
The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis


Department of Human and Molecular Genetics, VCU Institute of Molecular Medicine, VCU Massey Cancer Center, School of Medicine, Virginia Commonwealth University, School of Medicine, Richmond, VA 23298, USA
*
Author to whom correspondence should be addressed.
Cancers 2017, 9(10), 131; https://doi.org/10.3390/cancers9100131
Submission received: 7 September 2017
/
Revised: 22 September 2017
/
Accepted: 25 September 2017
/
Published: 27 September 2017
(This article belongs to the Special Issue The Epithelial-to-Mesenchymal Transition (EMT) in Cancer)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Polo-like kinase 1 (PLK1) is a serine/threonine kinase that plays a key role in the regulation of the cell cycle. PLK1 is overexpressed in a variety of human tumors, and its expression level often correlates with increased cellular proliferation and poor prognosis in cancer patients. It has been suggested that PLK1 controls cancer development through multiple mechanisms that include canonical regulation of mitosis and cytokinesis, modulation of DNA replication, and cell survival. However, emerging evidence suggests novel and previously unanticipated roles for PLK1 during tumor development. In this review, we will summarize the recent advancements in our understanding of the oncogenic functions of PLK1, with a focus on its role in epithelial-mesenchymal transition and tumor invasion. We will further discuss the therapeutic potential of these functions.
1. Introduction
Polo-like kinases (PLKs) belong to the polo subfamily of serine/threonine kinases, which are highly evolutionarily conserved, from yeast to humans [1]. PLKs have emerged as important regulators for cell cycle progression, cell proliferation, differentiation, and adaptive responses (see reviews; [1,2,3]). The prototypic founding member of the family was identified in Drosophila melanogaster in 1988 and was named “Polo” since the knockout of the gene induced abnormal spindle poles during mitosis [4]. Only one Plk has been reported in the genomes of Drosophila (Polo), budding yeast (Cdc5) and fission yeast (Plo1) [2], whereas vertebrates have many PLK family members [2]. In humans, five PLK members (PLK1-PLK5) have been identified and exhibit differential tissue distributions and distinct functions with no or partial overlap in substrates [1,2,5,6] (Figure 1). Among the human PLKs, PLK1 has been most extensively studied.
Sharing a similar domain topology with other PLKs, full-length PLK1 is composed of an N-terminal serine/threonine kinase domain and the characteristic polo-box domain (PBD) in the C-terminus [7] (Figure 1). The PBD is comprised of two polo boxes, polo box 1 and polo box 2, which fold together to form a functional PBD. The PBD binds phosphorylated serine/threonine motifs in PLK1’s substrates. The optimal binding motif of its substrates is Ser-[pSer/pThr]-[Pro/X], in which X represents any amino acid [8,9]. By binding with such motifs on its substrates, the PBD brings the enzyme to an array of substrates found at different subcellular structures, including centrosomes, kinetochores, the mitotic spindle, and the midbody. This confers diversity to PLK1’s function and allows exquisite regulation of the cell cycle [2,10]. A PBD mutant (H538A, K540M) that is deficient in phospho-binding delocalizes PLK1 and disrupts its function [11]. PLK1 also interacts with some of its binding partners in a phospho-independent or PBD-independent manner. For instance, aurora borealis (Bora), aurora kinase A activator, was reported to be capable of binding to a PLK1 deletion mutant that lacks the PBD [12]. In addition to the role of the PBD in interacting with PLK1’s substrates, the PBD also modulates PLK1’s kinase activity through intramolecular interaction [13,14]. The PBD inhibits the kinase domain by reducing its flexibility. Reciprocally, the kinase domain induces a conformational alteration of the PBD that renders it less capable of interacting with its binding targets. Phosphopeptide binding or activational phosphorylation of the T210 residue of PLK1 within the kinase activation loop relieves the inhibitory intramolecular interaction [9,15].
PLK1 mediates almost every stage of cell division, including mitotic entry, centrosome maturation, bipolar spindle formation, chromosome congression and segregation, mitotic exit, and cytokinesis execution [2]. In addition to its canonical role in mitosis and cytokinesis, recent studies suggest that PLK1 may have other important functions such as regulation of microtubule dynamics, DNA replication, chromosome dynamics, p53 activity, and recovery from DNA damage-induced G2 arrest [16,17].
PLK1 is overexpressed in a variety of human tumors, and its expression level often correlates with increased cellular proliferation and poor prognosis in cancer patients [18,19]. It has been suggested that PLK1 controls cancer development through multiple mechanisms that include the canonical regulation of mitosis and cytokinesis, as well as modulation of DNA replication and cell survival [20,21]. However, emerging evidence suggests that the oncogenic functions of PLK1 extend far beyond what is currently known [21]. Here, we will discuss the recent advances in the understanding of PLK1 as an oncogene, with a focus on its role in epithelial-mesenchymal transition (EMT) and tumor invasion. We will further discuss the potential for therapeutic targeting of these newly identified oncogenic actions of PLK1.
2. PLK1 in Tumor Development
2.1. PLK1 Expression in Human Cancers
Consistent with its role in mitosis, PLK1 is highly expressed in the late G2 and M phases of the cell cycle, and enhanced PLK1 activity is observed in cells with high mitotic rates, including tumor cells [22,23]. Increasing evidence suggests that PLK1 is closely linked to human cancer development. For example, PLK1 is overexpressed in a variety of cancers, including prostate cancer [24], non-small cell lung cancer [25], head and neck cancer [26,27], esophageal and gastric cancer [28], melanoma [29], breast cancer [30], ovarian cancer [31], endometrial cancer [32], colorectal cancer [33], glioma [34], thyroid cancer [35], and hepatocellular cancer [36]. More importantly, its expression level often correlates with poor patient prognosis [19,24,26,27,28,29,37,38,39,40,41,42,43,44], suggesting that PLK1 is essential for tumorigenesis. Indeed, emerging evidence supports the notion that PLK1 is actively involved throughout the course of human cancer development [18,35,45,46,47,48,49] (Figure 2).
2.2. PLK1 and Oncogenic Pathways
A defining characteristic of cancer is the uncontrolled, abnormal growth of cells [50]. Since PLK1 plays a major role in regulating the cell cycle and maintaining genomic stability, it is believed that PLK1 controls cancer development through multiple mechanisms, including classic regulation of mitosis and cytokinesis, as well as response to cellular stress and cell survival [20,21].
However, recent studies show that PLK1 is capable of contributing to carcinogenesis through interconnections with multiple cancer-associated pathways. Several interacting partners of PLK1 have been identified that are encoded by tumor suppressor genes and oncogenes. For instance, the tumor suppressor p53 is considered to be the “guardian of the genome” and plays an important role in antiproliferation. Recent studies suggest that PLK1 has the ability to control p53’s activity through multiple pathways: (1) PLK1 phosphorylates and inhibits p53-dependent transcriptional activation as well as p53’s pro-apoptotic activity [51]; (2) PLK1 phosphorylates MDM2, an E3 ubiquitin ligase for p53, to promote p53 turnover [52,53]; (3) PLK1-mediated phosphorylation of S718 on Topors, a ubiquitin and SUMO E3 ligase, inhibits Topors-mediated SUMOylation of p53 and enhances ubiquitin-mediated degradation of p53 [53]; (4) PLK1 also phosphorylates G2 and S-phase-expressed 1 (GTSE1), resulting in GTSE1’s translocation into the nucleus, where it binds to and shuttles p53 out of the nucleus for degradation [54].
Phosphatase and tensin homologue (PTEN) is one of the most commonly disrupted tumor suppressors in human cancers [55]. PLK1 has been identified as an important regulator of PTEN. PLK1 catalytic activity has been shown to phosphorylate PTEN near its C-terminal tail, which contributes to the mitotic function of PTEN [56]. Moreover, Li and colleagues showed that PLK1 phosphorylates PTEN and Nedd4-1, an E3 ubiquitin ligase of PTEN, which leads to the inactivation of PTEN and activation of the phosphatidylinositol 3-kinase (PI3K) pathway, thereby facilitating aerobic glycolysis and promoting tumorigenesis [57]. Analyses using prostate cancer cell lines, a prostate-specific PTEN-deletion mouse model, and a xenograft mouse model revealed that PLK1 is critical for PTEN-depleted cells to adapt to mitotic stress for survival, which assists the loss of PTEN-induced prostate cancer formation [58]. It has been reported that PLK1 also interacts with other tumor suppressors such as CHK2 [59], BRCA1/2 [60,61], ATM [62] and ATR [63], BUB1B or BUBR1 [64,65], CYLD [66], REST [67], and TSC1/2 [68,69]. The imbalance between these interactions and the resulting deregulation of oncogenic pathways could contribute to cancer development.
While negatively regulating tumor suppressors, PLK1 also intensively interplays with numerous oncogenes [70]. For instance, the Forkhead box protein M1 (FoxM1) transcription factor is a major mitotic transcription factor that is required for the proliferation of normal cells [71]. However, FoxM1 is frequently overexpressed in a wide spectrum of human cancers [72]. More importantly, overwhelming evidence reveals that FoxM1 is implicated in different phases of cancer development, and all major hallmarks of cancer delineated by Hanahan and Weinberg [50]. Our previous studies showed that PLK1 directly interacts with and phosphorylates FoxM1, leading to the activation of FoxM1’s transcriptional activity. Activated FoxM1 then transcribes multiple mitotic regulators, including PLK1, which generate a positive feedback loop to further increase PLK1 levels and FoxM1 activity [73].
The MYC family of oncogenes contains three members (c-Myc, L-Myc, and N-Myc), which have been implicated in the genesis of specific human cancers [74]. Several studies have shown that PLK1 induces c-Myc accumulation by direct phosphorylation [75,76]. PLK1 can also stabilize N-Myc via the PLK1-Fbw7-Myc signaling circuit [77]. PLK1 binds to and phosphorylates the specificity factor Fbw7 of SCFFbw7 ubiquitin ligase, which promotes Fbw7 auto-polyubiquitination and proteasomal degradation and in turn prevents SCFFbw7-mediated degradation of N-Myc. Stabilized N-Myc further activates the transcription of PLK1, resulting in a positive feed-forward regulatory loop that strengthens N-Myc-regulated oncogenic programs [77].
