PRIMA-1 and PRIMA-1Met (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies



 , ,
, ,
Abstract
1. Introduction
2. Effects of PRIMA-1 and APR-246 on Cancer Growth Inhibition and Mutant-p53 Reactivation
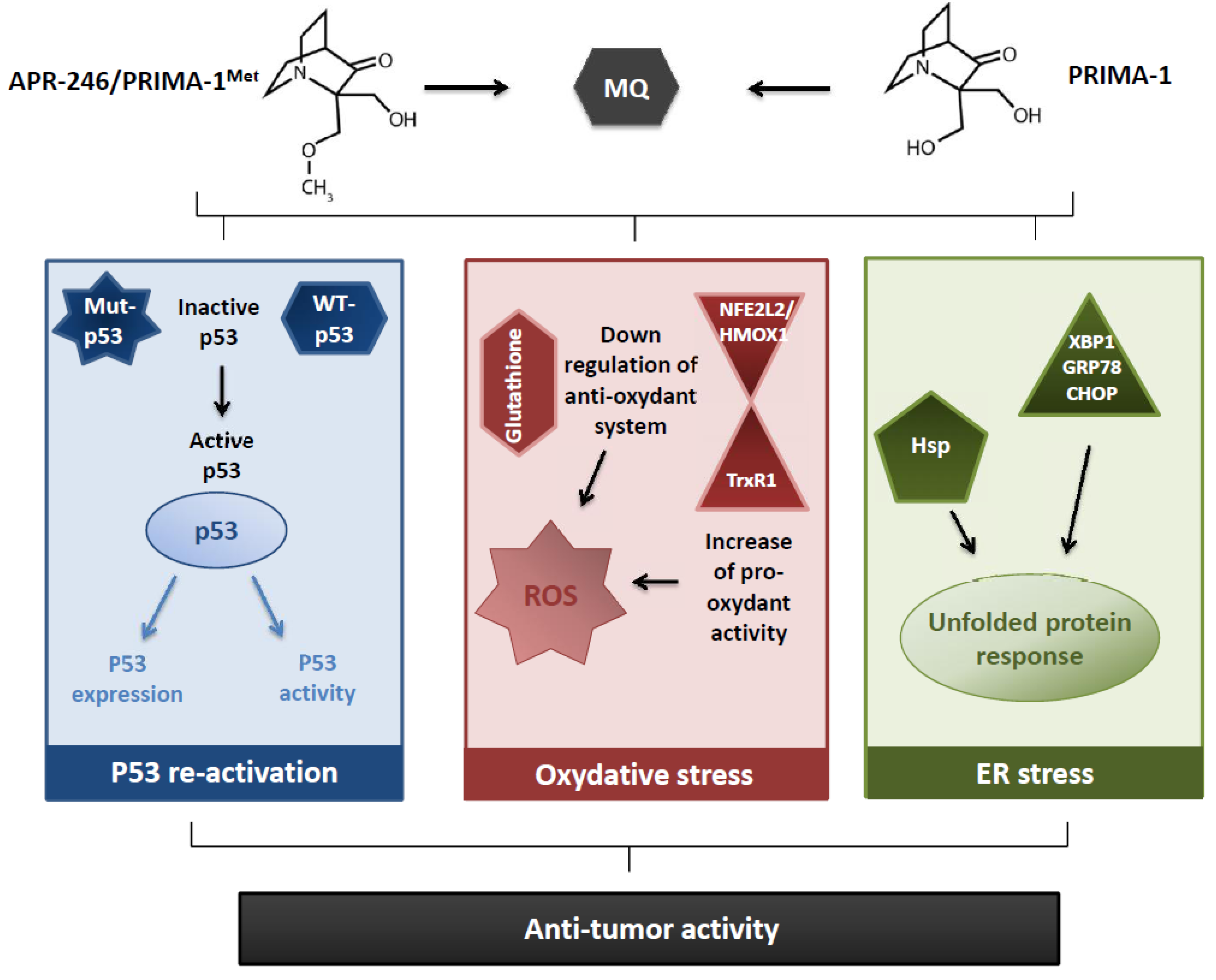
3. Unexpected Effects of PRIMA-1/APR-246 Driving New Hypothetical Mechanisms of Action
4. PRIMA-1 and APR-246 in Combination with Other Anti-Cancer Therapies
5. Conclusions, Perspectives, and Clinical Impacts
Acknowledgments
Conflicts of Interest
References
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Selivanova, G. Wild type p53 reactivation: From lab bench to clinic. FEBS Lett. 2014, 588, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Selivanova, G. Therapeutic targeting of p53 by small molecules. Semin. Cancer Biol. 2010, 20, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Wanzel, M.; Vischedyk, J.B.; Gittler, M.P.; Gremke, N.; Seiz, J.R.; Hefter, M.; Noack, M.; Savai, R.; Mernberger, M.; Charles, J.P.; et al. CRISPR-Cas9-based target validation for p53-reactivating model compounds. Nat. Chem. Biol. 2016, 12, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Zhang, Q.; Zhang, M.; Ceder, S.; Abrahmsen, L.; Wiman, K.G. Targeting of Mutant p53 and the Cellular Redox Balance by APR-246 as a Strategy for Efficient Cancer Therapy. Front. Oncol. 2016, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Maurer, U.; Green, D.R.; Schuler, M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell 2003, 4, 371–381. [Google Scholar] [CrossRef]
- Nahi, H.; Lehmann, S.; Mollgard, L.; Bengtzen, S.; Selivanova, G.; Wiman, K.G.; Paul, C.; Merup, M. Effects of PRIMA-1 on chronic lymphocytic leukaemia cells with and without hemizygous p53 deletion. Br. J. Haematol. 2004, 127, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Chahal, M.S.; Tang, X.; Bruce, J.E.; Pommier, Y.; Daoud, S.S. Proteomic identification of heat shock protein 90 as a candidate target for p53 mutation reactivation by PRIMA-1 in breast cancer cells. Breast Cancer Res. BCR 2005, 7, R765–R774. [Google Scholar] [CrossRef] [PubMed]
- Nahi, H.; Merup, M.; Lehmann, S.; Bengtzen, S.; Möllgård, L.; Selivanova, G.; Wiman, K.G.; Paul, C. PRIMA-1 induces apoptosis in acute myeloid leukaemia cells with p53 gene deletion. Br. J. Haematol. 2006, 132, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Wang, T.; Paszczynski, A.J.; Daoud, S.S. Expression proteomics to p53 mutation reactivation with PRIMA-1 in breast cancer cells. Biochem. Biophys. Res. Commun. 2006, 349, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lee, K.; Rehman, A.; Daoud, S.S. PRIMA-1 induces apoptosis by inhibiting JNK signaling but promoting the activation of Bax. Biochem. Biophys. Res. Commun. 2007, 352, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Rökaeus, N.; Klein, G.; Wiman, K.G.; Szekely, L.; Mattsson, K. PRIMA-1(MET) induces nucleolar accumulation of mutant p53 and PML nuclear body-associated proteins. Oncogene 2007, 26, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Magrini, R.; Russo, D.; Ottaggio, L.; Fronza, G.; Inga, A.; Menichini, P. PRIMA-1 synergizes with adriamycin to induce cell death in non-small cell lung cancer cells. J. Cell. Biochem. 2008, 104, 2363–2373. [Google Scholar] [CrossRef] [PubMed]
