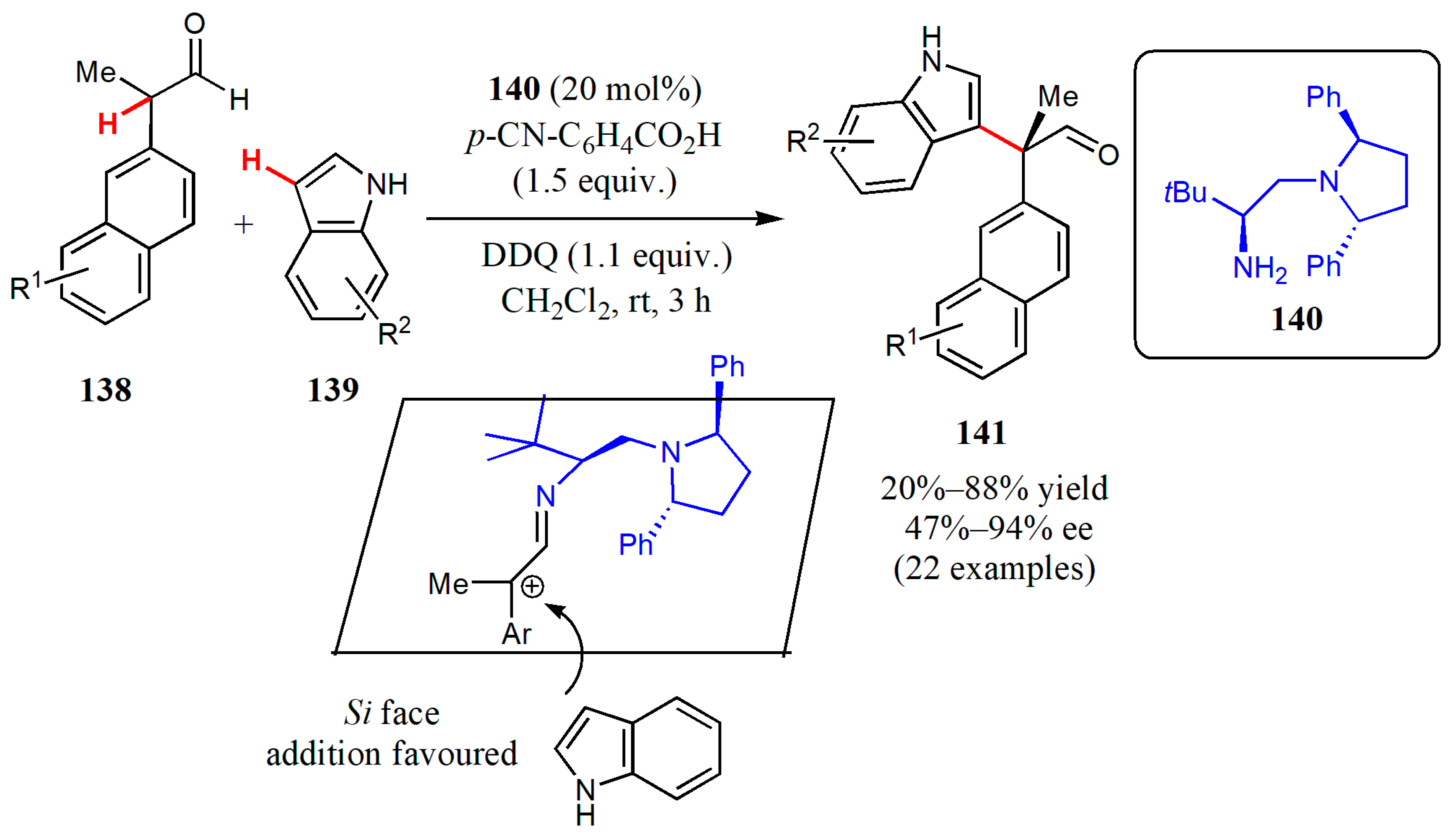{kind=link}
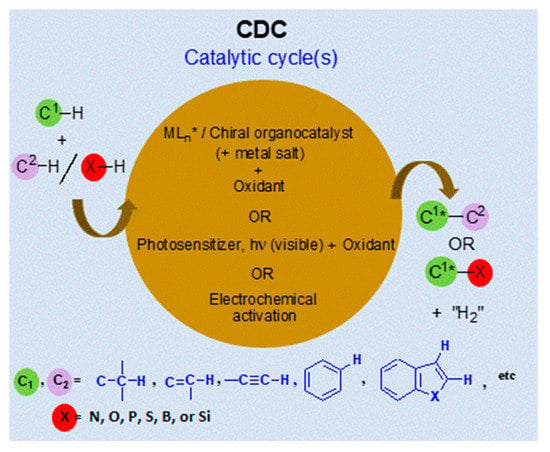
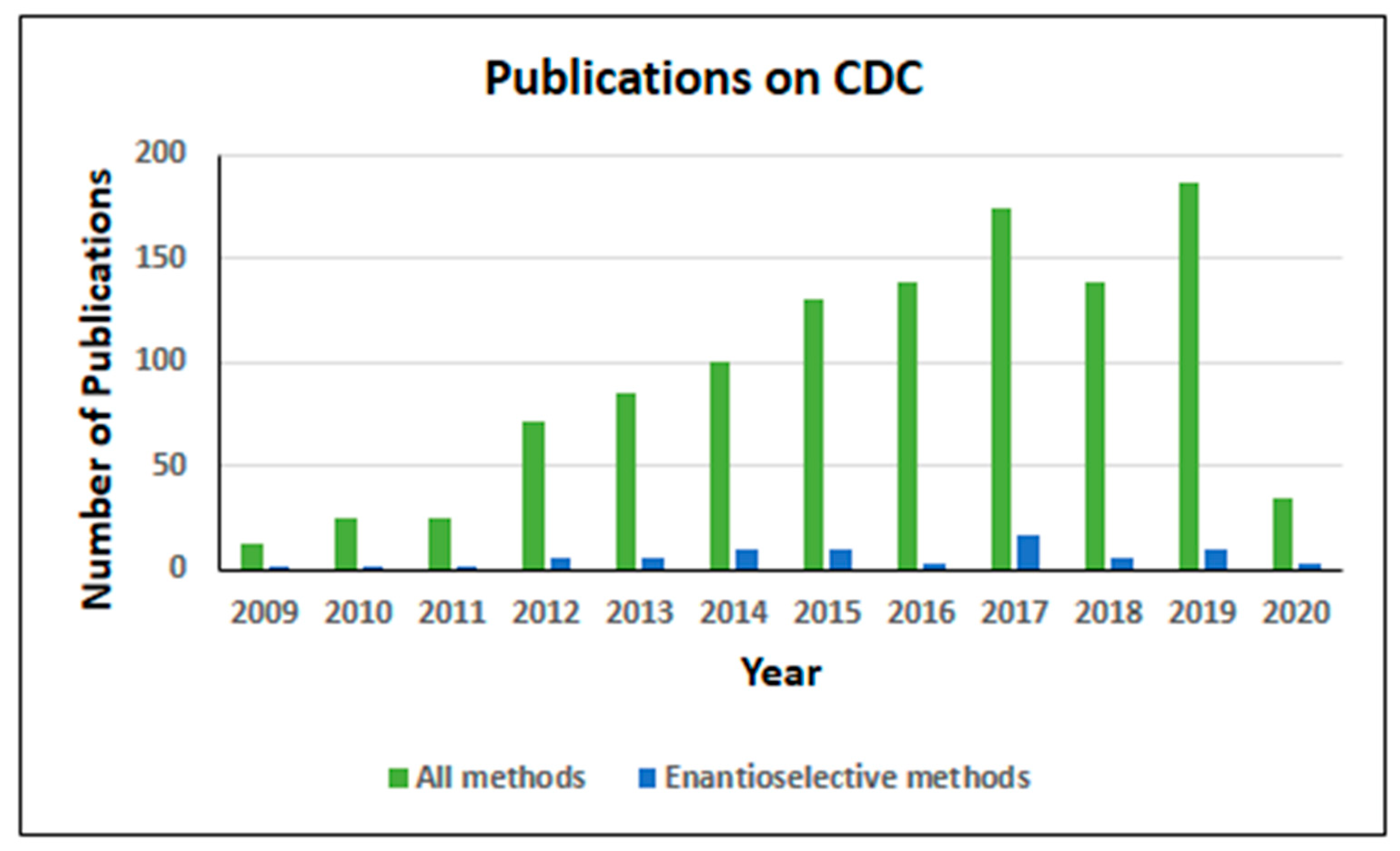
1. Introduction
2. Transition Metal-Catalyzed Enantioselective CDC
3. Cooperative/Synergistic Catalysis by a Metal-Organocatalyst Combination in Enantioselective CDC
3.1. Aminocatalysis
3.2. Brønsted Acid Catalysis
3.3. Photoredox Catalysis
3.4. Electrochemical Catalysis
4. Bimetallic Cooperative/Synergistic and Relay Catalysis in Enantioselective CDC
5. Organocatalyzed CDC
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Huang, C.-Y.; Kang, H.; Li, J.; Li, C. En route to intermolecular cross-dehydrogenative coupling reactions. J. Org. Chem. 2019, 84, 12705–12721. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yuan, J.; Gao, M.; Tang, S.; Li, W.; Shi, R.; Lei, A. Oxidative coupling between two hydrocarbons: An update of recent C–H functionalizations. Chem. Rev. 2015, 115, 12138–12204. [Google Scholar] [CrossRef] [PubMed]
- Faisca Phillips, A.M.; Pombeiro, A.J.L. Recent developments in transition metal-catalyzed cross-dehydrogenative coupling reactions of ethers and thioethers. ChemCatChem 2018, 10, 3354–3383. [Google Scholar] [CrossRef]
- Varun, B.V.; Dhineshkumar, J.; Bettadapur, K.R.; Siddaraju, Y.; Alagiri, K.; Prabhu, K.R. Recent advancements in dehydrogenative cross coupling reactions for C–C bond formation. Tetrahedron Lett. 2017, 58, 803–824. [Google Scholar] [CrossRef]
- Kozlowski, M.C. Oxidative coupling in complexity building transforms. Acc. Chem. Res. 2017, 50, 638–643. [Google Scholar] [CrossRef]
- Qin, Y.; Zhu, L.; Luo, S. Organocatalysis in inert C−H bond functionalization. Chem. Rev. 2017, 117, 9433–9520. [Google Scholar] [CrossRef]
- Yeung, C.S.; Dong, V.M. Catalytic dehydrogenative cross-coupling: Forming carbon−carbon bonds by oxidizing two carbon−hydrogen bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef]
- Roudesly, F.; Oble, J.; Poli, G. Metal-catalyzed CH activation/functionalization: The fundamentals. J. Mol. Catal. A Chem. 2017, 426, 275–296. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, N.-X.; Xing, Y. Advances in transition-metal-catalyzed direct sp3-carbon–hydrogen bond functionalization. Synlett 2015, 26, 2088–2098. [Google Scholar] [CrossRef]
- Fabry, D.C.; Rueping, M. Merging visible light photoredox catalysis with metal catalyzed C–H activations: On the role of oxygen and superoxide ions as oxidants. Acc. Chem. Res. 2016, 49, 1969–1979. [Google Scholar] [CrossRef]