2.3. PLK1 and Oncogenic Transformation
The constitutive expression of PLK1 in NIH/3T3 cells causes oncogenic foci formation and is tumorigenic in nude mice [78]. In contrast, depleting PLK1 in U2OS cells abrogates anchorage-independent growth [79]. These results highlight PLK1 as a possible driver of oncogenic transformation, although it remains unclear whether PLK1 itself is sufficient to induce tumor development. The oncogenic transformation potential of PLK1 has recently been documented in human cells. Our recent studies show that PLK1 overexpression in human prostate epithelial cells leads to cellular transformation in vitro and promotes tumor formation in NOD/SCID/γcnull (NSG) mice, which provides convincing evidence that PLK1 is directly involved in neoplastic transformation, and that PLK1 has a tumor-promoting role in the prostate [47].
2.4. PLK1 and EMT
A recent study from our group revealed an important additional function of PLK1 [47]. We documented an interesting observation that PLK1 overexpression in prostate epithelial cells causes the cells to change shape from an orthogonal epithelial cell morphology to a spindle-shaped fibroblast-like morphology, reminiscent of cells having undergone EMT. EMT is an important mechanism of tumor progression and metastasis [80,81]. It involves a loss of epithelial cell characteristics (cell–cell junctions, apicobasal cell polarity, and cobblestone morphology) and an acquisition of mesenchymal characteristics (fibroblast-like cell morphology, increased cell-matrix adhesions, and motility). On the molecular level, EMT can be easily recognized by the reduced expression of epithelial markers such as E-cadherin and some cytokeratin isoforms, and the elevated expression of mesenchymal markers such as N-cadherin and vimentin. Significantly, the loss of cell–cell contacts and the reorganization of the intracellular cytoskeleton during EMT result in increased cell migration and invasion [82], which allows cells to invade the surrounding stroma and vasculature, thereby leading to tumor dissemination and metastases [83]. In addition, EMT enables cancer cells to avoid apoptosis, anoikis, and oncogene addiction [84].
Indeed, forced overexpression of PLK1 in prostate epithelial cells led to the downregulation of epithelial markers (E-cadherin and cytokeratin 19) and upregulation of mesenchymal markers (N-cadherin, vimentin, fibronectin, and SM22) [47]. The switch from epithelial to mesenchymal markers did not depend on a specific stage of the cell cycle. Importantly, PLK1 overexpression in prostate epithelial cells disrupted the localization of E-cadherin, β-catenin, and junctional adhesion molecule (JAM)-A in areas of cell-cell contacts, which are indicative of the profound disassembly of adherens and tight junctions. In addition, this was accompanied by the dramatic reorganization of the actomyosin cytoskeleton manifested by the redistribution of non-muscle myosin IIB from perijunctional F-actin bundles into basal stress fibers. A comparison of EMT induction in cells expressing wild-type, constitutively active, or kinase-defective PLK1 suggests that a PLK1-mediated phosphorylation event contributes to the induction of EMT in prostate epithelial cells [47]. The role of PLK1 in EMT induction was further substantiated by the observation that PLK1 downregulation in metastatic prostate cancer cells enhances epithelial characteristics [47]. Moreover, an androgen-refractory cancer of the prostate (ARCaP) model was adopted for further validation [47]. ARCaP cells were derived from the ascites fluid of an 83-year-old Caucasian man diagnosed with metastatic prostate cancer [85]. Epithelium-like ARCaPE cells and mesenchymal-like ARCaPM cells are sublines of ARCaP cells that were isolated by single-cell dilution cloning [86]. Interestingly, PLK1 is not only differentially expressed and activated in these two cell lines (higher in the highly metastatic ARCaPM cells and lower in the less metastatic ARCaPE cells), it also controls the switch between EMT and mesenchymal-to-epithelial transition (MET) in those two cell lines (EMT induction in ARCaPE cells upon PLK1 overexpression, and MET induction in ARCaPM cells with PLK1 downregulation). Taken together, these results convincingly established a novel function of PLK1 as a critical regulator of EMT in prostate cancer.
Subsequently, the molecular mechanism underlying PLK1-mediated EMT was investigated [47]. We demonstrated that CRAF a member of the Raf kinase family of serine/threonine-specific protein kinases, is a physiological substrate of PLK1. CRAF consists of an N-terminal regulatory domain and a C-terminal catalytic domain. PLK1 directly interacts with and phosphorylates CRAF at S338 and S339 (the critical activating phosphorylation sites), resulting in CRAF activation. The activated CRAF undergoes autophosphorylation of S621, which hinders the proteasome-mediated degradation of CRAF, and thereby generates a positive feedback loop, leading to a further increase in the level and activity of CRAF. This activation event triggers the activation of downstream MEK1/2-ERK1/2 signaling in prostate epithelial cells overexpressing PLK1. Through a series of biochemical analyses, the events between PLK1-triggered MAPK signaling and EMT induction were elucidated [47]. ERK activation stimulates Fra1 expression; Fra1 belongs to the Fos gene family, whose protein products can dimerize with proteins of the JUN family, thereby forming the transcription factor complex AP-1. The ectopic expression of Fra1 in epithelioid cells resulted in morphologic changes that resembled fibroblastoid conversion, and increased motility and invasiveness [87]. Fra1 has been implicated as a potent regulator of anti-apoptosis, cell motility, and invasion in a variety of tumor cell types [88,89]. Enhanced expression of Fra1 then leads to the transcriptional activation of zinc finger E-box binding homeobox (ZEB) 1 and 2, two key transcription factors in EMT that orchestrate the EMT program [47]. Later, Cai et al. documented that PLK1 promotes EMT in gastric carcinoma cells through regulation of the AKT pathway [90], which suggests that regulating EMT is a general, and not a cell-type specific, function of PLK1, and the underlying mechanisms of PLK1-dependent EMT induction may vary dramatically from one setting to another. In addition to direct regulation, PLK1 may indirectly contribute to EMT induction through its substrates. For instance, FoxM1 has been linked to EMT in various tumor types, including pancreatic cancer [91,92], breast cancer [93], prostate cancer [94], gastric cancer [95], and lung cancer [96]. Several EMT regulators, such as Snail [97], Slug [93], and Twist [98] have been documented as direct targets of FoxM1. Given the aforementioned PLK1–FoxM1 regulatory circuit [73], it is likely that PLK1 may contribute to the EMT process by directly binding to and phosphorylating FoxM1, resulting in the activation of its transcriptional activity (Figure 3).
3. PLK1 in Tumor Invasion and Metastasis
Elevated PLK1 expression has been associated with an increased invasiveness of colorectal, breast, renal, and thyroid cancer cells [45,46,47,48,49]. PLK1 inhibition using either siRNA or pharmacological inhibitors caused significant reductions in the invasiveness of glioblastoma, bladder carcinoma, renal cell carcinoma, anaplastic thyroid carcinoma, and colorectal cancer cells [45,48,49,99,100]. Our recent study has provided direct evidence of the pro-invasive activity of PLK1 in tumor progression [47]. PLK1 was differentially expressed and/or activated in prostate cancer cells (higher in metastatic prostate cancer cell lines and lower in non-metastatic cell lines) [47]. In addition to EMT induction, PLK1 overexpression in prostate epithelial cells led to enhanced motility and invasiveness, as manifested by wound-healing scratch and Transwell invasion analyses [47]. The results were further validated by monitoring the random movement of the cells using time-lapse video microscopy and cell tracking, which indicated that PLK1 directly regulates the velocity of epithelial cell migration, independently of its effects on other cellular processes. Interestingly, NOD/SCID/γcnull (NSG) mice engrafted with PLK1-overexpressing prostate epithelial cells developed not only primary tumors, but also lung micrometastases, which suggests that PLK1 overexpression not only leads to the oncogenic transformation of prostate epithelial cells, but may also drive prostate cancer metastasis [47]. Consistently, PLK1 downregulation in metastatic prostate cancer cells inhibited cell motility [47].
Both the profound disassembly of adherens and tight junctions and the dramatic reorganization of the actomyosin cytoskeleton were observed in prostate epithelial cells undergoing PLK1-mediated EMT [47]. Therefore, the following mechanisms by which EMT induction promotes prostate cancer cell motility were proposed: (1) disassembly of epithelial junctions that weaken intercellular adhesions, thereby allowing cell dissemination [101,102], and (2) rearrangement of the actomyosin cytoskeleton from epithelia-specific perijunctional bundles to basal stress fibers that are characteristic of mesenchymal cells. This rearrangement enhances cell–matrix adhesion and enables more efficient cell migration [103,104].
In line with these findings, Rizki et al. showed that PLK1 mediates invasion through vimentin and β1 integrin in breast cancer cells, which is independent of its mitotic function [46]. PLK1 phosphorylates vimentin on S82, which regulates cell surface levels of β1 integrin and thereby promotes the invasiveness of breast cancer cells [46]. In addition, it has been reported that the downregulation of PLK1 in thyroid cancer cells led to a significant decrease in CD44v6, matrix metalloproteinase (MMP)-2, and MMP-9, which are all key players in tumor invasion and metastasis [49].
4. PLK1 as a Key Target for Cancer Therapy
PLK1 has been reported as widely overexpressed in tumor samples from cancer patients, and its overexpression has been validated as a biomarker of poor prognosis in a variety of human cancers [18,19]. Importantly, several studies have shown that inhibiting PLK1 expression or function by antibodies, RNAi, or small molecule inhibitors leads to mitotic arrest and apoptotic cell death in a wide range of human cancer cells, and is sufficient to prompt tumor regression in mouse xenograft models [105]. In contrast, toxicity modeling of PLK1-targeted therapies using primary human cells and various organs of adult Plk1 RNAi mice reveals that normal cells can tolerate up to a ~80% reduction in PLK1 level [106]. Thus, it has been repeatedly proposed that PLK1 could be a particularly attractive target for anti-cancer drug discovery [107]. Over the years, PLK1 has been the subject of an extensive effort in developing anti-mitotic agents that primarily target fast-growing mitotic cancer cells while leaving normal cells unscathed. To date, a large number of anti-PLK1 agents have been developed and tested under various preclinical and clinical settings, and several of them are currently in clinical trials, with varying degrees of success (for a comprehensive review, see [108,109]).
