
The Role of the C-Clamp in Wnt-Related Colorectal Cancers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
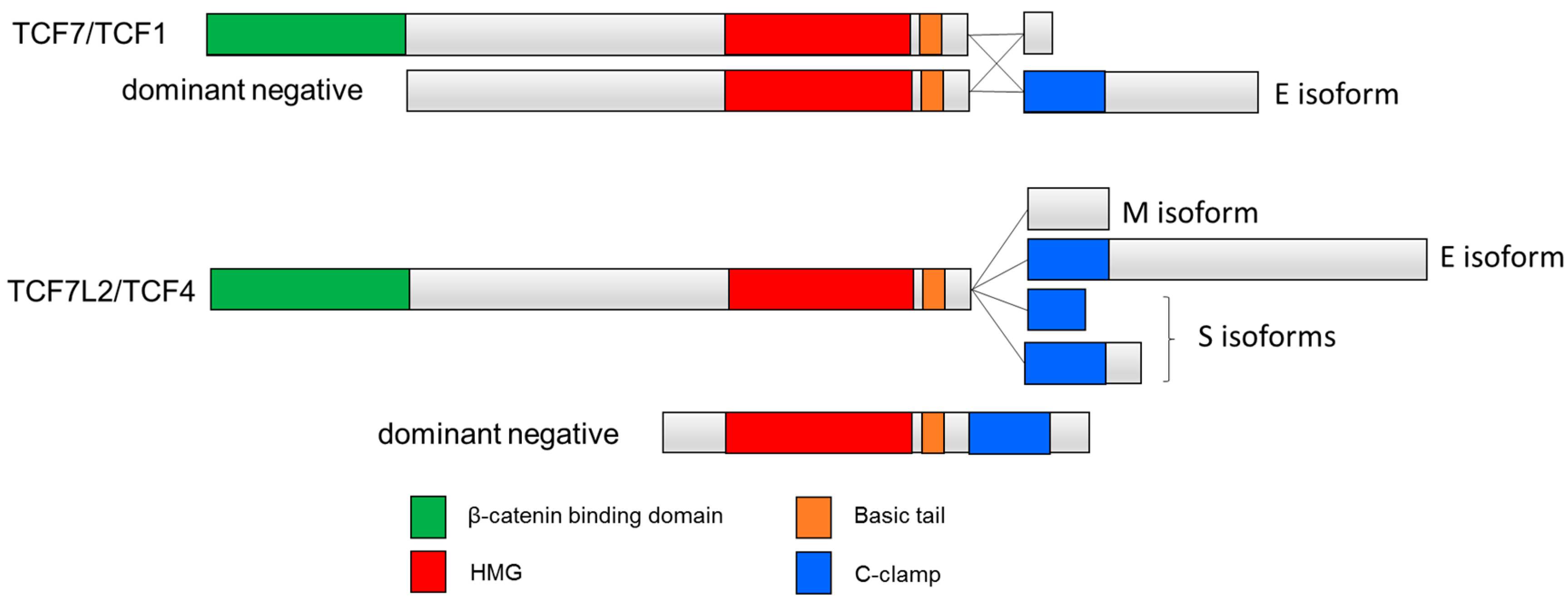
:1. Introduction to the TCF Family
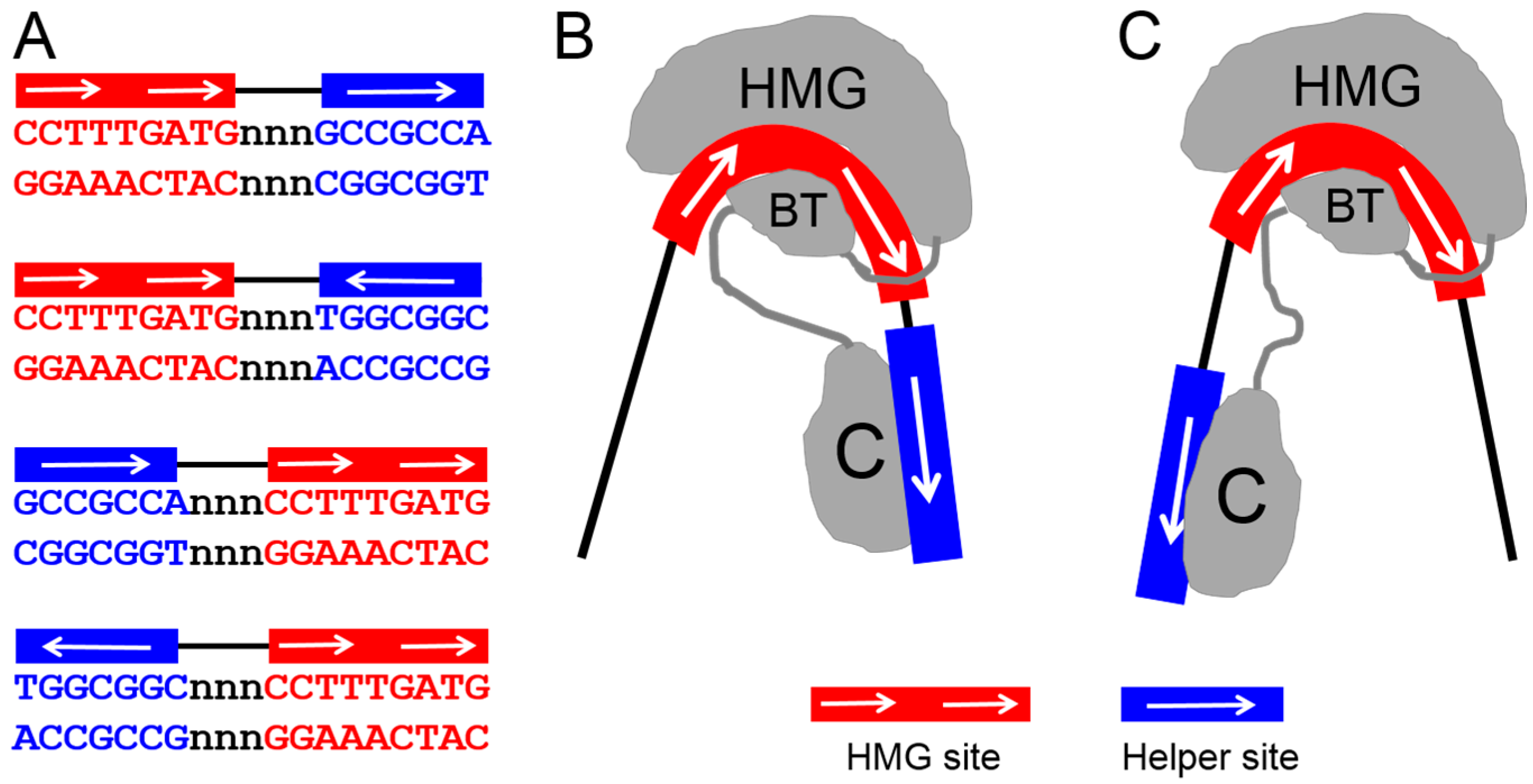
2. The C-Clamp: Biochemical Properties and Functional Roles
3. Other C-Clamp Containing Proteins
4. The Role of C-Clamp Containing TCFs in Colorectal Cancer
5. Final Perspective
Acknowledgments
Conflicts of Interest
References
- Archbold, H.C.; Yang, Y.X.; Chen, L.; Cadigan, K.M. How do they do Wnt they do? Regulation of transcription by the Wnt/beta-catenin pathway. Acta Physiol. (Oxf.) 2012, 204, 74–109. [Google Scholar] [CrossRef] [PubMed]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4, a007906. [Google Scholar] [CrossRef] [PubMed]
- Cadigan, K.M. TCFs and Wnt/beta-catenin signaling: More than one way to throw the switch. Curr. Top. Dev. Biol. 2012, 98, 1–34. [Google Scholar] [PubMed]
- Brunner, E.; Peter, O.; Schweizer, L.; Basler, K. Pangolin encodes a Lef-1 homologue that acts downstream of armadillo to transduce the wingless signal in Drosophila. Nature 1997, 385, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, L.; Nellen, D.; Basler, K. Requirement for Pangolin/dTCF in Drosophila wingless signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 5846–5851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Wetering, M.; Cavallo, R.; Dooijes, D.; van Beest, M.; van Es, J.; Loureiro, J.; Ypma, A.; Hursh, D.; Jones, T.; Bejsovec, A.; et al. Armadillo coactivates transcription driven by the product of the drosophila segment polarity gene dtcf. Cell 1997, 88, 789–799. [Google Scholar] [CrossRef]
- Lam, N.; Chesney, M.A.; Kimble, J. Wnt signaling and CEH-22/tinman/Nkx2.5 specify a stem cell niche in C. elegans. Curr. Biol. 2006, 16, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Herman, M. C. elegans pop-1/tcf functions in a canonical Wnt pathway that controls cell migration and in a noncanonical Wnt pathway that controls cell polarity. Development 2001, 128, 581–590. [Google Scholar] [PubMed]
- Huang, S.; Shetty, P.; Robertson, S.M.; Lin, R. Binary cell fate specification during C. elegans embryogenesis driven by reiterated reciprocal asymmetry of TCF POP-1 and its coactivator beta-catenin SYS-1. Development 2007, 134, 2685–2695. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Thompson, S.; Priess, J.R. POP-1 encodes an HMG box protein required for the specification of a mesoderm precursor in early C. elegans embryos. Cell 1995, 83, 599–609. [Google Scholar] [CrossRef]