- Wang, C.-S.; Dixneuf, P.H.; Soulé, J.-F. Catalysis for building C−C Bonds from C(sp2)−H bonds. Chem. Rev. 2018, 118, 7532–7585. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xia, W. Photochemical C–H bond coupling for (hetero) aryl C(sp2)–C(sp3) bond construction. Org. Biomol. Chem. 2019, 17, 4951–4963. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xu, K.; Zeng, C. Use of electrochemistry in the synthesis of heterocyclic structures. Chem. Rev. 2018, 118, 4485–4540. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Li, L.; Luo, S. Asymmetric electrochemical catalysis. Chem. Eur. J. 2019, 25, 10033–10044. [Google Scholar] [CrossRef]
- Funes-Ardoiz, I.; Maseras, F. Oxidative coupling mechanisms: Current state of understanding. ACS Catal. 2018, 8, 1161–1172. [Google Scholar] [CrossRef]
- Gandhi, S. Catalytic enantioselective cross dehydrogenative coupling of sp3 C–H of heterocycles. Org. Biomol. Chem. 2019, 17, 9683–9692. [Google Scholar] [CrossRef]
- Cheng, M.-X.; Yang, S.-D. Recent advances in the enantioselective oxidative α-C–H functionalization of amines. Synlett 2017, 28, 159–174. [Google Scholar]
- Zheng, C.; You, S.-L. Recent development of direct asymmetric functionalization of inert C–H bonds. RSC Adv. 2014, 4, 6173. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Wang, Y.; Luo, Y.-C.; Fu, X.-Z.; Xu, P.-F. Asymmetric C–H functionalization involving organocatalysis. Tetrahedron Lett. 2015, 56, 3703–3714. [Google Scholar] [CrossRef]
- Li, Z.; Li, C.-J. Catalytic enantioselective alkynylation of prochiral sp3 C−H bonds adjacent to a nitrogen atom. Org. Lett. 2004, 6, 4997–4999. [Google Scholar] [CrossRef]
- Wencel-Delord, J.; Panossian, A.; Leroux, F.R.; Coloberta, F. Recent advances and new concepts for the synthesis of axially stereoenriched biaryls. Chem. Soc. Rev. 2015, 44, 3418–3430. [Google Scholar] [CrossRef]
- Tkachenko, N.V.; Bryliakov, K.P. Transition metal catalyzed aerobic asymmetric coupling of 2-naphthols. Mini Rev. Org. Chem. 2019, 16, 392–398. [Google Scholar] [CrossRef]
- Nguyen, T.T. The use of chiral ortho-auxiliaries/substituents and remote stereogenic centers in atropselective biaryl synthesis. Eur. J. Org. Chem. 2020, 147–155. [Google Scholar]
- Mikami, K.; Aikawa, K.; Yusa, Y.; Jodry, J.J.; Yamanaka, M. Tropos or atropos? That is the question! Synlett 2002, 2002, 1561–1578. [Google Scholar] [CrossRef]
- Kočovský, P.; Vyskočil, S.; Smrčina, M. Non-symmetrically substituted 1,1’-binaphthyls in enantioselective catalysis. Chem. Rev. 2003, 103, 3213–3245. [Google Scholar] [CrossRef]
- San Segundo, M.; Correa, A. Cross-dehydrogenative coupling reactions for the functionalization, of α-amino acid derivatives and peptides. Synthesis 2018, 50, 2853–2866. [Google Scholar]