- Supiot, S.; Zhao, H.; Wiman, K.; Hill, R.P.; Bristow, R.G. PRIMA-1met radiosensitizes prostate cancer cells independent of their MTp53-status. Radiother. Oncol. 2008, 86, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Lambert, J.M.R.; Hautefeuille, A.; Bykov, V.J.N.; Wiman, K.G.; Hainaut, P.; Caron de Fromentel, C. In vitro and in vivo cytotoxic effects of PRIMA-1 on hepatocellular carcinoma cells expressing mutant p53ser249. Carcinogenesis 2008, 29, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Stuber, G.; Flaberg, E.; Petranyi, G.; Otvös, R.; Rökaeus, N.; Kashuba, E.; Wiman, K.G.; Klein, G.; Szekely, L. PRIMA-1MET induces nucleolar translocation of Epstein-Barr virus-encoded EBNA-5 protein. Mol. Cancer 2009, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.R.; Moshfegh, A.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene 2010, 29, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Russo, D.; Ottaggio, L.; Penna, I.; Foggetti, G.; Fronza, G.; Inga, A.; Menichini, P. PRIMA-1 cytotoxicity correlates with nucleolar localization and degradation of mutant p53 in breast cancer cells. Biochem. Biophys. Res. Commun. 2010, 402, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Rökaeus, N.; Shen, J.; Eckhardt, I.; Bykov, V.J.N.; Wiman, K.G.; Wilhelm, M.T. PRIMA-1(MET)/APR-246 targets mutant forms of p53 family members p63 and p73. Oncogene 2010, 29, 6442–6451. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Gao, L.; Wu, X.; Wang, L.; Nana-Sinkam, S.P.; Otterson, G.A.; Villalona-Calero, M.A. MicroRNA-34a is an important component of PRIMA-1-induced apoptotic network in human lung cancer cells. Int. J. Cancer 2010, 127, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.-L.; Kang, S.K.; Minn, I.; Califano, J.A.; Sidransky, D.; Koch, W.M. p53-Reactivating small molecules induce apoptosis and enhance chemotherapeutic cytotoxicity in head and neck squamous cell carcinoma. Oral Oncol. 2011, 47, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Besch-Williford, C.; Benakanakere, I.; Thorpe, P.E.; Hyder, S.M. Targeting mutant p53 protein and the tumor vasculature: An effective combination therapy for advanced breast tumors. Breast Cancer Res. Treat. 2011, 125, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Chen, M.; Zhao, X.; Kumar, R.; Spinnler, C.; Thullberg, M.; Issaeva, N.; Selivanova, G.; Strömblad, S. PRIMA-1Met/APR-246 induces wild-type p53-dependent suppression of malignant melanoma tumor growth in 3D culture and in vivo. Cell Cycle 2011, 10, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Ali, D.; Jönsson-Videsäter, K.; Deneberg, S.; Bengtzén, S.; Nahi, H.; Paul, C.; Lehmann, S. APR-246 exhibits anti-leukemic activity and synergism with conventional chemotherapeutic drugs in acute myeloid leukemia cells. Eur. J. Haematol. 2011, 86, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Messina, R.L.; Sanfilippo, M.; Vella, V.; Pandini, G.; Vigneri, P.; Nicolosi, M.L.; Gianì, F.; Vigneri, R.; Frasca, F. Reactivation of p53 mutants by prima-1 [corrected] in thyroid cancer cells. Int. J. Cancer 2012, 130, 2259–2270. [Google Scholar] [CrossRef] [PubMed]