- Maduro, M.F.; Kasmir, J.J.; Zhu, J.; Rothman, J.H. The Wnt effector POP-1 and the PAL-1/caudal homeoprotein collaborate with SKN-1 to activate C. elegans endoderm development. Dev. Biol. 2005, 285, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Shetty, P.; Lo, M.C.; Robertson, S.M.; Lin, R. C. elegans TCF protein, POP-1, converts from repressor to activator as a result of Wnt-induced lowering of nuclear levels. Dev. Biol. 2005, 285, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Dedhar, S.; Coetzee, G.A.; Nelson, C.C. Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr. Rev. 2005, 26, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of beta-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; van den Broek, O.; Destree, O.; Hoppler, S. Distinct roles for Xenopus Tcf/Lef genes in mediating specific responses to Wnt/beta-catenin signalling in mesoderm development. Development 2005, 132, 5375–5385. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, K.; Galceran, J.; Tontsch, S.; Roth, W.; Grosschedl, R. FGF4, a direct target of LEF1 and Wnt signaling, can rescue the arrest of tooth organogenesis in Lef1(−/−) mice. Genes Dev. 2002, 16, 3173–3185. [Google Scholar] [CrossRef] [PubMed]
- Merrill, B.J.; Pasolli, H.A.; Polak, L.; Rendl, M.; Garcia-Garcia, M.J.; Anderson, K.V.; Fuchs, E. Tcf3: A transcriptional regulator of axis induction in the early embryo. Development 2004, 131, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Dodge, M.; Gundapaneni, D.; Michnoff, C.; Roth, M.; Lum, L. A genome-wide rnai screen for Wnt/beta-catenin pathway components identifies unexpected roles for TCF transcription factors in cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 9697–9702. [Google Scholar] [CrossRef] [PubMed]
- Standley, H.J.; Destree, O.; Kofron, M.; Wylie, C.; Heasman, J. Maternal XTcf1 and XTcf4 have distinct roles in regulating Wnt target genes. Dev. Biol. 2006, 289, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Grumolato, L.; Liu, G.; Haremaki, T.; Mungamuri, S.K.; Mong, P.; Akiri, G.; Lopez-Bergami, P.; Arita, A.; Anouar, Y.; Mlodzik, M.; et al. Beta-catenin-independent activation of TCF1/LEF1 in human hematopoietic tumor cells through interaction with ATF2 transcription factors. PLoS Genet. 2013, 9, e1003603. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, A.; Yuen, H.F.; Chan, K.K.; Grills, C.; Fennell, D.A.; Lappin, T.R.; El-Tanani, M. Wnt-beta-catenin-Tcf-4 signalling-modulated invasiveness is dependent on osteopontin expression in breast cancer. Br. J. Cancer 2011, 105, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Slyper, M.; Shahar, A.; Bar-Ziv, A.; Granit, R.Z.; Hamburger, T.; Maly, B.; Peretz, T.; Ben-Porath, I. Control of breast cancer growth and initiation by the stem cell-associated transcription factor TCF3. Cancer Res. 2012, 72, 5613–5624. [Google Scholar] [CrossRef] [PubMed]