- Zhang, G.; Zhang, Y.; Wang, R. Catalytic asymmetric activation of a C(sp³)-H bond adjacent to a nitrogen atom: A versatile approach to optically active α-alkyl α-amino acids and C1-alkylated tetrahydroisoquinoline derivatives. Angew. Chem. Int. Ed. 2011, 50, 10429–10432. [Google Scholar] [CrossRef]
- Zhao, L.; Li, C.-J. Functionalizing glycine derivatives by direct C-C bond formation. Angew. Chem. Int. Ed. 2008, 47, 7075–7078. [Google Scholar] [CrossRef]
- Zhao, L.; Baslé, O.; Li, C.-J. Site-specific C-functionalization of free-(NH) peptides and glycine derivatives via direct C–H bond functionalization. Proc. Natl. Acad. Sci. USA 2009, 106, 4106–4111. [Google Scholar] [CrossRef]
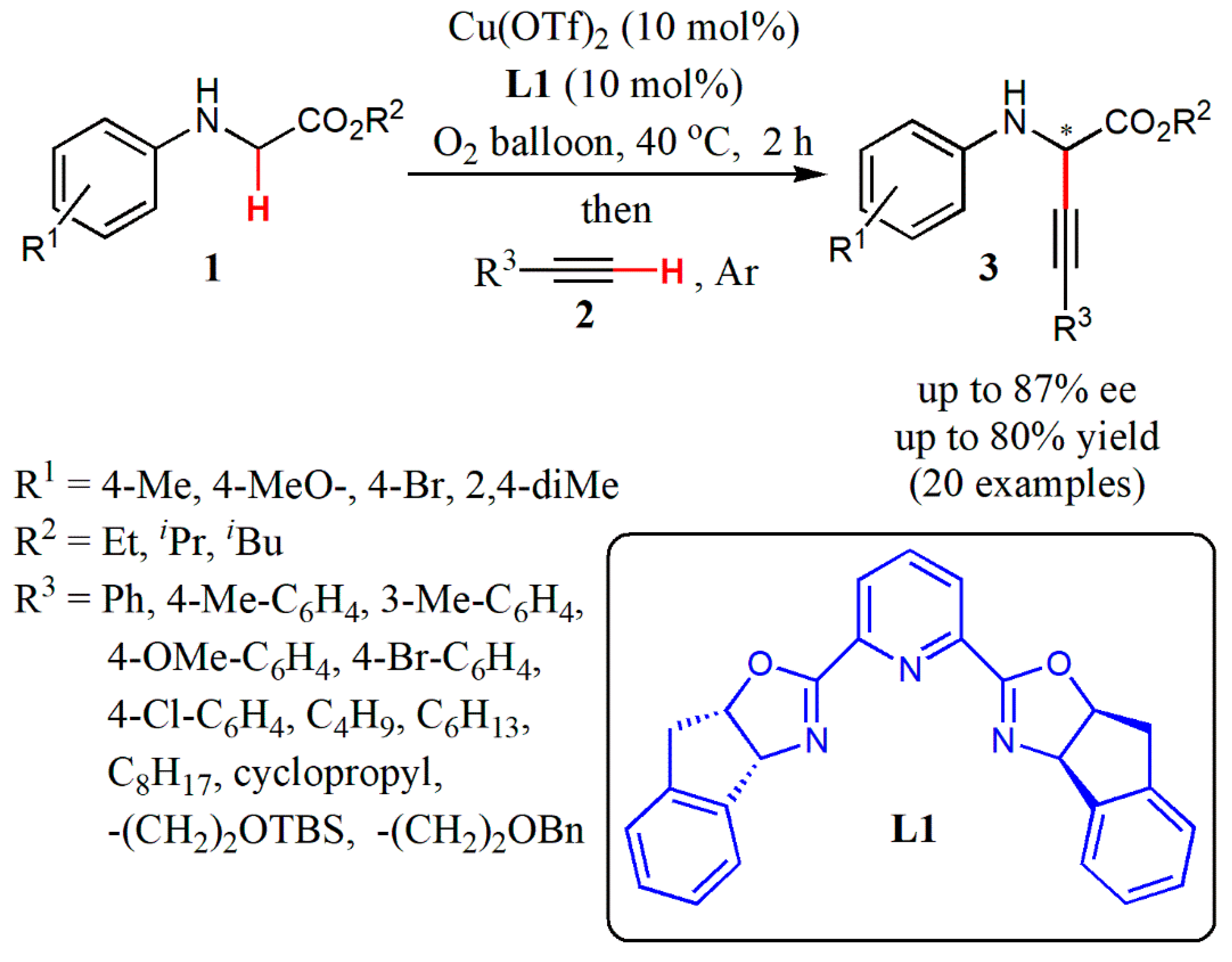
- Xie, Z.; Liu, X.; Liu, L. Copper-catalyzed aerobic enantioselective cross-dehydrogenative coupling of N-aryl glycine esters with terminal alkynes. Org. Lett. 2016, 18, 2982–2985. [Google Scholar] [CrossRef]
- Meng, Z.; Sun, S.; Yuan, H.; Lou, H.; Liu, L. Catalytic enantioselective oxidative cross-coupling of benzylic ethers with aldehydes. Angew. Chem. Int. Ed. 2014, 53, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Betori, R.C.; Crane, E.A.; Scheidt, K.A. An enantioselective cross-dehydrogenative coupling catalysis approach to substituted tetrahydropyrans. J. Am. Chem. Soc. 2018, 140, 6212–6216. [Google Scholar] [CrossRef] [PubMed]
- Mayr, H.; Ofial, A.R. Electrophilic scales for carbocations. In Carbocation Chemistry; Olah, G.A., Prakash, G.K.S., Eds.; Wiley: Hoboken, NJ, USA, 2004; Chapter 13; pp. 331–358. [Google Scholar]
- Cao, W.; Liu, X.; Peng, R.; He, P.; Lin, L.; Feng, X. Catalytic asymmetric cross-dehydrogenative coupling: Activation of C–H bonds by a cooperative bimetallic catalyst system. Chem. Commun. 2013, 49, 3470–3472. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Song, J.; Luo, S.-W.; Gong, L.-Z. Enantioselective oxidative cross-coupling reaction of 3-indolylmethyl C–H bonds with 1,3-dicarbonyls using a chiral Lewis acid-bonded nucleophile to control stereochemistry. Angew. Chem. Int. Ed. 2010, 49, 5558–5562. [Google Scholar] [CrossRef]
- Yu, J.; Ying, P.; Wang, H.; Xiang, K.; Su, W. Mechanochemical asymmetric cross-dehydrogenative coupling reaction: Liquid-assisted grinding enables reaction acceleration and enantioselectivity control. Adv. Synth. Catal. 2020, 362, 893–902. [Google Scholar] [CrossRef]
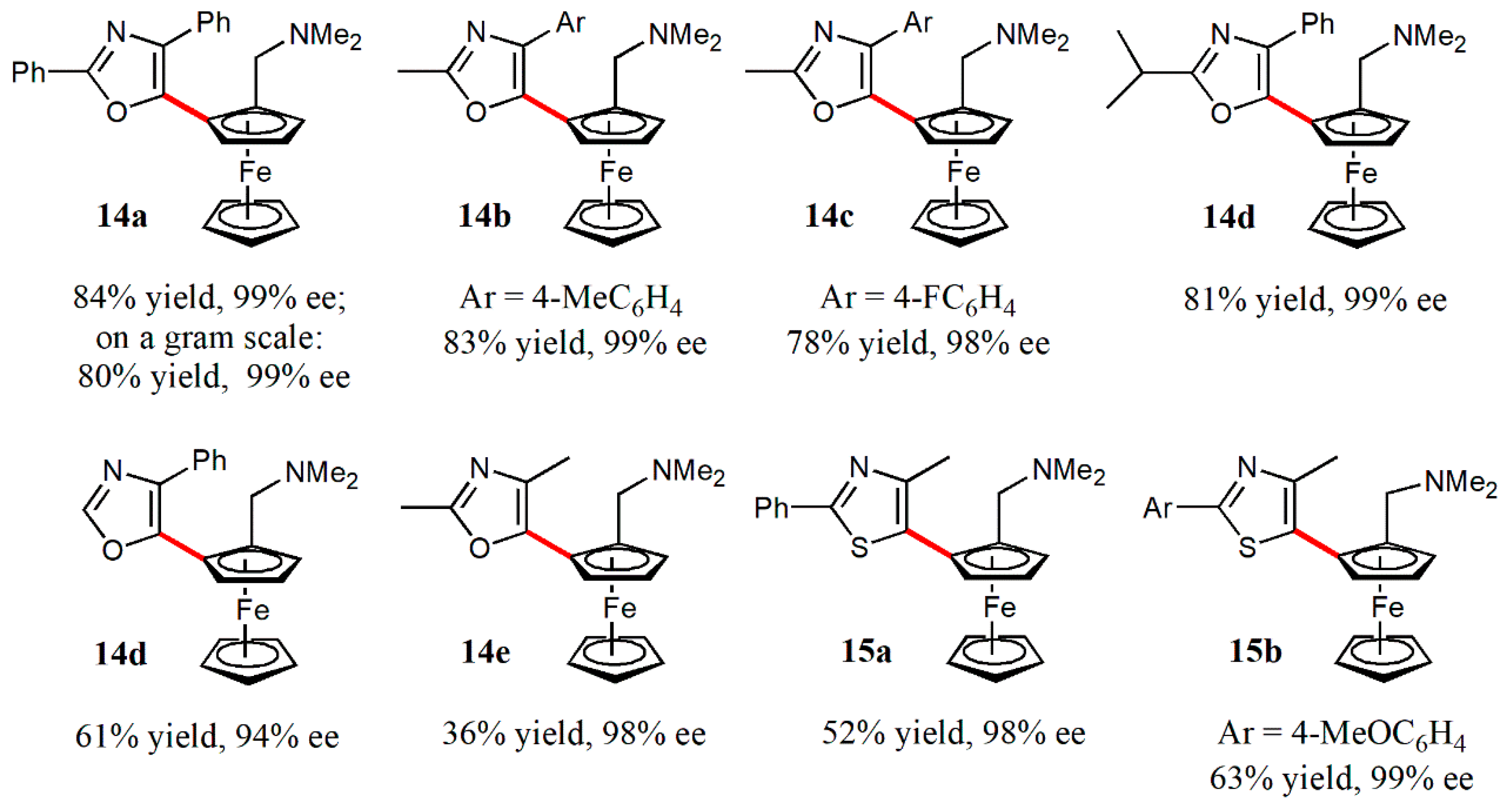
- Gao, D.-W.; Gu, Q.; You, S.-L. An enantioselective oxidative C−H/C−H cross-coupling reaction: Highly efficient method to prepare planar chiral ferrocenes. J. Am. Chem. Soc. 2016, 138, 2544–2547. [Google Scholar] [CrossRef]
- Schaarschmidt, D.; Lang, H. Selective synthesis of planar chiral ferrocenes. Organometallics 2013, 32, 5668–5704. [Google Scholar] [CrossRef]