There are two druggable domains of PLK1 that have been pursued extensively: the catalytic domain and the PBD domain [108,109] (Figure 1). Volasertib (BI6727, a dihydropteridine derivative; Boehringer Ingelheim) is the most advanced inhibitor in the class of ATP-competitive inhibitors directed against the catalytic activity of PLK1. Its anti-cancer efficacies have been evaluated and proven to be superior in multiple nude mouse xenograft models [110]. Considerably, volasertib has also shown significant clinical efficacies against advanced solid and hematologic cancers in phase I/II clinical trials. Subsequently, a phase III clinical trial in elderly patients with acute myeloid leukemia was undertaken. The initial outcome, however, turned out to be less than satisfactory (presented at the 21st Annual Congress of the European Hematology Association, 2016 [111]). One of the major problems associated with the currently available PLK1 ATP-competitive inhibitors is their low degree of selectivity against other kinases, and their toxicity could be partly due to their interference with other kinases [108]. In the case of volasertib, it inhibits PLK2 and PLK3, with similar IC50 values as PLK1 [110,112]. A new generation of anti-PLK1 agents that target the PBD domain of PLK are currently being tested pre-clinically and have demonstrated improved specificity towards PLK1 [113]. Among all of the published PBD-interfering compounds, the most potent and selective inhibitors of the PBD have been peptide-like molecules [114,115,116]. However, they are often associated with poor cell membrane permeability due to their large size and the presence of charged groups [115,117]. Therefore, further efforts towards anti-PLK1 drug discovery will need to find new compounds with increased potency and specificity and improved pharmacokinetic properties to achieve better clinical outcomes.
The current rationale behind targeting PLK1 for anti-cancer therapy lies in its multifaceted functions throughout the cell cycle that target cancer’s sustaining proliferative signaling. Our recent study showed that PLK1 overexpression induces EMT and promotes cell motility and invasiveness in human prostate epithelial cells; whereas the attenuation of PLK1 expression reduces the invasiveness of human prostate cancer cells [47]. These novel findings not only provide mechanistic insight into the important role of PLK1 overexpression in human cancer development and metastasis, but will also aid the advancement of the prevention and treatment of advanced prostate cancer human cancers. In this regard, PLK1 can serve as a molecular biomarker to improve the stratification of cancer patients at high-, intermediate-, or low-risk of metastatic progression. In addition, PLK1 inhibition could potentially be a promising strategy to prevent prostate cancer dissemination. Consistent with this notion, a recent study reported that PLK1 depletion, mediated by PLK1 siRNA delivered by an antioxidant nanoparticle platform, inhibits lung metastasis and prolongs overall survival in a mouse model of breast cancer metastasis [118]. PLK1 inhibition by a small molecule inhibitor hindered brain metastases and prolonged survival in a mouse model of breast cancer brain metastasis [119]. These encouraging findings provide a proof of concept to substantiate the hypothesis above. Since the pro-metastatic properties of PLK1 overexpression were also reported in other different types of cancers, including breast and thyroid cancers [46,49], this suggests that targeting cancer’s ability to activate invasion and metastasis by PLK1 inhibition could be generalized to a growing list of a variety of cancers that have PLK1 overexpression implicated in their metastatic progression.
In recent years, EMT has also emerged to be a major driver of resistance to anti-cancer therapies, manifested not only in experimental models, but also in clinical settings [120]. Remnant cancer cells that survive after different types of therapies (including chemotherapy, molecularly targeted therapy, and immunotherapy), recurrently display signs of EMT activation [120]. Gene expression profiling of tumor samples revealed a strong correlation between an EMT gene signature and resistance to chemotherapy [121,122]. However, the mechanisms by which the activation of the EMT program triggers the development of resistance to therapeutics in cancer cells remain elusive. Nonetheless, these findings suggest that targeting cancer cells that have activated the EMT program might significantly improve the efficiency of therapeutic modalities in generating durable clinical responses. In this regard, targeting signaling pathways that are critical for the activation and subsequent maintenance of the EMT program have been demonstrated to be an effective therapeutic approach to prevent and/or reverse the EMT process, thereby overcoming therapeutic resistance [120]. As a major driving force of the EMT process in prostate cancer [47], PLK1 overexpression might contribute to therapeutic resistance to anti-cancer therapies through EMT. Indeed, PLK1 has been reported to be closely associated with drug resistance in cancer cells to a number of chemotherapy drugs, including doxorubicin, paclitaxel, and gemcitabine [108]. Therefore, the inhibition of PLK1 might reverse the drug resistance and increase sensitivity to chemotherapy. In this line of thinking, PLK1 inhibition has been reported to be effective in treating EGFR-inhibitor resistant non-small cell lung cancer through EMT [123,124]. Furthermore, PLK1 inhibition has been documented to enhance anti-cancer drug efficacy in a variety of types of human cancers [125,126,127,128]. Taken together, the emerging evidence for the novel oncogenic roles of PLK1 (ranging from neoplastic transformation, EMT induction, tumor invasion and metastasis, and therapeutic resistance) further highlights PLK1 as a fascinating anti-cancer target, and may substantially aid in developing and deploying anti-PLK1 therapeutics. It is reasonable to speculate that targeting PLK1 has the potential for multi-dimensional actions against cancer, and ultimately paves the way for curative cancer treatments.
5. Conclusions and Outlook
PLK1 is a fascinating multifaceted protein that targets many binding partners to ensure proper cell cycle progression and cell proliferation, and its deregulation contributes to the genesis of a broad range of human cancers. The differential requirement of PLK1 levels in cancer versus normal cells for survival makes PLK1 a particularly attractive target for anti-cancer drug discovery. Recently, a wealth of data has shed new light on the additional biochemical functions of PLK1 proteins and on the mechanisms through which they function in neoplastic transformation, tumor progression and dissemination, and the development of therapeutic resistance. The identification of the diverse roles of PLK1 throughout the course of tumor development highlights PLK1 as one of the most appealing anti-cancer drug targets.
Anti-PLK1 drug discovery has reached an advanced stage of development. A number of PLK1 inhibitors have been developed. The major problems commonly associated with currently available PLK1 inhibitors are insufficient specificity and cancer cell-selective killing. Further studies are needed to identify new compounds with increased potency and specificity and improved pharmacokinetic properties. Furthermore, there is no doubt that additional functions of PLK1 will be uncovered in the near future. Additional studies aimed at disclosing all of the molecular mechanisms of PLK1 signaling in cancers are needed to achieve the full therapeutic potential of an anti-PLK1 drug. In other words, better understanding of the oncogenic action of PLK1 overexpression will greatly facilitate the optimization of treatment regimens targeting PLK1 signaling to significantly enhance therapeutic efficacy.
Acknowledgments
This work was supported in part by grants from the American Cancer Society (ACS Research Scholar Grant 127626-RSG-15-005-01-CCG to Zheng Fu), and the National Institutes of Health (NIH R01 CA191002 to Zheng Fu). The authors thank Heidi Sankala Bauer for editorial assistance with the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Archambault, V.; Glover, D.M. Polo-like kinases: Conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell Biol. 2009, 10, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.A.; Sillje, H.H.; Nigg, E.A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004, 5, 429–440. [Google Scholar] [CrossRef] [PubMed]
- van de Weerdt, B.C.; Medema, R.H. Polo-like kinases: A team in control of the division. Cell Cycle 2006, 5, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Sunkel, C.E.; Glover, D.M. polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J. Cell Sci. 1988, 89, 25–38. [Google Scholar] [PubMed]
- de Carcer, G.; Manning, G.; Malumbres, M. From Plk1 to Plk5: Functional evolution of polo-like kinases. Cell Cycle 2011, 10, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Winkles, J.A.; Alberts, G.F. Differential regulation of polo-like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues. Oncogene 2005, 24, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.; Maloid, S.; Smith, M.R.; O’Connell, C.D.; Longo, D.L.; Ferris, D.K. Cloning and characterization of human and murine homologues of the Drosophila polo serine-threonine kinase. Cell Growth Differ. 1994, 5, 249–257. [Google Scholar] [PubMed]
- Elia, A.E.; Cantley, L.C.; Yaffe, M.B. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 2003, 299, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.; Rellos, P.; Haire, L.F.; Chao, J.W.; Ivins, F.J.; Hoepker, K.; Mohammad, D.; Cantley, L.C.; Smerdon, S.J.; Yaffe, M.B. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 2003, 115, 83–95. [Google Scholar] [CrossRef]
- Donaldson, M.M.; Tavares, A.A.; Hagan, I.M.; Nigg, E.A.; Glover, D.M. The mitotic roles of Polo-like kinase. J. Cell Sci. 2001, 114, 2357–2358. [Google Scholar] [PubMed]
- Lee, K.S.; Grenfell, T.Z.; Yarm, F.R.; Erikson, R.L. Mutation of the polo-box disrupts localization and mitotic functions of the mammalian polo kinase Plk. Proc. Natl. Acad. Sci. USA 1998, 95, 9301–9306. [Google Scholar] [CrossRef] [PubMed]