- Izetti, P.; Hautefeuille, A.; Abujamra, A.L.; de Farias, C.B.; Giacomazzi, J.; Alemar, B.; Lenz, G.; Roesler, R.; Schwartsmann, G.; Osvaldt, A.B.; et al. PRIMA-1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Investig. New Drugs 2014, 32, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Russo, D.; Ottaggio, L.; Foggetti, G.; Masini, M.; Masiello, P.; Fronza, G.; Menichini, P. PRIMA-1 induces autophagy in cancer cells carrying mutant or wild type p53. Biochim. Biophys. Acta 2013, 1833, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhang, M.-Q.-Z.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.N.; Arnér, E.S.J.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Abedini, M.; Sakuragi, N.; Tsang, B.K. PRIMA-1 increases cisplatin sensitivity in chemoresistant ovarian cancer cells with p53 mutation: A requirement for Akt down-regulation. J. Ovarian Res. 2013, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Jiang, H.; Yang, Y.; Reece, D.; Chang, H. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol. Cancer Ther. 2013, 12, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Yang, Q.; Guan, H.; Shi, B.; Hou, P.; Ji, M. PRIMA-1, a mutant p53 reactivator, restores the sensitivity of TP53 mutant-type thyroid cancer cells to the histone methylation inhibitor 3-Deazaneplanocin A. J. Clin. Endocrinol. Metab. 2014, 99, E962–E970. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Jin, T.; Yang, Q.; Liu, W.; Liu, S.; Ji, M.; He, N.; Chen, C.; Shi, B.; Hou, P. PRIMA-1 selectively induces global DNA demethylation in p53 mutant-type thyroid cancer cells. J. Biomed. Nanotechnol. 2014, 10, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Mohell, N.; Alfredsson, J.; Fransson, Å.; Uustalu, M.; Byström, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.N.; Björklund, U.; Wiman, K.G. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef] [PubMed]
- Grellety, T.; Laroche-Clary, A.; Chaire, V.; Lagarde, P.; Chibon, F.; Neuville, A.; Italiano, A. PRIMA-1(MET) induces death in soft-tissue sarcomas cell independent of p53. BMC Cancer 2015, 15, 684. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Zhou, J.; Chan, Z.-L.; Chooi, J.-Y.; Chen, Z.-R.; Chng, W.-J. PRIMA-1met (APR-246) inhibits growth of colorectal cancer cells with different p53 status through distinct mechanisms. Oncotarget 2015, 6, 36689–36699. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, M.; Abdi, J.; Manujendra, S.N.; Chen, C.; Chang, H. PRIMA-1Met induces apoptosis in Waldenström’s Macroglobulinemia cells independent of p53. Cancer Biol. Ther. 2015, 16, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Konstantakou, E.G.; Voutsinas, G.E.; Velentzas, A.D.; Basogianni, A.