- Cai, Z.-J.; Liu, C.-X.; Gu, Q.; Zheng, C.; You, S.-L. PdII-catalyzed regio-and enantioselective oxidative C-H/C-H cross-coupling reaction between ferrocenes and azoles. Angew. Chem. Int. Ed. 2019, 58, 2149–2153. [Google Scholar] [CrossRef]
- Chu, C.-Y.; Hwang, D.-R.; Wang, S.-K.; Uang, B.-J. Chiral oxovanadium complex catalyzed enantioselective oxidative coupling of 2-naphthols. Chem. Commun. 2001, 980–981. [Google Scholar] [CrossRef]
- Egami, H.; Katsuki, T. Iron-catalyzed asymmetric aerobic oxidation: Oxidative coupling of 2-naphthols. J. Am. Chem. Soc. 2009, 131, 6082–6083. [Google Scholar] [CrossRef]
- Li, X.; Yang, J.; Kozlowski, M.C. Enantioselective oxidative biaryl coupling reactions catalyzed by 1,5-diazadecalin metal complexes. Org. Lett. 2001, 3, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.-X.; Wu, Z.-J.; Luo, Z.-B.; Liu, Q.-Z.; Ye, J.-L.; Luo, S.-W.; Cun, L.-F.; Gong, L.-Z. Highly enantioselective oxidative couplings of 2-naphthols catalyzed by chiral bimetallic oxovanadium complexes with either oxygen or air as oxidant. J. Am. Chem. Soc. 2007, 129, 13927–13938. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Takizawa, S.; Sasai, H.; Oh, K. Reversal of enantioselectivity approach to BINOLs via single and dual 2-naphthol activation modes. Org. Lett. 2017, 19, 3867–3870. [Google Scholar] [CrossRef] [PubMed]
- Smrčina, M.; Vyskočil, S.; Máca, B.; Polášek, M.; Claxton, T.A.; Abbott, A.P.; Kočovský, P. Selective cross-coupling of 2-naphthol and 2-naphthylamine derivatives. A facile synthesis of 2,2’,3-trisubstituted and 2,2’,3,3’-tetrasubstituted 1,1’-binaphthyls. J. Org. Chem. 1994, 59, 2156–2163. [Google Scholar]
- Temma, T.; Habaue, S. Highly selective oxidative cross-coupling of 2-naphthol derivatives with chiral copper(I)–bisoxazoline catalysts. Tetrahedron Lett. 2005, 46, 5655–5657. [Google Scholar] [CrossRef]
- Narute, S.; Parnes, R.; Toste, F.D.; Pappo, D. Enantioselective oxidative homocoupling and cross-coupling of 2-naphthols catalyzed by chiral iron phosphate complexes. J. Am. Chem. Soc. 2016, 138, 16553–16560. [Google Scholar] [CrossRef]
- Meca, L.; Reha, D.; Havlas, Z. Racemization barriers of 1,1′-binaphthyl and 1,1′-binaphthalene-2,2′-diol: A DFT study. J. Org. Chem. 2003, 68, 5677–5680. [Google Scholar] [CrossRef]
- Wang, H. Recent advances in asymmetric oxidative coupling of 2-naphthol and its derivatives. Chirality 2010, 22, 827–837. [Google Scholar] [CrossRef]
- Narute, S.; Pappo, D. Iron phosphate catalyzed asymmetric cross-dehydrogenative coupling of 2-naphthols with β-ketoesters. Org. Lett. 2017, 19, 2917–2920. [Google Scholar] [CrossRef]
- Forkosh, H.-; Vershinin, V.; Reiss, H.; Pappo, D. Stereoselective synthesis of optically pure 2-amino-2′-hydroxy-1,1′-binaphthyls. Org. Lett. 2018, 20, 2459–2463. [Google Scholar] [CrossRef]