- Seki, A.; Coppinger, J.A.; Jang, C.Y.; Yates, J.R.; Fang, G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008, 320, 1655–1658. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Soung, N.K.; Johmura, Y.; Kang, Y.H.; Liao, C.; Lee, K.H.; Park, C.H.; Nicklaus, M.C.; Lee, K.S. Polo-box domain: A versatile mediator of polo-like kinase function. Cell Mol. Life Sci. 2010, 67, 1957–1970. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, W.; Raaijmakers, J.A.; Medema, R.H. Switching Polo-like kinase-1 on and off in time and space. Trends Biochem. Sci. 2012, 37, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Lin, C.Y.; Ma, S.; Erikson, R.L. Functional studies on the role of the C-terminal domain of mammalian polo-like kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 1984–1989. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Song, B.; Liu, X. The substrates of Plk1, beyond the functions in mitosis. Protein Cell 2010, 1, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Liu, X.S.; Davis, K.; Liu, X. Plk1 phosphorylation of Orc2 promotes, D.N.A replication under conditions of stress. Mol. Cell. Biol. 2011, 31, 4844–4856. [Google Scholar] [CrossRef] [PubMed]
- Cholewa, B.D.; Liu, X.; Ahmad, N. The role of polo-like kinase 1 in carcinogenesis: Cause or consequence? Cancer Res. 2013, 73, 6848–6855. [Google Scholar] [CrossRef] [PubMed]
- Takai, N.; Hamanaka, R.; Yoshimatsu, J.; Miyakawa, I. Polo-like kinases (Plks) and cancer. Oncogene 2005, 24, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Deeraksa, A.; Pan, J.; Sha, Y.; Liu, X.D.; Eissa, N.T.; Lin, S.H.; Yu-Lee, L.Y. Plk1 is upregulated in androgen-insensitive prostate cancer cells and its inhibition leads to necroptosis. Oncogene 2012, 32, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Liu, X. Polo-like kinase 1, on the rise from cell cycle regulation to prostate cancer development. Protein Cell 2012, 3, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Golsteyn, R.M.; Mundt, K.E.; Fry, A.M.; Nigg, E.A. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J. Cell Biol. 1995, 129, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Yuan, Y.L.; Kuriyama, R.; Erikson, R.L. Plk is an M-phase-specific protein kinase and interacts with a kinesin-like protein, CHO1/MKLP-1. Mol. Cell. Biol. 1995, 15, 7143–7151. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Schmidt, M.; Gekeler, V.; Denkert, C.; Stephan, C.; Jung, K.; Loening, S.; Dietel, M.; Kristiansen, G. Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. Prostate 2004, 60, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Elez, R.; Doermer, A.; Holtrich, U.; Ackermann, H.; Stutte, H.J.; Altmannsberger, H.M.; Rubsamen-Waigmann, H.; Strebhardt, K. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 1997, 14, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Knecht, R.; Elez, R.; Oechler, M.; Solbach, C.; von Ilberg, C.; Strebhardt, K. Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res. 1999, 59, 2794–2797. [Google Scholar] [PubMed]
- Knecht, R.; Oberhauser, C.; Strebhardt, K. PLK (polo-like kinase), a new prognostic marker for oropharyngeal carcinomas. Int. J. Cancer 2000, 89, 535–536. [Google Scholar] [CrossRef]
- Tokumitsu, Y.; Mori, M.; Tanaka, S.; Akazawa, K.; Nakano, S.; Niho, Y. Prognostic significance of polo-like kinase expression in esophageal carcinoma. Int. J. Oncol. 1999, 15, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Kneisel, L.; Linhart, C.; Bernd, A.; Kaufmann, R. Prognostic value of pololike kinase expression in melanomas. JAMA 2000, 283, 479–480. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Hildenbrand, R.; Schwar, C.; Grobholz, R.; Kaufmann, M.; Stutte, H.J.; Strebhardt, K.; Bleyl, U. Polo-like kinase: A novel marker of proliferation: Correlation with estrogen-receptor expression in human breast cancer. Pathol. Res. Pract. 2000, 196, 753–759. [Google Scholar] [PubMed]
- Weichert, W.; Denkert, C.; Schmidt, M.; Gekeler, V.; Wolf, G.; Kobel, M.; Dietel, M.; Hauptmann, S. Polo-like kinase isoform expression is a prognostic factor in ovarian carcinoma. Br. J. Cancer 2004, 90, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Takai, N.; Miyazaki, T.; Fujisawa, K.; Nasu, K.; Hamanaka, R.; Miyakawa, I. Polo-like kinase (PLK) expression in endometrial carcinoma. Cancer Lett. 2001, 169, 41–49. [Google Scholar] [CrossRef]
- Takahashi, T.; Sano, B.; Nagata, T.; Kato, H.; Sugiyama, Y.; Kunieda, K.; Kimura, M.; Okano, Y.; Saji, S. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci. 2003, 94, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Dietzmann, K.; Kirches, E.; von, B.; Jachau, K.; Mawrin, C. Increased human polo-like kinase-1 expression in gliomas. J. Neurooncol. 2001, 53, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Miyoshi, E.; Sasaki, N.; Kakudo, K.; Yoshida, H.; Tomoda, C.; Uruno, T.; Takamura, Y.; Miya, A.; Kobayashi, K.; et al. Polo-like kinase 1 overexpression is an early event in the progression of papillary carcinoma. Br. J. Cancer 2004, 90, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Mok, W.C.; Wasser, S.; Tan, T.; Lim, S.G. Polo-like kinase 1, a new therapeutic target in hepatocellular carcinoma. World J. Gastroenterol. 2012, 18, 3527–3536. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.B.; Lin, D.C.; Shi, Z.Z.; Wang, X.C.; Shen, X.M.; Zhang, Y.; Du, X.L.; Luo, M.L.; Xu, X.; Han, Y.L.; et al. Overexpression of PLK1 is associated with poor survival by inhibiting apoptosis via enhancement of survivin level in esophageal squamous cell carcinoma. Int J. Cancer 2009, 124, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Kanaji, S.; Saito, H.; Tsujitani, S.; Matsumoto, S.; Tatebe, S.; Kondo, A.; Ozaki, M.; Ito, H.; Ikeguchi, M. Expression of polo-like kinase 1 (PLK1) protein predicts the survival of patients with gastric carcinoma. Oncology 2006, 70, 126–133. [Google Scholar] [CrossRef] [PubMed]
- King, S.I.; Purdie, C.A.; Bray, S.E.; Quinlan, P.R.; Jordan, L.B.; Thompson, A.M.; Meek, D.W. Immunohistochemical detection of Polo-like kinase-1 (PLK1) in primary breast cancer is associated with TP53 mutation and poor clinical outcom. Breast Cancer Res. 2012, 14, R40. [Google Scholar] [CrossRef] [PubMed]
- Tut, T.G.; Lim, S.H.; Dissanayake, I.U.; Descallar, J.; Chua, W.; Ng, W.; de Souza, P.; Shin, J.S.; Lee, C.S. Upregulated Polo-Like Kinase 1 Expression Correlates with Inferior Survival Outcomes in Rectal Cancer. PLoS ONE 2015, 10, e0129313. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Kristiansen, G.; Schmidt, M.; Gekeler, V.; Noske, A.; Niesporek, S.; Dietel, M.; Denkert, C. Polo-like kinase 1 expression is a prognostic factor in human colon cancer. World J. Gastroenterol. 2005, 11, 5644–5650. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Kristiansen, G.; Winzer, K.J.; Schmidt, M.; Gekeler, V.; Noske, A.; Muller, B.M.; Niesporek, S.; Dietel, M.; Denkert, C. Polo-like kinase isoforms in breast cancer: Expression patterns and prognostic implications. Virchows Arch. 2005, 446, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Ohira, M.; Horie, H.; Ando, K.; Takayasu, H.; Suzuki, Y.; Sugano, S.; Hirata, T.; Goto, T.; Matsunaga, T.; et al. Expression profiling and differential screening between hepatoblastomas and the corresponding normal livers: Identification of high expression of the PLK1 oncogene as a poor-prognostic indicator of hepatoblastomas. Oncogene 2004, 23, 5901–5911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, G.; Kong, C. High expression of polo-like kinase 1 is associated with the metastasis and recurrence in urothelial carcinoma of bladder. Urol. Oncol. 2013, 31, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Han, D.P.; Zhu, Q.L.; Cui, J.T.; Wang, P.X.; Qu, S.; Cao, Q.F.; Zong, Y.P.; Feng, B.; Zheng, M.H.; Lu, A.G. Polo-like kinase 1 is overexpressed in colorectal cancer and participates in the migration and invasion of colorectal cancer cells. Med. Sci. Monit. 2012, 18, BR237–BR246. [Google Scholar] [CrossRef] [PubMed]
- Rizki, A.; Mott, J.D.; Bissell, M.J. Polo-like kinase 1 is involved in invasion through extracellular matrix. Cancer Res. 2007, 67, 11106–11110. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ivanov, A.I.; Fisher, P.B.; Fu, Z. Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. Elife 2016, 5, e10734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, Z.; Liu, Z. Polo-like kinase 1 is overexpressed in renal cancer and participates in the proliferation and invasion of renal cancer cells. Tumour Biol. 2013, 34, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.G.; Lu, X.F.; Jiao, X.M.; Chen, B.; Wu, J.X. PLK1 gene suppresses cell invasion of undifferentiated thyroid carcinoma through the inhibition of CD44v6, MMP-2 and MMP-9. Exp. Ther. Med. 2012, 4, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Ozaki, T.; Yamamoto, H.; Furuya, K.; Hosoda, M.; Hayashi, S.; Fukuzawa, M.; Nakagawara, A. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J. Biol. Chem. 2004, 279, 25549–25561. [Google Scholar] [CrossRef] [PubMed]
- Dias, S.S.; Hogan, C.; Ochocka, A.M.; Meek, D.W. Polo-like kinase-1 phosphorylates MDM2 at Ser260 and stimulates MDM2-mediated p53 turnover. FEBS Lett. 2009, 583, 3543–3548. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, H.; Zhou, Z.; Wang, W.H.; Deng, A.; Andrisani, O.; Liu, X. Plk1-mediated phosphorylation of Topors regulates p53 stability. J. Biol. Chem. 2009, 284, 18588–18592. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Li, H.; Song, B.; Liu, X. Polo-like kinase 1 phosphorylation of G2 and S-phase-expressed 1 protein is essential for p53 inactivation during G2 checkpoint recovery. EMBO Rep. 2010, 11, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell. Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Pagano, M.; Dai, W. Plk1 protein phosphorylates phosphatase and tensin homolog (PTEN) and regulates its mitotic activity during the cell cycle. J. Biol. Chem. 2014, 289, 14066–14074. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, J.; Bi, P.; Lu, Y.; Burcham, G.; Elzey, B.D.; Ratliff, T.; Konieczny, S.F.; Ahmad, N.; Kuang, S.; et al. Plk1 phosphorylation of PTEN causes a tumor-promoting metabolic state. Mol. Cell. Biol. 2014, 34, 3642–3661. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Song, B.; Elzey, B.D.; Ratliff, T.L.; Konieczny, S.F.; Cheng, L.; Ahmad, N.; Liu, X. Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J. Biol. Chem. 2011, 286, 35795–35800. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, L.; Xu, X.; Li, J.; Stern, D.F. Polo-like kinase 1 and Chk2 interact and co-localize to centrosomes and the midbody. J. Biol. Chem. 2003, 278, 8468–8475. [Google Scholar] [CrossRef] [PubMed]