-S.; Paronis, E.; Balafas, E.; Kostomitsopoulos, N.; Syrigos, K.N.; Anastasiadou, E.; Stravopodis, D.J. 3-BrPA eliminates human bladder cancer cells with highly oncogenic signatures via engagement of specific death programs and perturbation of multiple signaling and metabolic determinants. Mol. Cancer 2015, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Marzec, K.A.; Lin, M.Z.; Martin, J.L.; Baxter, R.C. Involvement of p53 in insulin-like growth factor binding protein-3 regulation in the breast cancer cell response to DNA damage. Oncotarget 2015, 6, 26583–26598. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, B.; Jacquot, C.; Le Palabe, J.; Malleter, M.; Tomasoni, C.; Boutard, T.; Sakanyan, V.; Roussakis, C. TP53 transcription factor for the NEDD9/HEF1/Cas-L gene: Potential targets in Non-Small Cell Lung Cancer treatment. Sci. Rep. 2015, 5, 10356. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; D’Orazi, V.; Crispini, A.; D’Orazi, G. Zn(II)-curc targets p53 in thyroid cancer cells. Int. J. Oncol. 2015, 47, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.H.; Read, M.; Cullinane, C.; Azar, W.J.; Fennell, C.M.; Montgomery, K.G.; Haupt, S.; Haupt, Y.; Wiman, K.G.; Duong, C.P.; et al. APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut 2015, 64, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yi, B.; Wang, C.; Chen, D.; Bae, S.; Wei, S.; Guo, R.-J.; Lu, C.; Nguyen, L.L.H.; Yang, W.-H.; et al. Silencing of CD24 Enhances the PRIMA-1-Induced Restoration of Mutant p53 in Prostate Cancer Cells. Clin. Cancer Res. 2016, 22, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Deben, C.; Lardon, F.; Wouters, A.; Op de Beeck, K.; Van den Bossche, J.; Jacobs, J.; Van Der Steen, N.; Peeters, M.; Rolfo, C.; Deschoolmeester, V.; et al. APR-246 (PRIMA-1(MET)) strongly synergizes with AZD2281 (olaparib) induced PARP inhibition to induce apoptosis in non-small cell lung cancer cell lines. Cancer Lett. 2016, 375, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Fransson, Å.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.; Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant High-Grade Serous ovarian cancer. J. Ovarian Res. 2016, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, N.; Kajiyama, H.; Nakamura, K.; Utsumi, F.; Niimi, K.; Mitsui, H.; Sekiya, R.; Suzuki, S.; Shibata, K.; Callen, D.; et al. PRIMA-1MET induces apoptosis through accumulation of intracellular reactive oxygen species irrespective of p53 status and chemo-sensitivity in epithelial ovarian cancer cells. Oncol. Rep. 2016, 35, 2543–2552. [Google Scholar] [CrossRef] [PubMed]
- Ali, D.; Mohammad, D.K.; Mujahed, H.; Jonson-Videsäter, K.; Nore, B.; Paul, C.; Lehmann, S. Anti-leukaemic effects induced by APR-246 are dependent on induction of oxidative stress and the NFE2L2/HMOX1 axis that can be targeted by PI3K and mTOR inhibitors in acute myeloid leukaemia cells. Br. J. Haematol. 2016, 174, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Teoh, P.J.; Bi, C.; Sintosebastian, C.; Tay, L.S.; Fonseca, R.; Chng, W.J. PRIMA-1 targets the vulnerability of multiple myeloma of deregulated protein homeostasis through the perturbation of ER stress via p73 demethylation. Oncotarget 2016, 7, 61806–61819. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Abdi, J.; Yang, Y.; Chang, H. MiRNA-29a as a tumor suppressor mediates PRIMA-1Met-induced anti-myeloma activity by targeting c-Myc. Oncotarget 2016, 7, 7149–7160. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Zou, Y.; Xu, G.; Potter, J.A.; Taylor, G.L.; Duan, Q.; Yang, Q.; Xiong, H.; Qiu, H.; Ye, D.; et al. PRIMA-1Met suppresses colorectal cancer independent of p53 by targeting MEK. Oncotarget 2016, 7, 83017–83030. [Google Scholar] [CrossRef] [PubMed]
- Aryee, D.N.T.; Niedan, S.; Ban, J.; Schwentner, R.; Muehlbacher, K.; Kauer, M.; Kofler, R.; Kovar, H. Variability in functional p53 reactivation by PRIMA-1(Met)/APR-246 in Ewing sarcoma. Br. J. Cancer 2013, 109, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Najem, A.; Salès, F.; van Kempen, L.C.; Sibille, C.; Awada, A.; Marine, J.-C.; et al. p53 Reactivation by PRIMA-1(Met) (APR-246) sensitises (V600E/K)BRAF melanoma to vemurafenib. Eur. J. Cancer 2016, 55, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Orue, A.; Chavez, V.; Strasberg-Rieber, M.; Rieber, M. Hypoxic resistance of KRAS mutant tumor cells to 3-Bromopyruvate is counteracted by Prima-1 and reversed by N-acetylcysteine. BMC Cancer 2016, 16, 902. [Google Scholar] [CrossRef] [PubMed]
- Patyka, M.; Sharifi, Z.; Petrecca, K.; Mansure, J.; Jean-Claude, B.; Sabri, S. Sensitivity to PRIMA-1MET is associated with decreased MGMT in human glioblastoma cells and glioblastoma stem cells irrespective of p53 status. Oncotarget 2016, 7, 60245–60269. [Google Scholar] [CrossRef] [PubMed]
- Synnott, N.C.; Murray, A.; McGowan, P.M.; Kiely, M.; Kiely, P.A.; O’Donovan, N.; O’Connor, D.P.; Gallagher, W.M.; Crown, J.; Duffy, M.J. Mutant p53: A novel target for the treatment of patients with triple-negative breast cancer? Int. J. Cancer 2017, 140, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, E.; Safa, M.; Sharifi, A.M.; Bashash, D. PRIMA-1 induces caspase-mediated apoptosis in acute promyelocytic leukemia NB4 cells by inhibition of nuclear factor-κB and downregulation of Bcl-2, XIAP, and c-Myc. Anticancer. Drugs 2017, 28, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Najem, A.; Krayem, M.; Salès, F.; Hussein, N.; Badran, B.; Robert, C.; Awada, A.; Journe, F.; Ghanem, G.E. P53 and MITF/Bcl-2 identified as key pathways in the acquired resistance of NRAS-mutant melanoma to MEK inhibition. Eur. J. Cancer 2017, 83, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Rieber, M.; Strasberg-Rieber, M. Hypoxia, Mn-SOD and H2O2 regulate p53 reactivation and PRIMA-1 toxicity irrespective of p53 status in human breast cancer cells. Biochem. Pharmacol. 2012, 84, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Idogawa, M.; Ohashi, T.; Sugisaka, J.; Sasaki, Y.; Suzuki, H.; Tokino, T. Array-based genome-wide RNAi screening to identify shRNAs that enhance p53-related apoptosis in human cancer cells. Oncotarget 2014, 5, 7540–7548. [Google Scholar] [CrossRef] [PubMed]
- Piantino, C.B.; Reis, S.T.; Viana, N.I.; Silva, I.A.; Morais, D.R.; Antunes, A.A.; Dip, N.; Srougi, M.; Leite, K.R. Prima-1 induces apoptosis in bladder cancer cell lines by activating p53. Clin. Sao Paulo Braz. 2013, 68, 297–303. [Google Scholar] [CrossRef]