- Tian, J.-M.; Wang, A.-F.; Yang, J.-S.; Zhao, X.-J.; Tu, Y.-Q.; Zhang, S.-Y.; Chen, Z.-M. Copper-complex-catalyzed asymmetric aerobic oxidative cross-coupling of 2-naphthols: Enantioselective synthesis of 3,3’-substituted C1-symmetric BINOLs. Angew. Chem. Int. Ed. 2019, 58, 11023–11027. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Yakeishi, S.; Hamada, D.; Shibata, T. Functionalized BINOL-mono-PHOS for multinuclear Cu-catalysts in asymmetric conjugate addition of organozinc reagents. Chem. Lett. 2013, 42, 547–549. [Google Scholar] [CrossRef]
- Oguma, T.; Doiuchi, D.; Fujitomo, C.; Kim, C.; Hayashi, H.; Uchida, T.; Katsuki, T. Iron-catalyzed asymmetric inter-and intramolecular aerobic oxidative dearomatizing spirocyclization of 2-naphthols. Asian, J. Org. Chem. 2020, 9, 404–415. [Google Scholar] [CrossRef]
- Appendino, G.; Ottino, M.; Marquez, N.; Bianchi, F.; Giana, A.; Ballero, M.; Sterner, O.; Fiebich, B.L.; Munoz, E.; Appendino, G.; et al. Aezanol, an anti-inflammatory and anti-HIV-1 phloroglucinol α-pyrone from Helichrysum. Ital. Ssp. Microphyllum. J. Nat. Prod. 2007, 70, 608–612. [Google Scholar] [CrossRef]
- Wang, G.; Xin, X.; Wang, Z.; Lu, G.; Ma, Y.; Liu, L. Catalytic enantioselective oxidative coupling of saturated ethers with carboxylic acid derivatives. Nature Commun. 2019, 10, 559. [Google Scholar] [CrossRef]
- Pan, X.; Liu, X.; Sun, S.; Meng, Z.; Liu, L. Catalytic asymmetric cross-dehydrogenative coupling of 2H-chromenes and aldehydes. Chin. J. Chem. 2018, 36, 1187–1190. [Google Scholar] [CrossRef]
- Hong, L.; Sun, W.; Yang, D.; Li, G.; Wang, R. Additive effects on asymmetric catalysis. Chem. Rev. 2016, 116, 4006–4123. [Google Scholar] [CrossRef]
- Becica, J.; Dobereiner, G.E. The roles of Lewis acidic additives in organotransition metal catalysis. Org. Biomol. Chem. 2019, 17, 2055–2069. [Google Scholar] [CrossRef]
- Xin, X.; Pan, X.; Meng, Z.; Liu, X.; Liu, L. Catalytic enantioselective cross-dehydrogenative coupling of 3,6-dihydro-2H-pyrans with aldehydes. Org. Chem. Front. 2019, 6, 1448–1452. [Google Scholar] [CrossRef]
- Yang, B.; Qiu, Y.; Jiang, T.; Wulff, W.D.; Yin, X.; Zhu, C.; Bäckvall, J.-E. Enantioselective palladium-catalyzed carbonylative carbocyclization of enallenes via cross-dehydrogenative coupling with terminal alkynes: Efficient construction of α-chirality of ketones. Angew. Chem. Int. Ed. 2017, 56, 4535–4539. [Google Scholar] [CrossRef]