- Chabalier-Taste, C.; Brichese, L.; Racca, C.; Canitrot, Y.; Calsou, P.; Larminat, F. Polo-like kinase 1 mediates BRCA1 phosphorylation and recruitment at DNA double-strand breaks. Oncotarget 2016, 7, 2269–2283. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Daniels, M.J.; Venkitaraman, A.R. Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated by DNA damage and mitotic progression. Oncogene 2004, 23, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Hwang, H.I.; Jang, Y.J. Mitotic DNA damage response: Polo-like kinase-1 is dephosphorylated through ATM-Chk1 pathway. Cell Cycle 2010, 9, 2389–2398. [Google Scholar] [CrossRef] [PubMed]
- Deming, P.B.; Flores, K.G.; Downes, C.S.; Paules, R.S.; Kaufmann, W.K. ATR enforces the topoisomerase II-dependent G2 checkpoint through inhibition of Plk1 kinase. J. Biol. Chem. 2002, 277, 36832–36838. [Google Scholar] [CrossRef] [PubMed]
- Elowe, S.; Hummer, S.; Uldschmid, A.; Li, X.; Nigg, E.A. Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev. 2007, 21, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Matsumoto, Y.; Ikeuchi, T.; Saya, H.; Kajii, T.; Matsuura, S. BubR1 localizes to centrosomes and suppresses centrosome amplification via regulating Plk1 activity in interphase cells. Oncogene 2009, 28, 2806–2820. [Google Scholar] [CrossRef] [PubMed]
- Stegmeier, F.; Sowa, M.E.; Nalepa, G.; Gygi, S.P.; Harper, J.W.; Elledge, S.J. The tumor suppressor CYLD regulates entry into mitosis. Proc. Natl. Acad. Sci. USA 2007, 104, 8869–8874. [Google Scholar] [CrossRef] [PubMed]
- Karlin, K.L.; Mondal, G.; Hartman, J.K.; Tyagi, S.; Kurley, S.J.; Bland, C.S.; Hsu, T.Y.; Renwick, A.; Fang, J.E.; Migliaccio, I.; et al. The oncogenic STP axis promotes triple-negative breast cancer via degradation of the REST tumor suppressor. Cell Rep. 2014, 9, 1318–1332. [Google Scholar] [CrossRef] [PubMed]
- Astrinidis, A.; Senapedis, W.; Henske, E.P. Hamartin, the tuberous sclerosis complex 1 gene product, interacts with polo-like kinase 1 in a phosphorylation-dependent manner. Hum. Mol. Genet. 2006, 15, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.; Hanneder, M.; Siegel, N.; Valli, A.; Hengstschlager, M. The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partners. Mutat. Res. 2008, 658, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Kooistra, M.R.; Bras, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Gartel, A.L. FOX(M1) news—It is cancer. Mol. Cancer Ther. 2013, 12, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Malureanu, L.; Huang, J.; Wang, W.; Li, H.; van Deursen, J.M.; Tindall, D.J.; Chen, J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat. Cell. Biol. 2008, 10, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Li, X.; Bieberich, C.J. Protein kinase A regulates MYC protein through transcriptional and post-translational mechanisms in a catalytic subunit isoform-specific manner. J. Biol. Chem. 2013, 288, 14158–14169. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Li, Z.; Lee, P.L.; Guan, P.; Aau, M.Y.; Lee, S.T.; Feng, M.; Lim, C.Z.; Lee, E.Y.; Wee, Z.N.; et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 2013, 3, 1156–1171. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Yue, M.; Su, H.; Ren, P.; Jiang, J.; Li, F.; Hu, Y.; Du, H.; Liu, H.; Qing, G. Polo-like Kinase-1 Regulates Myc Stabilization and Activates a Feedforward Circuit Promoting Tumor Cell Survival. Mol. Cell 2016, 64, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Wilson, M.L.; Hamanaka, R.; Chase, D.; Kung, H.; Longo, D.L.; Ferris, D.K. Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem. Biophys. Res. Commun. 1997, 234, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Yuan, J.; Strebhardt, K. Polo-like kinases and oncogenesis. Oncogene 2005, 24, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef] [PubMed]
- Hugo, H.; Ackland, M.L.; Blick, T.; Lawrence, M.G.; Clements, J.A.; Williams, E.D.; Thompson, E.W. Epithelial—Mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J. Cell Physiol. 2007, 213, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, N.; Gheldof, A.; Tatari, M.; Christofori, G. EMT as the ultimate survival mechanism of cancer cells. Semin. Cancer Biol. 2012, 22, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Zhau, H.Y.; Chang, S.M.; Chen, B.Q.; Wang, Y.; Zhang, H.; Kao, C.; Sang, Q.A.; Pathak, S.J.; Chung, L.W. Androgen-repressed phenotype in human prostate cancer. Proc. Natl. Acad. Sci. USA 1996, 93, 15152–15157. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, R.; Xie, Z.H.; Odero-Marah, V.; Pathak, S.; Multani, A.; Chung, L.W.; Zhau, H.E. Prostate cancer metastasis: Role of the host microenvironment in promoting epithelial to mesenchymal transition and increased bone and adrenal gland metastasis. Prostate 2006, 66, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Kustikova, O.; Kramerov, D.; Grigorian, M.; Berezin, V.; Bock, E.; Lukanidin, E.; Tulchinsky, E. Fra-1 induces morphological transformation and increases in vitro invasiveness and motility of epithelioid adenocarcinoma cells. Mol. Cell. Biol. 1998, 18, 7095–7105. [Google Scholar] [CrossRef] [PubMed]
- Milde-Langosch, K. The Fos family of transcription factors and their role in tumourigenesis. Eur. J. Cancer 2005, 41, 2449–2461. [Google Scholar] [CrossRef] [PubMed]
- Young, M.R.; Colburn, N.H. Fra-1 a target for cancer prevention or intervention. Gene 2006, 379, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.P.; Chen, L.D.; Song, H.B.; Zhang, C.X.; Yuan, Z.W.; Xiang, Z.X. PLK1 promotes epithelial-mesenchymal transition and metastasis of gastric carcinoma cells. Am. J. Transl. Res. 2016, 8, 4172–4183. [Google Scholar] [PubMed]
- Bao, B.; Wang, Z.; Ali, S.; Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Azmi, A.S.; Miele, L.; Sarkar, F.H. Over-expression of FoxM1 leads to epithelial-mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J. Cell Biochem. 2011, 112, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Xie, D.; Cui, J.; Li, Q.; Gao, Y.; Xie, K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clin. Cancer Res. 2014, 20, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, H.; Tan, G.; Gao, W.; Cheng, L.; Jiang, X.; Yu, L.; Tan, Y. FOXM1 promotes the epithelial to mesenchymal transition by stimulating the transcription of Slug in human breast cancer. Cancer Lett. 2013, 340, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yao, B.; Zhang, M.; Fu, S.; Gao, H.; Peng, R.; Zhang, L.; Tang, J. Increased FoxM1 expression is a target for metformin in the suppression of EMT in prostate cancer. Int. J. Mol. Med. 2014, 33, 1514–1522. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Xiong, X.; Lin, Y.; Cheng, Y.; Lu, J.; Zhang, J.; Cheng, N. Down-regulation of FoxM1 leads to the inhibition of the epithelial-mesenchymal transition in gastric cancer cells. Cancer Genet. 2014, 207, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.F.; Qu, Z.Q.; Yuan, H.H.; Wang, J.Y.; Zhao, M.; Guo, Y.H.; Shi, J.; Gong, X.D.; Zhu, Y.L.; Liu, F.; et al. Overexpression of FOXM1 is associated with EMT and is a predictor of poor prognosis in non-small cell lung cancer. Oncol Rep. 2014, 31, 2660–2668. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Zhang, N.; Wang, Y.; Li, D.; Wang, L.; Sun, X.; Shen, C.; Yang, Y.; Zhou, X.; Du, X. FOXM1 promotes lung adenocarcinoma invasion and metastasis by upregulating SNAIL. Int. J. Biol. Sci. 2015, 11, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Wu, S.; Wu, H.; Li, L.; Guo, T. CD44 is functionally crucial for driving lung cancer stem cells metastasis through Wnt/beta-catenin-FoxM1-Twist signaling. Mol. Carcinog. 2016, 55, 1962–1973. [Google Scholar] [CrossRef] [PubMed]
- Brassesco, M.S.; Pezuk, J.A.; Morales, A.G.; de Oliveira, J.C.; Roberto, G.M.; da Silva, G.N.; Francisco de Oliveira, H.; Scrideli, C.A.; Tone, L.G. In vitro targeting of Polo-like kinase 1 in bladder carcinoma: Comparative effects of four potent inhibitors. Cancer Biol. Ther. 2013, 14, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Pezuk, J.A.; Brassesco, M.S.; Morales, A.G.; de Oliveira, J.C.; de Paula Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Neder, L.; Scrideli, C.A.; Tone, L.G. Polo-like kinase 1 inhibition causes decreased proliferation by cell cycle arrest, leading to cell death in glioblastoma. Cancer Gene Ther. 2013, 20, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Godde, N.J.; Galea, R.C.; Elsum, I.A.; Humbert, P.O. Cell polarity in motion: Redefining mammary tissue organization through EMT and cell polarity transitions. J. Mammary Gland Biol. Neoplasia 2010, 15, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adh. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Kamelgarn, M.; Kyprianou, N. Cytoskeleton targeting value in prostate cancer treatment. Am. J. Clin. Exp. Urol. 2014, 2, 15–26. [Google Scholar] [PubMed]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.; Efferth, T. Polo-like kinase 1 as target for cancer therapy. Exp. Hematol. Oncol. 2012, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Kappel, S.; Kramer, A.; Sanhaji, M.; Matthess, Y.; Kurunci-Csacsko, E.; Calzada-Wack, J.; Rathkolb, B.; Rozman, J.; Adler, T.; et al. Toxicity modelling of Plk1-targeted therapies in genetically engineered mice and cultured primary mammalian cells. Nat. Commun. 2011, 2, 395. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, Y.; Lampkin, T. Targeting Polo-like kinase in cancer therapy. Clin. Cancer Res. 2010, 16, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, R.E.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Hymel, D.; Burke, T.R., Jr.; Lee, K.S. Current progress and future perspectives in the development of anti-polo-like kinase 1 therapeutic agents. F1000Res 2017, 6, 1024. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef] [PubMed]
- Results of Phase III Study of Volasertib for the Treatment of Acute Myeloid Leukemia Presented at European Hematology Association Annual Meeting. Available online: http://www.evaluategroup.com/Universal/View.aspx?type=Story&id=649494 (accessed on September 2017).