- Angus-Hill, M.L.; Elbert, K.M.; Hidalgo, J.; Capecchi, M.R. T-cell factor 4 functions as a tumor suppressor whose disruption modulates colon cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 4914–4919. [Google Scholar] [CrossRef] [PubMed]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-tcf complex in APC−/− colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Najdi, R.; Syed, A.; Arce, L.; Theisen, H.; Ting, J.H.; Atcha, F.; Nguyen, A.V.; Martinez, M.; Holcombe, R.F.; Edwards, R.A.; et al. A Wnt kinase network alters nuclear localization of TCF-1 in colon cancer. Oncogene 2009, 28, 4133–4146. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.P.; et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [Google Scholar] [CrossRef]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destree, O.; Clevers, H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Roose, J.; Huls, G.; van Beest, M.; Moerer, P.; van der Horn, K.; Goldschmeding, R.; Logtenberg, T.; Clevers, H. Synergy between tumor suppressor APC and the beta-catenin-Tcf4 target Tcf1. Science 1999, 285, 1923–1926. [Google Scholar] [CrossRef] [PubMed]
- Kennell, J.A.; O’Leary, E.E.; Gummow, B.M.; Hammer, G.D.; MacDougald, O.A. T-cell factor 4N (TCF-4N), a novel isoform of mouse TCF-4, synergizes with beta-catenin to coactivate C/EBPalpha and steroidogenic factor 1 transcription factors. Mol. Cell. Biol. 2003, 23, 5366–5375. [Google Scholar] [CrossRef] [PubMed]
- Vacik, T.; Lemke, G. Dominant-negative isoforms of Tcf/Lef proteins in development and disease. Cell Cycle 2011, 10, 4199–4200. [Google Scholar] [CrossRef] [PubMed]
- Vacik, T.; Stubbs, J.L.; Lemke, G. A novel mechanism for the transcriptional regulation of Wnt signaling in development. Genes Dev. 2011, 25, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Malarkey, C.S.; Churchill, M.E. The high mobility group box: The ultimate utility player of a cell. Trends Biochem. Sci. 2012, 37, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Stros, M. HMGB proteins: Interactions with DNA and chromatin. Biochim. Biophys. Acta 2010, 1799, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Love, J.J.; Li, X.; Case, D.A.; Giese, K.; Grosschedl, R.; Wright, P.E. Structural basis for DNA bending by the architectural transcription factor LEF-1. Nature 1995, 376, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Giese, K.; Amsterdam, A.; Grosschedl, R. DNA-binding properties of the hmg domain of the lymphoid-specific transcriptional regulator LEF-1. Genes Dev. 1991, 5, 2567–2578. [Google Scholar] [CrossRef] [PubMed]
- Love, J.J.; Li, X.; Chung, J.; Dyson, H.J.; Wright, P.E. The LEF-1 high-mobility group domain undergoes a disorder-to-order transition upon formation of a complex with cognate DNA. Biochemistry 2004, 43, 8725–8734. [Google Scholar] [CrossRef] [PubMed]
- Van Beest, M.; Dooijes, D.; van De Wetering, M.; Kjaerulff, S.; Bonvin, A.; Nielsen, O.; Clevers, H. Sequence-specific high mobility group box factors recognize 10-12-base pair minor groove motifs. J. Biol. Chem. 2000, 275, 27266–27273. [Google Scholar] [PubMed]