- Zhu, C.; Yang, B.; Bäckvall, J.-E. Highly selective cascade C−C bond formation via palladium-catalyzed oxidative carbonylation−carbocyclization−carbonylation−alkynylation of enallenes. J. Am. Chem. Soc. 2015, 137, 11868–11871. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Tian, Y.; Meng, X.; Gu, Q.-S.; Liu, X.-Y. Enantioselective copper(I)/chiral phosphoric acid catalyzed intramolecular amination of allylic and benzylic C.–H. bonds. Angew. Chem. Int. Ed. 2020, 59, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Panda, G. Synthetic methodologies of achiral diarylmethanols, diaryl and triarylmethanes (TRAMs) and medicinal properties of diaryl and triarylmethanes-an overview. RSC Adv. 2014, 4, 28317–28358. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, Y.; Pan, X.; Wang, G.; Liu, L. Synthesis of chiral triarylmethanes bearing all-carbon quaternary stereocenters: Catalytic asymmetric oxidative cross-coupling of 2,2-diarylacetonitriles and (hetero)arenes. Angew. Chem. Int. Ed. 2020, 59, 3053–3057. [Google Scholar] [CrossRef] [PubMed]
- McAtee, R.C.; McClain, E.J.; Stephenson, C.R.J. Illuminating photoredox catalysis. Trends Chem. 2019, 1, 111–125. [Google Scholar] [CrossRef]
- Jin, J.; MacMillan, D.W.C. Direct α-arylation of ethers through the combination of photoredox-mediated C−H functionalization and the Minisci reaction. Angew. Chem. Int. Ed. 2015, 54, 1565–1569. [Google Scholar] [CrossRef]
- Larionov, E.; Mastandrea, M.M.; Pericàs, M.A. Asymmetric visible-light photoredox cross-dehydrogenative coupling of aldehydes with xanthenes. ACS Catal. 2017, 7008–7013. [Google Scholar] [CrossRef]
- Hou, H.; Zhu, S.; Atodiresei, I.; Rueping, M. Asymmetric organocatalysis and photoredox catalysis for the α-functionalization of tetrahydroisoquinolines. Eur. J. Org. Chem. 2018, 1277–1280. [Google Scholar] [CrossRef]
- Scott, J.D.; Williams, R.M. Chemistry and biology of the tetrahydroisoquinoline antitumour antibiotics. Chem. Rev. 2002, 102, 1669–1730. [Google Scholar] [CrossRef]
- Welsch, M.E.; Snyder, S.A.; Stockwell, B.R. Priviledged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010, 14, 347–361. [Google Scholar] [CrossRef]
- Reddy, R.J.; Kawai, N.; Uenishi, J. Synthesis of the 1-phenethyltetrahydroisoquinoline alkaloids (+)-dysoxyline, (+)-colchiethanamine, and (+)-colchiethine. J. Org. Chem. 2012, 77, 11101–11108. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Vila, C.; Koenigs, R.M.; Poscharny, K.; Fabry, D.C. Dual catalysis: Combining photoredox and Lewis base catalysis for direct Mannich reactions. Chem. Commun. 2011, 47, 2360–2362. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Zoller, J.; Fabry, D.C.; Poscharny, K.; Koenigs, R.M.; Weirich, T.E.; Mayer, J. Light-Mediated Heterogeneous Cross Dehydrogenative Coupling Reactions: Metal Oxides as Efficient, Recyclable, Photoredox Catalysts in C—C Bond-Forming Reactions. Chem. Eur. J. 2012, 18, 3478–3481. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, L.; Ye, C.; Luo, S.; Wu, L.-Z.; Tung, C.-H. Visible-light-promoted asymmetric cross-dehydrogenative coupling of tertiary amines to ketones by synergistic multiple catalysis. Angew. Chem. Int. Ed. 2017, 56, 3694–3698. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Sharma, A. Recent advances in photocatalytic manipulations of Rose Bengal in organic synthesis. Org. Biomol. Chem. 2019, 17, 4384–4405. [Google Scholar] [CrossRef] [PubMed]
- Kumara, G.; Vermaa, S.; Ansaria, A.; Khana, N.-u.H.; Kureshya, R.I. Enantioselective cross dehydrogenative coupling reaction catalyzed by Rose Bengal incorporated-Cu(I)-dimeric chiral complexes. Catal. Commun. 2017, 99, 94–99. [Google Scholar] [CrossRef]
- Wei, G.; Zhang, C.; Bures, F.; Ye, X.; Tan, C.-H.; Jiang, Z. Enantioselective aerobic oxidative C(sp3)-H olefination of amines via cooperative photoredox and asymmetric catalysis. Acs Catal. 2016, 3708–3712. [Google Scholar] [CrossRef]
- Jensen, K.L.; Franke, P.T.; Nielsen, L.T.; Daasbjerg, K.; Jørgensen, K.A. Anodic Oxidation and Organocatalysis: Direct regio-and stereoselective access to meta-substituted anilines by a-arylation of aldehydes. Angew. Chem. Int. Ed. 2010, 49, 129–133. [Google Scholar] [CrossRef]
- Ho, X.; Mho, S.; Kang, H.; Jang, H. Electro-organocatalysis: Enantioselective α-alkylation of aldehydes. Eur. J. Org. Chem. 2010, 4436–4441. [Google Scholar] [CrossRef]
- Fu, N.; Li, L.; Yang, Q.; Luo, S. Catalytic asymmetric electrochemical oxidative coupling of tertiary amines with simple ketones. Org. Lett. 2017, 19, 2122–2125. [Google Scholar] [CrossRef]
- Sun, S.; Li, C.; Floreancig, P.E.-; Lou, H.; Liu, L. Highly enantioselective catalytic cross-dehydrogenative coupling of N-carbamoyl tetrahydroisoquinolines and terminal alkynes. Org. Lett. 2015, 17, 1684–1687. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; MacLeod, P.D.; Li, C.-J. Studies on Cu-catalyzed asymmetric alkynylation of tetrahydroisoquinoline derivatives. Tetrahedron: Asymmetry 2006, 17, 590–597. [Google Scholar] [CrossRef]
- Christian, D.; Yoshitaka, H.; Naoki, S.; Seidel, T.M.; Suzuki, S.; Hashizume, D.; Sodeoka, M. Mechanistic studies on the catalytic asymmetric mannich-type reaction with dihydroisoquinolines and development of oxidative Mannich-type reactions starting from tetrahydroisoquinolines. J. Org. Chem. 2008, 73, 5859–5871. [Google Scholar]
- Huang, T.; Liu, X.; Lang, J.; Xu, J.; Lin, L.; Feng, X. Asymmetric aerobic oxidative cross-coupling of tetrahydroisoquinolines with alkynes. ACS Catal. 2017, 7, 5654–5660. [Google Scholar] [CrossRef]