- Gjertsen, B.T.; Schoffski, P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 2015, 29, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archambault, V.; Normandin, K. Several inhibitors of the Plk1 Polo-Box Domain turn out to be non-specific protein alkylators. Cell Cycle 2017, 16, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Burke, T.R., Jr.; Park, J.E.; Bang, J.K.; Lee, E. Recent Advances and New Strategies in Targeting Plk1 for Anticancer Therapy. Trends Pharmacol. Sci. 2015, 36, 858–877. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Park, J.E.; Qian, W.J.; Lim, D.; Graber, M.; Berg, T.; Yaffe, M.B.; Lee, K.S.; Burke, T.R., Jr. Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel. Nat. Chem. Biol. 2011, 7, 595–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Park, J.E.; Qian, W.J.; Lim, D.; Scharow, A.; Berg, T.; Yaffe, M.B.; Lee, K.S.; Burke, T.R., Jr. Identification of high affinity polo-like kinase 1 (Plk1) polo-box domain binding peptides using oxime-based diversification. ACS Chem. Biol. 2012, 7, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.J.; Park, J.E.; Lim, D.; Lai, C.C.; Kelley, J.A.; Park, S.Y.; Lee, K.W.; Yaffe, M.B.; Lee, K.S.; Burke, T.R., Jr. Mono-anionic phosphopeptides produced by unexpected histidine alkylation exhibit high Plk1 polo-box domain-binding affinities and enhanced antiproliferative effects in HeLa cells. Biopolymers 2014, 102, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Morry, J.; Ngamcherdtrakul, W.; Gu, S.; Reda, M.; Castro, D.J.; Sangvanich, T.; Gray, J.W.; Yantasee, W. Targeted Treatment of Metastatic Breast Cancer by, PLK1 siRNA Delivered by an Antioxidant Nanoparticle Platform. Mol. Cancer Ther. 2017, 16, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Hua, E.; Bisht, K.; Woditschka, S.; Skordos, K.W.; Liewehr, D.J.; Steinberg, S.M.; Brogi, E.; Akram, M.M.; Killian, J.K.; et al. Inhibition of Polo-like kinase 1 prevents the growth of metastatic breast cancer cells in the brain. Clin. Exp. Metastasis 2011, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Farmer, P.; Bonnefoi, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Becette, V.; Andre, S.; Piccart, M.; Campone, M.; Brain, E.; et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Singh, R.; Wang, L.; Nilsson, M.; Goonatilake, R.; Tong, P.; Li, L.; Giri, U.; Villalobos, P.; Mino, B.; et al. Polo-like kinase 1 inhibition diminishes acquired resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer with T790M mutations. Oncotarget 2016, 7, 47998–48010. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Rubio-Viqueira, B.; Rajeshkumar, N.V.; Chan, A.; sSolomon, A.; Hidalgo, M. A fine-needle aspirate-based vulnerability assay identifies polo-like kinase 1 as a mediator of gemcitabine resistance in pancreatic cancer. Mol. Cancer Ther. 2010, 9, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Spankuch, B.; Heim, S.; Kurunci-Csacsko, E.; Lindenau, C.; Yuan, J.; Kaufmann, M.; Strebhardt, K. Down-regulation of Polo-like kinase 1 elevates drug sensitivity of breast cancer cells in vitro and in vivo. Cancer Res. 2006, 66, 5836–5846. [Google Scholar] [CrossRef] [PubMed]
- Spankuch, B.; Kurunci-Csacsko, E.; Kaufmann, M.; Strebhardt, K. Rational combinations of siRNAs targeting Plk1 with breast cancer drugs. Oncogene 2007, 26, 5793–5807. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhang, X.; Sun, G.; Guo, X.; Li, H.; You, Y.; Jacobs, J.L.; Gardner, K.; Yuan, D.; Xu, Z.; et al. RNA interference-mediated silencing of the polo-like kinase 1 gene enhances chemosensitivity to gemcitabine in pancreatic adenocarcinoma cells. J. Cell. Mol. Med. 2008, 12, 2334–2349. [Google Scholar] [CrossRef] [PubMed]
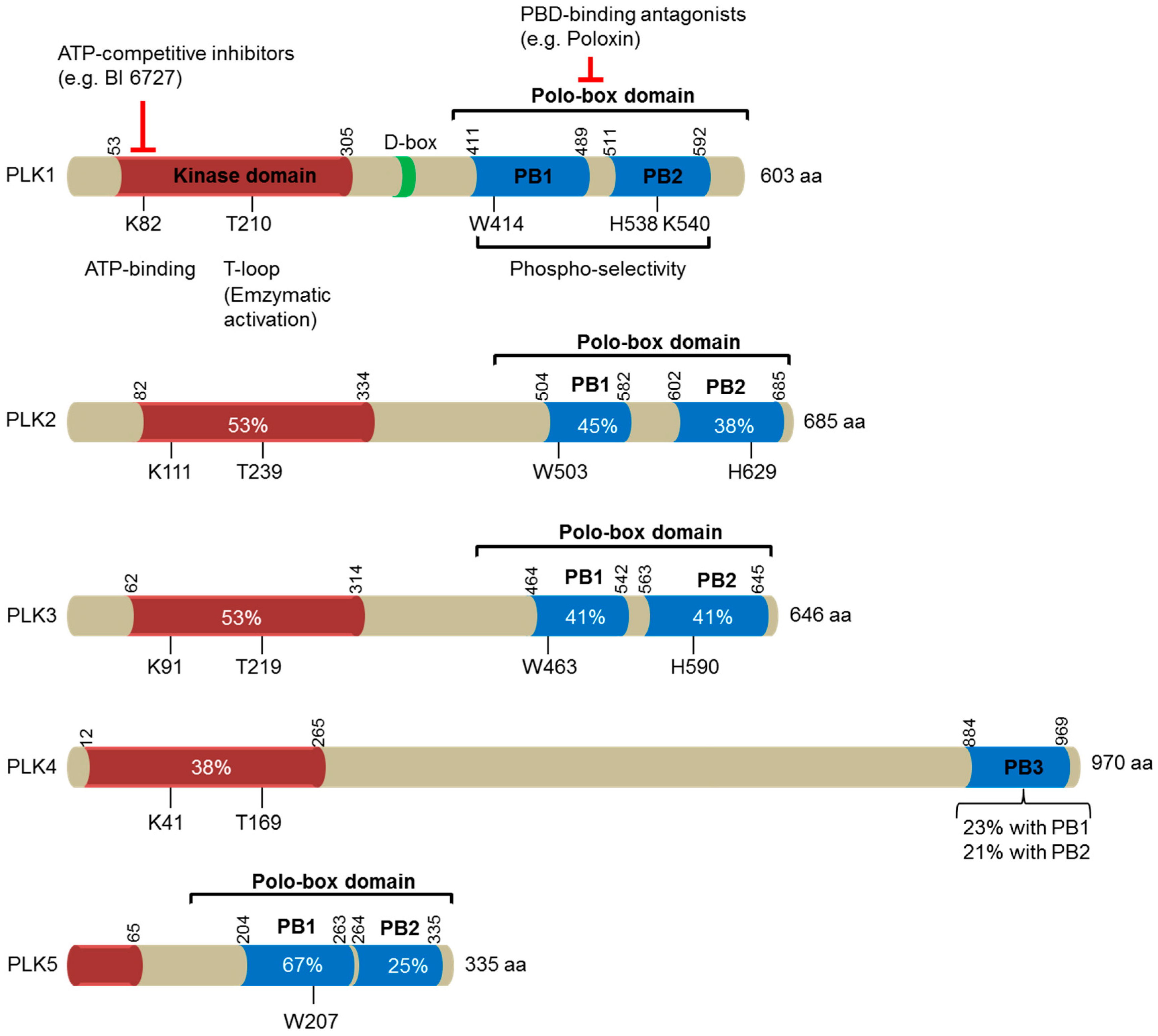
Figure 1.
A schematic diagram illustrating the domain structures of the human polo-like kinase (PLK) family of proteins (PLK1-5). The number of amino acids in each family member is indicated on the right. The location of the kinase domains is shown in orange, whereas the polo-box domains (PBD), made of two polo-boxes (PB), are represented in blue. These two domains are separated by the interdomain linker, which comprises a destruction box (D-Box) indicated in green. The numbers indicate the first and the last residues of these domains in human PLKs. Residues that are essential for ATP-binding and enzymatic activation (T-loop) within the kinase domains, and for phosphoselectivity within the polo-box domains, are depicted. Sequence identities with the corresponding domains in PLK1 are provided in percentages. Two distinct strategies for targeting PLK1 are included: ATP-competitive inhibitors targeting the catalytic activity of PLK1, and PBD-binding antagonists competitively inhibiting the function of PBD.
Figure 1.
A schematic diagram illustrating the domain structures of the human polo-like kinase (PLK) family of proteins (PLK1-5). The number of amino acids in each family member is indicated on the right. The location of the kinase domains is shown in orange, whereas the polo-box domains (PBD), made of two polo-boxes (PB), are represented in blue. These two domains are separated by the interdomain linker, which comprises a destruction box (D-Box) indicated in green. The numbers indicate the first and the last residues of these domains in human PLKs. Residues that are essential for ATP-binding and enzymatic activation (T-loop) within the kinase domains, and for phosphoselectivity within the polo-box domains, are depicted. Sequence identities with the corresponding domains in PLK1 are provided in percentages. Two distinct strategies for targeting PLK1 are included: ATP-competitive inhibitors targeting the catalytic activity of PLK1, and PBD-binding antagonists competitively inhibiting the function of PBD.