- Zache, N.; Lambert, J.M.R.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cell. Oncol. 2008, 30, 411–418. [Google Scholar] [PubMed]
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.-K.; Hall, L.V.; Salehi, F.; Lin, D.-W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef] [PubMed]
- Shalom-Feuerstein, R.; Serror, L.; Aberdam, E.; Müller, F.-J.; van Bokhoven, H.; Wiman, K.G.; Zhou, H.; Aberdam, D.; Petit, I. Impaired epithelial differentiation of induced pluripotent stem cells from ectodermal dysplasia-related patients is rescued by the small compound APR-246/PRIMA-1MET. Proc. Natl. Acad. Sci. USA 2013, 110, 2152–2156. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; van den Bogaard, E.H.; Kouwenhoven, E.N.; Bykov, V.J.N.; Rinne, T.; Zhang, Q.; Tjabringa, G.S.; Gilissen, C.; van Heeringen, S.J.; Schalkwijk, J.; et al. APR-246/PRIMA-1(MET) rescues epidermal differentiation in skin keratinocytes derived from EEC syndrome patients with p63 mutations. Proc. Natl. Acad. Sci. USA 2013, 110, 2157–2162. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, C.; Wang, G.; Liu, Z.; Chen, P.; Yang, Q.; Dong, N.; Wu, H.; Liu, Z.; Li, W. APR-246/PRIMA-1Met Inhibits and Reverses Squamous Metaplasia in Human Conjunctival Epithelium. Investig. Ophthalmol. Vis. Sci. 2016, 57, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Faggio, C.; Alzoubi, K.; Calabrò, S.; Lang, F. Stimulation of suicidal erythrocyte death by PRIMA-1. Cell. Physiol. Biochem. 2015, 35, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Bykov, V.J.N.; Ali, D.; Andrén, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Reference | Molecule | Cancer Type | Reported IC50 (µmol/L) | Established Cell Line | Primary Culture (n) | Xenograft | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Wild-Type 53 | Mutant p53 | p53 Null | p53 KD | Wild-Type p53 | Mutant p53 | p53 Null | |||||
| [7] | PRIMA-1 | Colorectal, lung, ovary, burkitt lymphoma, osteosarcoma | (0.75–65) | HCT116, HDF, IARC-171 EW36, Seraphin, KH39, Caki1 | A461, SW480, Ramos, BL60, CW678, BL41, TK-10, KRC/Y+ (Saos-2-273, H1299-175, SKOV-175, SKOV-273, SKOV-175-22/23) a | H1299, Saos-2, SKOV | HCT116 | Yes | |||
| [10] | PRIMA-1 | Lung (non-small cell) | H460 | H23 | H1299 | ||||||
| [11] | PRIMA-1 | B cell chronic leukemia | 9 b | 5 c | |||||||
| [12] | PRIMA-1 | breast | (51–122) | MCF7 | MDA-MB-231, GI-101A | ||||||
| [8] | PRIMA-1 and APR-246 | Colorectal, lung, osteosarcoma | PRIMA-1: (14–24) APR-246: (9–19) | HCT116 | SW480+ (Saos-2-273, H1299-175) a | H1299, Saos-2 | HCT116 | Yes | |||
| [13] | PRIMA-1 | Acute myeloid leukemia | 52 b | 8 c | |||||||
| [14] | PRIMA-1 | Breast | MCF7 | DA-MB-231 | |||||||
| [15] | PRIMA-1 | Breast | MCF7 | MDA-MB-231, GI-101A | MDA-MB-231, GI-101A | ||||||
| [16] | PRIMA-1 | Colorectal, lung, breast, osteosarcoma | MCF7, U205 | SW480 + (H1299-175) a | H1299 | ||||||
| [17] | PRIMA-1 | Lung (non-small cell) | (60–175) * | A549 | LX1, SKMes | ||||||