- Atcha, F.A.; Syed, A.; Wu, B.; Hoverter, N.P.; Yokoyama, N.N.; Ting, J.H.; Munguia, J.E.; Mangalam, H.J.; Marsh, J.L.; Waterman, M.L. A unique DNA binding domain converts T-cell factors into strong wnt effectors. Mol. Cell. Biol. 2007, 27, 8352–8363. [Google Scholar] [CrossRef] [PubMed]
- Barolo, S. Transgenic Wnt/TCF pathway reporters: All you need is Lef? Oncogene 2006, 25, 7505–7511. [Google Scholar] [CrossRef] [PubMed]
- Bottomly, D.; Kyler, S.L.; McWeeney, S.K.; Yochum, G.S. Identification of {beta}-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Res. 2010, 38, 5735–5745. [Google Scholar] [CrossRef] [PubMed]
- Hatzis, P.; van der Flier, L.G.; van Driel, M.A.; Guryev, V.; Nielsen, F.; Denissov, S.; Nijman, I.J.; Koster, J.; Santo, E.E.; Welboren, W.; et al. Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol. Cell. Biol. 2008, 28, 2732–2744. [Google Scholar] [CrossRef] [PubMed]
- Norton, L.; Fourcaudot, M.; Abdul-Ghani, M.A.; Winnier, D.; Mehta, F.F.; Jenkinson, C.P.; Defronzo, R.A. Chromatin occupancy of transcription factor 7-like 2 (TCF7L2) and its role in hepatic glucose metabolism. Diabetologia 2011, 54, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Duval, A.; Rolland, S.; Tubacher, E.; Bui, H.; Thomas, G.; Hamelin, R. The human T-cell transcription factor-4 gene: Structure, extensive characterization of alternative splicings, and mutational analysis in colorectal cancer cell lines. Cancer Res. 2000, 60, 3872–3879. [Google Scholar] [PubMed]
- Van de Wetering, M.; Castrop, J.; Korinek, V.; Clevers, H. Extensive alternative splicing and dual promoter usage generate Tcf-1 protein isoforms with differential transcription control properties. Mol. Cell. Biol. 1996, 16, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Weise, A.; Bruser, K.; Elfert, S.; Wallmen, B.; Wittel, Y.; Wohrle, S.; Hecht, A. Alternative splicing of TCF7L2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/beta-catenin targets. Nucleic Acids Res. 2010, 38, 1964–1981. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, A.J.; Cadigan, K.M. Structure-function analysis of the C-clamp of TCF/Pangolin in Wnt/ss-catenin signaling. PLoS ONE 2014, 9, e86180. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.V.; Chang, J.L.; Gangopadhyay, A.; Shearer, A.; Cadigan, K.M. Activation of wingless targets requires bipartite recognition of DNA by TCF. Curr. Biol. 2008, 18, 1877–1881. [Google Scholar] [CrossRef] [PubMed]
- Hoverter, N.P.; Ting, J.H.; Sundaresh, S.; Baldi, P.; Waterman, M.L. A Wnt/p21 circuit directed by the C-clamp, a sequence-specific DNA binding domain in TCFs. Mol. Cell. Biol. 2012, 32, 3648–3662. [Google Scholar] [CrossRef] [PubMed]