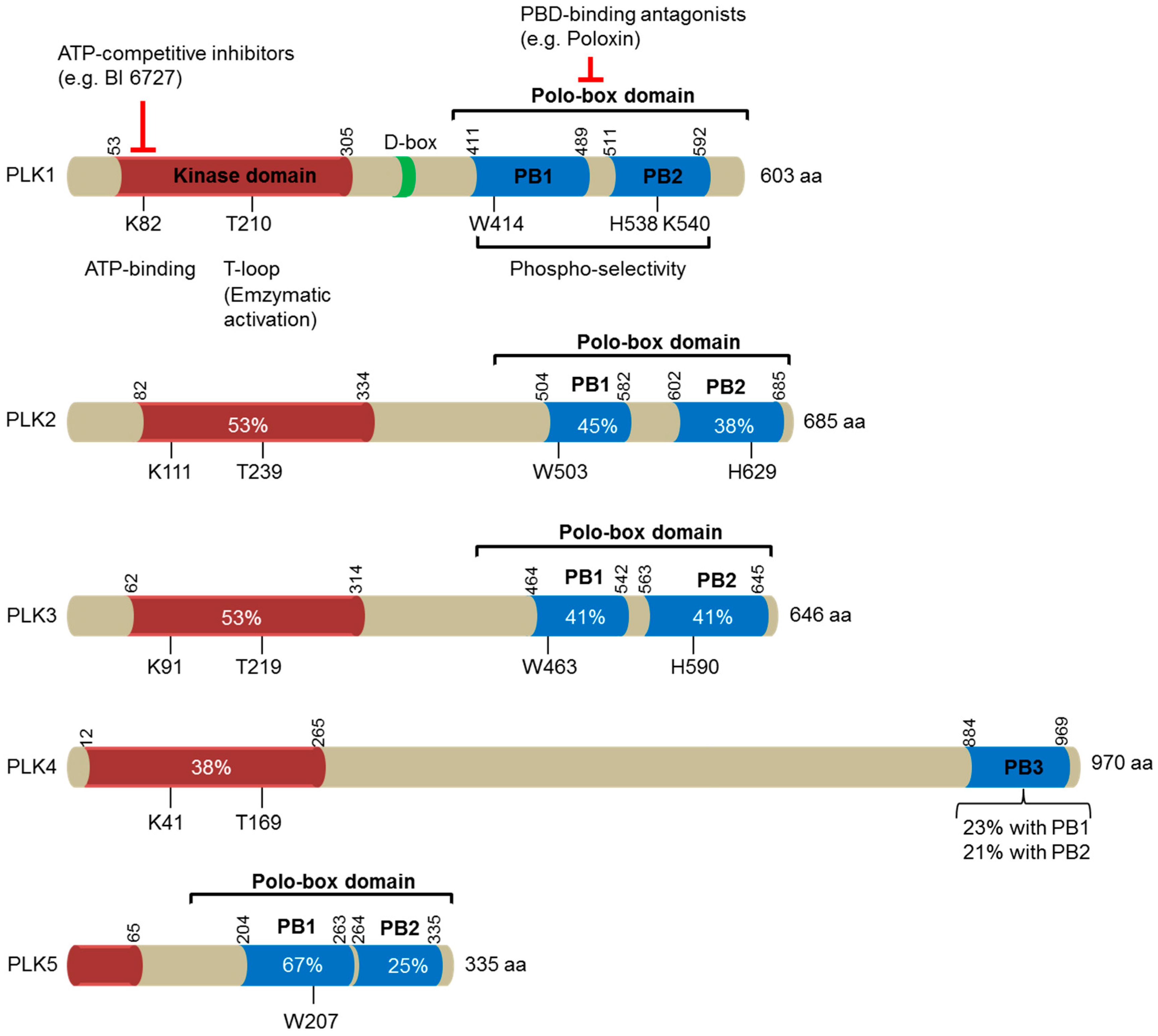
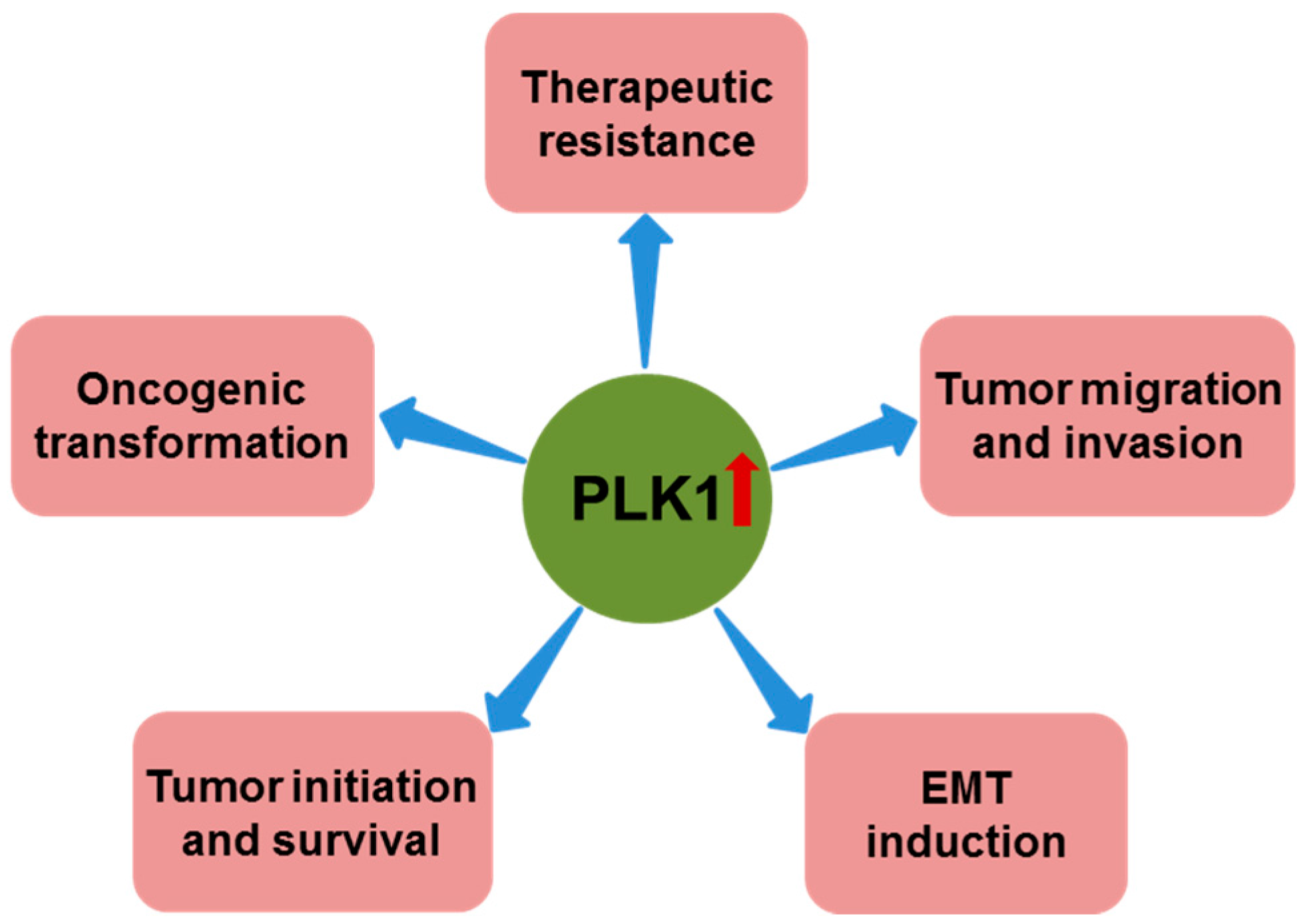
Figure 2.
Role of polo-like kinase 1 (PLK1) overexpression in cancer. In addition to its role in promoting cancer cell proliferation and suppressing apoptosis, PLK1 overexpression has also been reported to have important roles in oncogenic transformation, tumor initiation and survival, epithelial-mesenchymal transition (EMT) induction, tumor migration and invasion, and therapeutic resistance.
Figure 2.
Role of polo-like kinase 1 (PLK1) overexpression in cancer. In addition to its role in promoting cancer cell proliferation and suppressing apoptosis, PLK1 overexpression has also been reported to have important roles in oncogenic transformation, tumor initiation and survival, epithelial-mesenchymal transition (EMT) induction, tumor migration and invasion, and therapeutic resistance.