| [18] | APR-246 | Prostate | 22RV1 | DU145 | PC3 | ||||||
| [19] | PRIMA-1 | Hepato-cellular carcinoma | Mahlavu, PLC5/PRF/5 + (Hep3B-249, Hep3B-248) a | Hep3B | PLC5/PRF/5 | Yes | |||||
| [20] | APR-246 | Colorectal, lung, osteosarcoma, lymphoma | MQ: (14.8;20.6) | HCT116 + (H1299, BL41) d | SBL41 + (Saos-2-273, H1299-175) a | H1299, Saos-2 | HCT116 | ||||
| [21] | APR-246 | Colorectal, lung, breast, lymphocyte | MCF7 | SW480 + (H1299-175) a | H1299 | ||||||
| [22] | APR-246 | Colon, lung, osteosarcoma | HCT116 + (Saos-2) d | SW480 + (Saos-2-273, H1299-175, HCT-116-248) a | H1299, Saos-2 | HCT116 | |||||
| [23] | PRIMA-1 | Breast | MDA-MB-231 | MDA-MB-231 | |||||||
| [24] | APR-246 | Colorectal, lung, osteosarcoma | (17–27) * | H1299, Saos-2-TA-p63γ, H1299-TA-p73α, H1299-TA-p73β, H1299-TA-p63γ | HCT116 | ||||||
| [25] | PRIMA-1 | Lung | A549 | H211, H1155 | H1299 | ||||||
| [26] | PRIMA-1 | Head and neck | JHU-028 | UMSCC-22a, JHU-029, Fadu | |||||||
| [27] | APR-246 | Lung (small cell) | (CCD32Lu) e | DMS456, DMS406, DMS273, DMS153, DMS114, DMS92, DMS79, DMS53, NCIH69, MAR24h, MAR86MI, GLC28, GLC26, GLC19, GLC16, GLC14, GLC3, GLC2 + (MDA-MB-231) f | H1299 | DMS273, DMS53, GLC16 | Yes | ||||
| [28] | PRIMA-1 | Breast | BT-474, HCC-1428 | Yes | |||||||
| [29] | APR-246 | Melanoma | AA, FM88 | C8161 + (M21) g | Yes | ||||||
| [30] | APR-246 | Acute myeloid leukemia | Mean = 5 | KBM3 | 25 | 7 | |||||
| [31] | PRIMA-1 | Thyroid | (10–75) * | TPC-1 | BC-PAP, Hth-74, FTC-133, C-643, 8305-C, FF-1 | SW1736 | Hth-74 | Data not found | |||
| [32] | PRIMA-1 | Breast | MCF7 | SKBR3 + (MCF7) a | |||||||
| [33] | PRIMA-1 | Breast | MCF7, MRC5 + (HCT116) j | MDA-MB-231, DLD-1 | T1 + (HCT116) j | ||||||
| [34] | APR-246 | Lung, osteosarcoma, burkitt lymphoma | (BL41) d | BL41 + (Saos-2-273, H1299-175) a | H1299, Saos-2 | ||||||
| [35] | PRIMA-1 | Ovary | A2781cp | A2781cp | |||||||
| [36] | APR-246 | Myeloma | Cell line: (5–20) * Primary culture: (4–30) * | MM1S, H929 | LP1, U266, 8266 | 8266R5 | MM1S, U266 | 6 | 3 c | Yes | |
| [32] | PRIMA-1 | Pancreas | (65–70) | Capan-2 | PANC-1, BxPC-3 | PANC-1 | |||||
| [37] | APR-246 | Myeloma | (3–200) | XG6, XG3, XG7, BCN, NAN9, H929, MDN, MM1S, AMO | NAN10, SKMM2, U266, XG1, XG11, XG5, 8226, JIM3, LP1, OPM2, XG2 + (NAN3, KMN1) h | JJN3, KMS11, NAN1, L363 | XG6, H929, XG5 | 16 b | 7 c | Yes | |
| [38] | PRIMA-1 | Thyroid | TPC-1, K1, IHH4 | FTC-133, WRO, 8505C, C-643, BC-PAP | |||||||
| [39] | PRIMA-1 | Thyroid | (28.7–128.5) | K1, IHH4 | C-643, BC-PAP | ||||||
| [40] | APR-246 | Ovary | (11–37) | A2780, A2780cis, A2780adr | OVCAR-3, A2780-CP20, IGROV-1/CDDP + (H1770, H1975, H596, H378) i | (H1417) i | 1 | 4 | Yes | ||
| [41] | APR-246 | Sarcoma | (7.9–26.1) | IB139 + (HCT116) j | IB130, IB134, IB138 + (HT-29) j | IB136, IB117 | |||||
| [42] | APR-246 | Colorectal | (7–58.6) | HCT116, RKO, LOVO | DLD-1, SW480, SW620, Colo320, Caco2, HT29 | HCT116 | Yes | ||||
| [43] | APR-246 | Waldenström | (10–30) | BCWM-1 | MWCL-1 | 2 | |||||
| [44] | PRIMA-1 | Bladder | RT4 | T24, T24-X | |||||||