- Grishin, N.V. Treble clef finger—a functionally diverse zinc-binding structural motif. Nucleic Acids Res. 2001, 29, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.S.; Majumdar, I.; Grishin, N.V. Structural classification of zinc fingers: Survey and summary. Nucleic Acids Res. 2003, 31, 532–550. [Google Scholar] [CrossRef] [PubMed]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc finger proteins: New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Hoverter, N.P.; Zeller, M.D.; McQuade, M.M.; Garibaldi, A.; Busch, A.; Selwan, E.M.; Hertel, K.J.; Baldi, P.; Waterman, M.L. The TCF C-clamp DNA binding domain expands the Wnt transcriptome via alternative target recognition. Nucleic Acids Res. 2014, 42, 13615–13632. [Google Scholar] [CrossRef] [PubMed]
- Bhambhani, C.; Ravindranath, A.J.; Mentink, R.A.; Chang, M.V.; Betist, M.C.; Yang, Y.X.; Koushika, S.P.; Korswagen, H.C.; Cadigan, K.M. Distinct DNA binding sites contribute to the TCF transcriptional switch in C. elegans and drosophila. PLoS Genet. 2014, 10, e1004133. [Google Scholar] [CrossRef] [PubMed]
- Archbold, H.C.; Broussard, C.; Chang, M.V.; Cadigan, K.M. Bipartite recognition of DNA by Tcf/Pangolin is remarkably flexible and contributes to transcriptional responsiveness and tissue specificity of wingless signaling. PLoS Genet. 2014, 10, e1004591. [Google Scholar] [CrossRef] [PubMed]
- Hecht, A.; Stemmler, M.P. Identification of a promoter-specific transcriptional activation domain at the c terminus of the wnt effector protein T-cell factor 4. J. Biol. Chem. 2003, 278, 3776–3785. [Google Scholar] [CrossRef] [PubMed]
- Cadigan, K.M.; Peifer, M. Wnt signaling from development to disease: Insights from model systems. Cold Spring Harb. Perspect. Biol. 2009, 1, a002881. [Google Scholar] [CrossRef] [PubMed]
- Hikasa, H.; Ezan, J.; Itoh, K.; Li, X.; Klymkowsky, M.W.; Sokol, S.Y. Regulation of TCF3 by Wnt-dependent phosphorylation during vertebrate axis specification. Dev. Cell 2010, 19, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Oda, T.; Itoh, M.; Jiang, D.; Artinger, K.B.; Chandrasekharappa, S.C.; Driever, W.; Chitnis, A.B. Repressor activity of Headless/Tcf3 is essential for vertebrate head formation. Nature 2000, 407, 913–916. [Google Scholar] [PubMed]
- Shah, M.; Rennoll, S.A.; Raup-Konsavage, W.M.; Yochum, G.S. A dynamic exchange of TCF3 and TCF4 transcription factors controls myc expression in colorectal cancer cells. Cell Cycle 2015, 14, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Pereira, L.; Hoffman, J.A.; Shy, B.R.; Yuen, C.M.; Liu, D.R.; Merrill, B.J. Opposing effects of Tcf3 and Tcf1 control wnt stimulation of embryonic stem cell self-renewal. Nat. Cell Biol. 2011, 13, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Blauwkamp, T.A.; Chang, M.V.; Cadigan, K.M. Novel TCF-binding sites specify transcriptional repression by Wnt signalling. EMBO J. 2008, 27, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.U.; Blauwkamp, T.A.; Burby, P.E.; Cadigan, K.M. Wnt-mediated repression via bipartite DNA recognition by TCF in the drosophila hematopoietic system. PLoS Genet. 2014, 10, e1004509. [Google Scholar] [CrossRef] [PubMed]