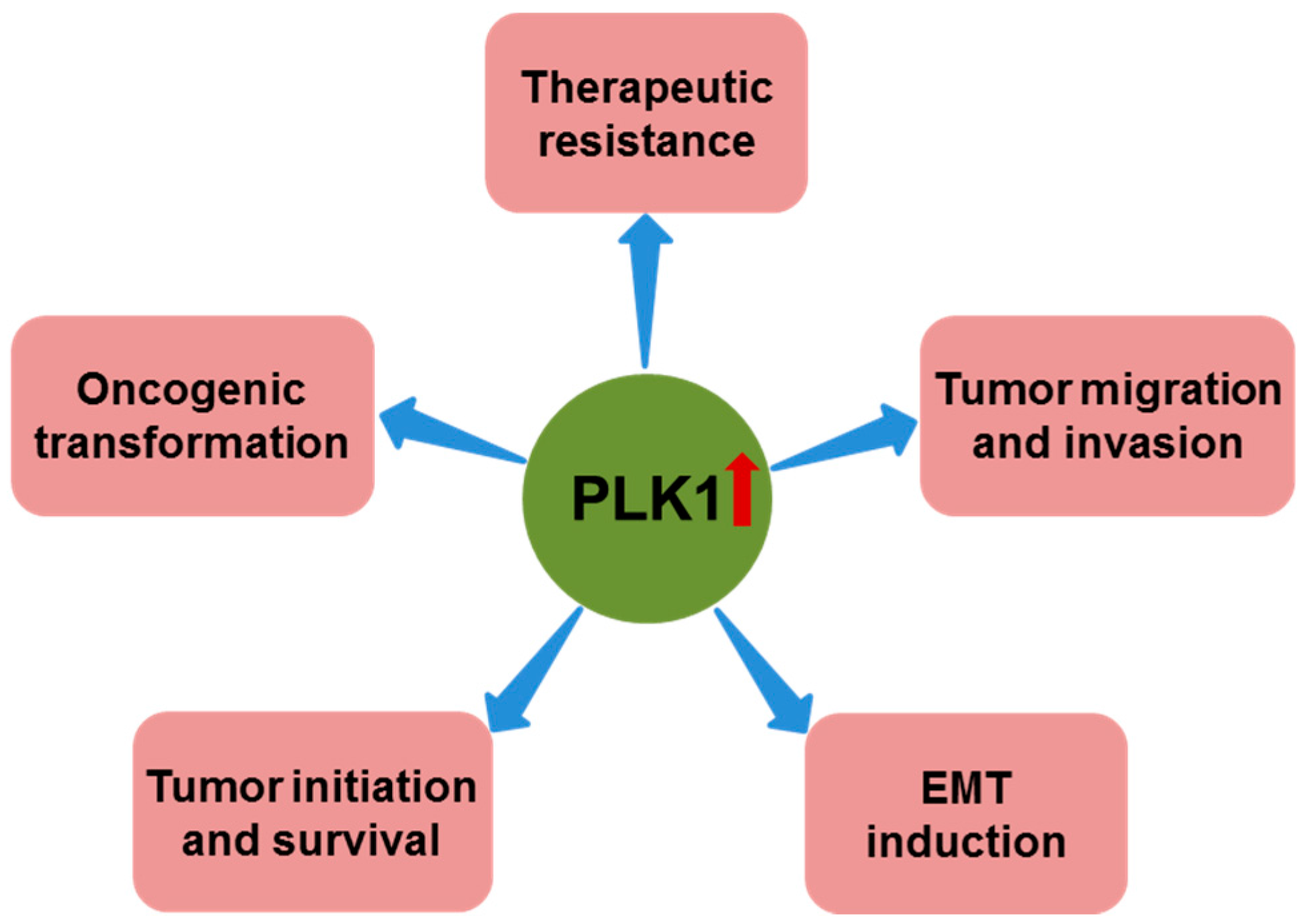
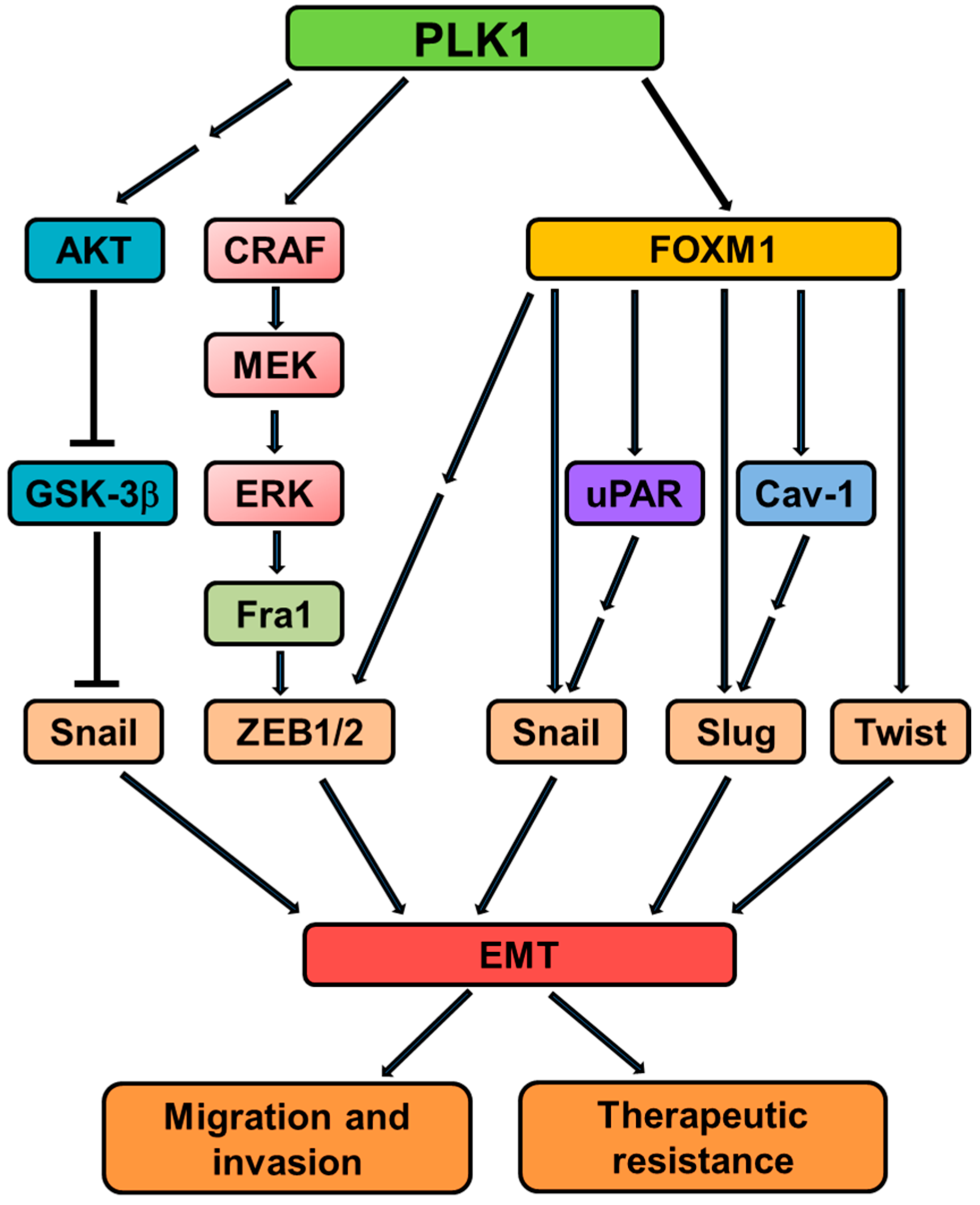
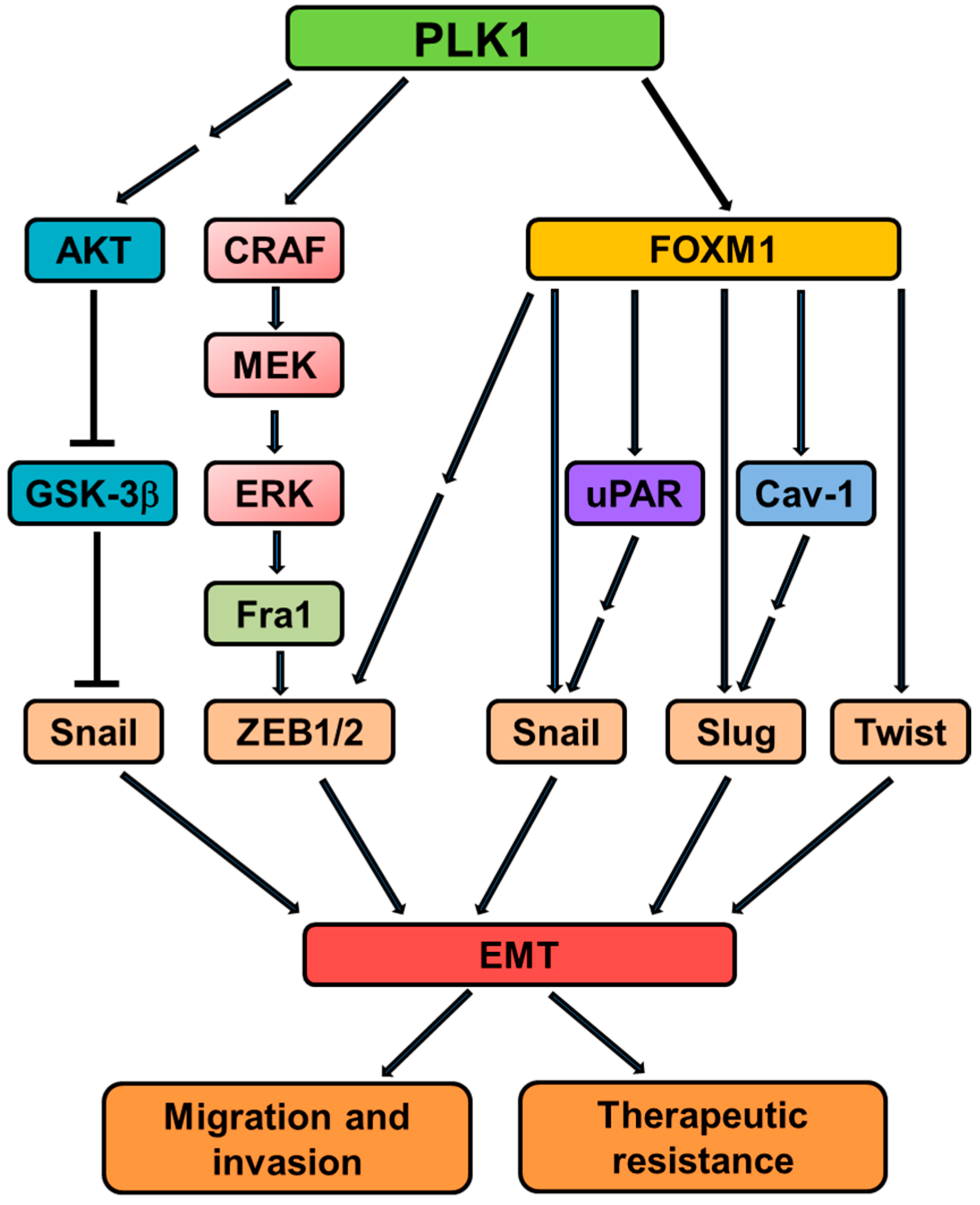
Figure 3.
An overview of signaling cascades involved in PLK1-induced EMT. PLK1 activates the MAPK pathway by directly binding and phosphorylating CRAF. The activated MAPK pathway causes transcriptional upregulation of Fra1, which in turn triggers the accumulation of ZEB1/2, thus orchestrating the transcriptional network necessary for the EMT program. PLK1 also induces EMT through AKT or FoxM1-dependent pathways. Together, these signaling events contribute to EMT induction and associated events (such as invasion and therapeutic resistance) in tumor cells overexpressing PLK1.
Figure 3.
An overview of signaling cascades involved in PLK1-induced EMT. PLK1 activates the MAPK pathway by directly binding and phosphorylating CRAF. The activated MAPK pathway causes transcriptional upregulation of Fra1, which in turn triggers the accumulation of ZEB1/2, thus orchestrating the transcriptional network necessary for the EMT program. PLK1 also induces EMT through AKT or FoxM1-dependent pathways. Together, these signaling events contribute to EMT induction and associated events (such as invasion and therapeutic resistance) in tumor cells overexpressing PLK1.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fu, Z.; Wen, D. The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis. Cancers 2017, 9, 131. https://doi.org/10.3390/cancers9100131
AMA Style
Fu Z, Wen D. The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis. Cancers. 2017; 9(10):131. https://doi.org/10.3390/cancers9100131
Chicago/Turabian StyleFu, Zheng, and Donghua Wen. 2017. "The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis" Cancers 9, no. 10: 131. https://doi.org/10.3390/cancers9100131
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.