- Ratnikov, M.O.; Xu, X.; Doyle, M.P. Simple and sustainable iron-catalyzed aerobic C–H functionalization of N,N-dialkylanilines. J. Am. Chem. Soc. 2013, 135, 9475–9479. [Google Scholar] [CrossRef]
- Beeson, T.D.; Mastracchio, A.; Hong, J.-B.; Ashton, K.; MacMillan, D.W.C. Enantioselective organocatalysis using SOMO activation. Science 2007, 316, 582. [Google Scholar] [CrossRef]
- Conrad, J.C.; Kong, J.; Laforteza, B.N.; MacMillan, D.W.C. Enantioselective α-arylation of aldehydes via organo-SOMO catalysis. An ortho-selective arylation reaction based on an open-shell pathway. J. Am. Chem. Soc. 2009, 131, 11640–11641. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Reingruber, R.; Sarlah, D.; Bräse, S. Enantioselective intramolecular Friedel−Crafts-type α-arylation of aldehydes. J. Am. Chem. Soc. 2009, 131, 2086–2087. [Google Scholar] [CrossRef]
- Vetica, F.; Bailey, S.; Chauhan, P.; Turberg, M.; Ghaur, A.; Raabe, G.; Enders, D. Desymmetrization of cyclopentenediones via organocatalytic cross-dehydrogenative coupling. Adv. Synth. Catal. 2017, 359, 3729–3734. [Google Scholar] [CrossRef]
- Chauhan, P.; Mahajan, S.; Enders, D. Asymmetric synthesis of pyrazoles and pyrazolones employing the reactivity of pyrazolin-5-one derivatives. Chem. Commun. 2015, 51, 12890–12907. [Google Scholar] [CrossRef]
- Schmidt, A.; Dreger, A. Recent advances in the chemistry of pyrazoles. Properties, biological activities, and syntheses. Curr. Org. Chem. 2011, 15, 1423–1463. [Google Scholar] [CrossRef]
- Rezayee, N.M.; Lauridsen, V.H.; Næsborg, L.; Nguyen, T.V.Q.; Tobiesen, H.N.; Jørgensen, K.A. Oxidative organocatalysed enantioselective coupling of indoles with aldehydes that forms quaternary carbon stereocentres. Chem. Sci. 2019, 10, 3586–3591. [Google Scholar] [CrossRef]















































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faisca Phillips, A.M.; C. Guedes da Silva, M.d.F.; Pombeiro, A.J.L. New Trends in Enantioselective Cross-Dehydrogenative Coupling. Catalysts 2020, 10, 529. https://doi.org/10.3390/catal10050529
Faisca Phillips AM, C. Guedes da Silva MdF, Pombeiro AJL. New Trends in Enantioselective Cross-Dehydrogenative Coupling. Catalysts. 2020; 10(5):529. https://doi.org/10.3390/catal10050529
Chicago/Turabian StyleFaisca Phillips, Ana Maria, Maria de Fátima C. Guedes da Silva, and Armando J. L. Pombeiro. 2020. "New Trends in Enantioselective Cross-Dehydrogenative Coupling" Catalysts 10, no. 5: 529. https://doi.org/10.3390/catal10050529
APA StyleFaisca Phillips, A. M., C. Guedes da Silva, M. d. F., & Pombeiro, A. J. L. (2020). New Trends in Enantioselective Cross-Dehydrogenative Coupling. Catalysts, 10(5), 529. https://doi.org/10.3390/catal10050529