- Wallmen, B.; Schrempp, M.; Hecht, A. Intrinsic properties of Tcf1 and Tcf4 splice variants determine cell-type-specific Wnt/beta-catenin target gene expression. Nucleic Acids Res. 2012, 40, 9455–9469. [Google Scholar] [CrossRef] [PubMed]
- Boeckle, S.; Pfister, H.; Steger, G. A new cellular factor recognizes e2 binding sites of papillomaviruses which mediate transcriptional repression by e2. Virology 2002, 293, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, R.; Tomaru, Y.; de Hoon, M.; Suzuki, H.; Hayashizaki, Y.; Shin, J.W. Identification of ZNF395 as a novel modulator of adipogenesis. Exp. Cell Res. 2013, 319, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Shouguchi-Miyata, J.; Miyamoto, N.; Ikeda, J.E. Novel nuclear shuttle proteins, HDBP1 and HDBP2, bind to neuronal cell-specific cis-regulatory element in the promoter for the human Huntington’s disease gene. J. Biol. Chem. 2004, 279, 7275–7286. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Watt, C.D.; Schmidt, R.J.; Kuscuoglu, U.; Miesfeld, R.L.; Goldhamer, D.J. Cloning and characterization of a novel myod enhancer-binding factor. Mech. Dev. 2007, 124, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.B.; Eyster, C.A.; Griesel, B.A.; Olson, A.L. Regulation of the human GLUT4 gene promoter: Interaction between a transcriptional activator and myocyte enhancer factor 2a. Proc. Natl. Acad. Sci. USA 2003, 100, 14725–14730. [Google Scholar] [CrossRef] [PubMed]
- Sparling, D.P.; Griesel, B.A.; Weems, J.; Olson, A.L. GLUT4 enhancer factor (GEF) interacts with MEF2A and HDAC5 to regulate the GLUT4 promoter in adipocytes. J. Biol. Chem. 2008, 283, 7429–7437. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; McGovern, D.P.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, U.; Huang, Z.; Terman, J.R. The glucose transporter (GLUT4) enhancer factor is required for normal wing positioning in Drosophila. Genetics 2008, 178, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Jordanovski, D.; Herwartz, C.; Pawlowski, A.; Taute, S.; Frommolt, P.; Steger, G. The hypoxia-inducible transcription factor ZNF395 is controlled by IkB kinase-signaling and activates genes involved in the innate immune response and cancer. PLoS ONE 2013, 8, e74911. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, H.; Zhang, S.; Buss, C.G.; Fish, L.; Tavazoie, S.; Tavazoie, S.F. Metastasis-suppressor transcript destabilization through TARBP2 binding of mRNA hairpins. Nature 2014, 513, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Pang, F.; Zha, R.; Zhao, Y.; Wang, Q.; Chen, D.; Zhang, Z.; Chen, T.; Yao, M.; Gu, J.; He, X. MiR-525-3p enhances the migration and invasion of liver cancer cells by downregulating ZNF395. PLoS ONE 2014, 9, e90867. [Google Scholar] [CrossRef] [PubMed]