| [45] | PRIMA-1 | Breast | MCF-10A | T47D, MDA-MB-468 | |||||||
| [46] | PRIMA-1 | Lung (non-small cell) | A549 | NSCLL-N6 | |||||||
| [47] | PRIMA-1 | Thyroid | WRO | FTC-133 | |||||||
| [48] | APR-246 | Oesophageal | (10–100) * | (NES) k, l | FLO-1, Eso26, OE19, OANC1, JH-EsoAd1, SKGT4, OE33, OACM5.1 + (H1299) a,i | OACP4C, TE7 m + (H1299) i | OACM5.1, OANC1, FLO-1, OE19, JH-EsoAd | Yes | |||
| [49] | PRIMA-1 | Prostate | LNCap + (PC3) d | DU145 + (PC3) a | PC3 | Yes | |||||
| [50] | APR-246 | Lung (non-small cell) | (9.56–29.35) | A549 | 1975, H2228, H596 | ||||||
| [51] | APR-246 | Ovary | (5.2–56) | 1 | 9 n | ||||||
| [52] | APR-246 | Ovary | (2.6–20.1) | NOS2, TOV21G, A2780 | NOS3, OVCAR-3, CAOV-3, OV-90, ES-2 | SKOV-3 | |||||
| [53] | APR-246 | Acute myeloid leukemia | (HCT116) j | KBM3 + (HCT116) a,j | (HCT116) j | 5 | |||||
| [54] | PRIMA-1 and APR-246 | Myeloma | PRIMA-1: (16.3–88.9) APR-246: (2–24.5) | H929 + (XG6, KMS18) h | U266, 8226, KMS28 | KMS11 + (JJN3) m | H929 | ||||
| [55] | APR-246 | Myelome | MM1S, H929 | U266, 8226, LP1 | 5 | Yes | |||||
| [56] | APR-246 | Colorectal | HCT 116, LOVO | SW480, DLD-1, HT29 | HCT116 | SW480, DLD-1 | Yes | ||||
| [57] | APR-246 | Ewing sarcoma | TC252 | STA-ET-7.2, RDES, IARC-EW2, RM82, SK-ES1, STA-ET-2.2 + (MDA-MB-468) f | A673, SK-N-MC | STA-ET-7.2 | (+3) o | ||||
| [58] | APR-246 | Melanoma | MM070, MM034, MM050,MM133, MM032, MM043, MM074-R, MM029, MM074, MM054 | Sk-MEL-28, MM164 | Yes | ||||||
| [59] | PRIMA-1 | Melanoma | MelJuso, C8161 + (A549) i | ||||||||
| [60] | APR-246 | Glioblastoma | (60–100) | U87MG, U87/EV, U87/MGMT | T98/EV, T98/shRNA, U138, LN-18, A172 | ||||||
| [61] | PRIMA-1 and APR-246 | Breast | PRIMA-1: (1.4–15.1) APR-246: (0.9–31.1) | UACC812, Hs878T(i8), ZR-75-1,MCF7,BT474 + (MCF12A, MCF10A) k | Hs878T(i8)2, HCC70, Hs578T, CAL-85-1, HCC1143, BT474, HCC1937, HDQ-P1, BT20, JimT1, Cama1, T47D, BT549,MDA-MB-468, MDA-MB-453 | ||||||
| [62] | APR-246 | Oesophageal | (NES) k,l | FLO-1, Eso26, OE19, OANC1, JH-EsoAd1, SKGT4, OE33, OACM5.1 + (H1299) a,i | OACP4C, TE7 m + (H1299) i | OACM5.1, OANC1, FLO-1, OE19, JH-EsoAd | Yes | ||||
| [63] | PRIMA-1 | Acute promyelocytic leukemia | NB4 | ||||||||
| [64] | APR-246 | Melanoma | MM161, MM057, MM165, MM052, MM167 | MM125 | |||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers 2017, 9, 172. https://doi.org/10.3390/cancers9120172
Perdrix A, Najem A, Saussez S, Awada A, Journe F, Ghanem G, Krayem M. PRIMA-1 and PRIMA-1Met (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers. 2017; 9(12):172. https://doi.org/10.3390/cancers9120172
Chicago/Turabian StylePerdrix, Anne, Ahmad Najem, Sven Saussez, Ahmad Awada, Fabrice Journe, Ghanem Ghanem, and Mohammad Krayem. 2017. "PRIMA-1 and PRIMA-1Met (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies" Cancers 9, no. 12: 172. https://doi.org/10.3390/cancers9120172
APA StylePerdrix, A., Najem, A., Saussez, S., Awada, A., Journe, F., Ghanem, G., & Krayem, M. (2017). PRIMA-1 and PRIMA-1Met (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers, 9(12), 172. https://doi.org/10.3390/cancers9120172