- Minster, R.L.; Sanders, J.L.; Singh, J.; Kammerer, C.M.; Barmada, M.M.; Matteini, A.M.; Zhang, Q.; Wojczynski, M.K.; Daw, E.W.; Brody, J.A.; et al. Genome-wide association study and linkage analysis of the healthy aging index. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Najdi, R.; Holcombe, R.F.; Waterman, M.L. Wnt signaling and colon carcinogenesis: Beyond APC. J. Carcinog. 2011, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Schuijers, J.; Mokry, M.; Hatzis, P.; Cuppen, E.; Clevers, H. Wnt-induced transcriptional activation is exclusively mediated by TCF/LEF. EMBO J. 2014, 33, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Korinek, V.; Barker, N.; Moerer, P.; van Donselaar, E.; Huls, G.; Peters, P.J.; Clevers, H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking TCF-4. Nat. Genet. 1998, 19, 379–383. [Google Scholar] [PubMed]
- Van Es, J.H.; Haegebarth, A.; Kujala, P.; Itzkovitz, S.; Koo, B.K.; Boj, S.F.; Korving, J.; van den Born, M.; van Oudenaarden, A.; Robine, S.; et al. A critical role for the Wnt effector Tcf4 in adult intestinal homeostatic self-renewal. Mol. Cell. Biol. 2012, 32, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Xiang, D.B.; Wang, H.; Zhao, C.; Chen, J.; Xiong, F.; Li, T.Y.; Wang, X.L. Inhibition of Tcf-4 induces apoptosis and enhances chemosensitivity of colon cancer cells. PLoS ONE 2012, 7, e45617. [Google Scholar] [CrossRef] [PubMed]
- Bass, A.J.; Lawrence, M.S.; Brace, L.E.; Ramos, A.H.; Drier, Y.; Cibulskis, K.; Sougnez, C.; Voet, D.; Saksena, G.; Sivachenko, A.; et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat. Genet. 2011, 43, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Atcha, F.A.; Munguia, J.E.; Li, T.W.; Hovanes, K.; Waterman, M.L. A new beta-catenin-dependent activation domain in T cell factor. J. Biol. Chem. 2003, 278, 16169–16175. [Google Scholar] [CrossRef] [PubMed]
- Nakano, N.; Kato, M.; Itoh, S. Regulation of the tmepai promoter by TCF7L2: The c-terminal tail of TCF7L2 is essential to activate the TMEPAI gene. J. Biochem. 2016, 159, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Nakano, N.; Itoh, S.; Watanabe, Y.; Maeyama, K.; Itoh, F.; Kato, M. Requirement of TCF7L2 for TGF-beta-dependent transcriptional activation of the TMEPAI gene. J. Biol. Chem. 2010, 285, 38023–38033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, Y.; Itoh, S.; Goto, T.; Ohnishi, E.; Inamitsu, M.; Itoh, F.; Satoh, K.; Wiercinska, E.; Yang, W.; Shi, L.; et al. TMEPAI, a transmembrane TGF-beta-inducible protein, sequesters smad proteins from active participation in tgf-beta signaling. Mol. Cell. Biol. 2010, 37, 123–134. [Google Scholar]
- Reichling, T.; Goss, K.H.; Carson, D.J.; Holdcraft, R.W.; Ley-Ebert, C.; Witte, D.; Aronow, B.J.; Groden, J. Transcriptional profiles of intestinal tumors in APC(Min) mice are unique from those of embryonic intestine and identify novel gene targets dysregulated in human colorectal tumors. Cancer Res. 2005, 65, 166–176. [Google Scholar] [PubMed]
- Brunschwig, E.B.; Wilson, K.; Mack, D.; Dawson, D.; Lawrence, E.; Willson, J.K.; Lu, S.; Nosrati, A.; Rerko, R.M.; Swinler, S.; et al. PMEPA1, a transforming growth factor-beta-induced marker of terminal colonocyte differentiation whose expression is maintained in primary and metastatic colon cancer. Cancer Res. 2003, 63, 1568–1575. [Google Scholar] [PubMed]
- Giannini, G.; Ambrosini, M.I.; Di Marcotullio, L.; Cerignoli, F.; Zani, M.; MacKay, A.R.; Screpanti, I.; Frati, L.; Gulino, A. EGF- and cell-cycle-regulated STAG1/PMEPA1/ERG1.2 belongs to a conserved gene family and is overexpressed and amplified in breast and ovarian cancer. Mol. Carcinog. 2003, 38, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Rae, F.K.; Hooper, J.D.; Nicol, D.L.; Clements, J.A. Characterization of a novel gene, STAG1/PMEPA1, upregulated in renal cell carcinoma and other solid tumors. Mol. Carcinog. 2001, 32, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Vo Nguyen, T.T.; Watanabe, Y.; Shiba, A.; Noguchi, M.; Itoh, S.; Kato, M. TMEPAI/PMEPA1 enhances tumorigenic activities in lung cancer cells. Cancer Sci. 2014, 105, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Tillo, E.; de Barrios, O.; Valls, E.; Darling, D.S.; Castells, A.; Postigo, A. ZEB1 and TCF4 reciprocally modulate their transcriptional activities to regulate Wnt target gene expression. Oncogene 2015, 34, 5760–5770. [Google Scholar] [CrossRef] [PubMed]
- Gregorieff, A.; Grosschedl, R.; Clevers, H. Hindgut defects and transformation of the gastro-intestinal tract in tcf4(−/−)/tcf1(−/−) embryos. EMBO J. 2004, 23, 1825–1833. [Google Scholar] [CrossRef] [PubMed]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Hinoi, T.; Akyol, A.; Theisen, B.K.; Ferguson, D.O.; Greenson, J.K.; Williams, B.O.; Cho, K.R.; Fearon, E.R. Mouse model of colonic adenoma-carcinoma progression based on somatic APC inactivation. Cancer Res. 2007, 67, 9721–9730. [Google Scholar] [CrossRef] [PubMed]
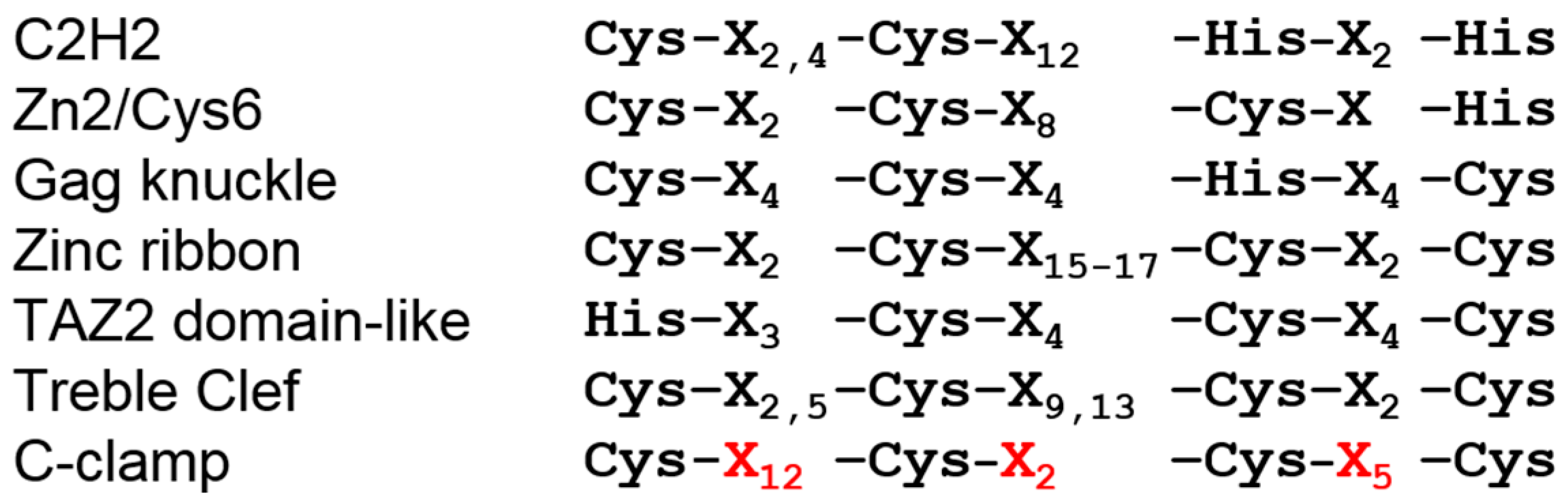
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravindranath, A.J.; Cadigan, K.M. The Role of the C-Clamp in Wnt-Related Colorectal Cancers. Cancers 2016, 8, 74. https://doi.org/10.3390/cancers8080074
Ravindranath AJ, Cadigan KM. The Role of the C-Clamp in Wnt-Related Colorectal Cancers. Cancers. 2016; 8(8):74. https://doi.org/10.3390/cancers8080074
Chicago/Turabian StyleRavindranath, Aditi J., and Ken M. Cadigan. 2016. "The Role of the C-Clamp in Wnt-Related Colorectal Cancers" Cancers 8, no. 8: 74. https://doi.org/10.3390/cancers8080074
APA StyleRavindranath, A. J., & Cadigan, K. M. (2016). The Role of the C-Clamp in Wnt-Related Colorectal Cancers. Cancers, 8(8), 74. https://doi.org/10.3390/cancers8080074