
Long Non-Coding RNAs and RNA-Binding Proteins in Pancreatic Cancer Development and Progression
 , , , , ,
, , , , ,
Simple Summary
Abstract
1. Introduction
2. Role of lncRNAs in PDAC Development and Progression
2.1. LncRNAs Associated with PDAC Initiation and Early Development
2.2. LncRNAs Associated with Oncogenic KRAS Signalling
2.3. LncRNAs Associated with Oncogenic and Tumour-Suppressive TGF-β Signalling
3. Role of RNA-Binding Proteins in PDAC
3.1. ELAV-Like RNA Binding Protein 1 (ELAVL1)
3.2. Insulin-Like Growth Factor II mRNA-Binding Proteins (IGF2BPs)
3.3. BicC Family RNA-Binding Protein 1 (BICC1)
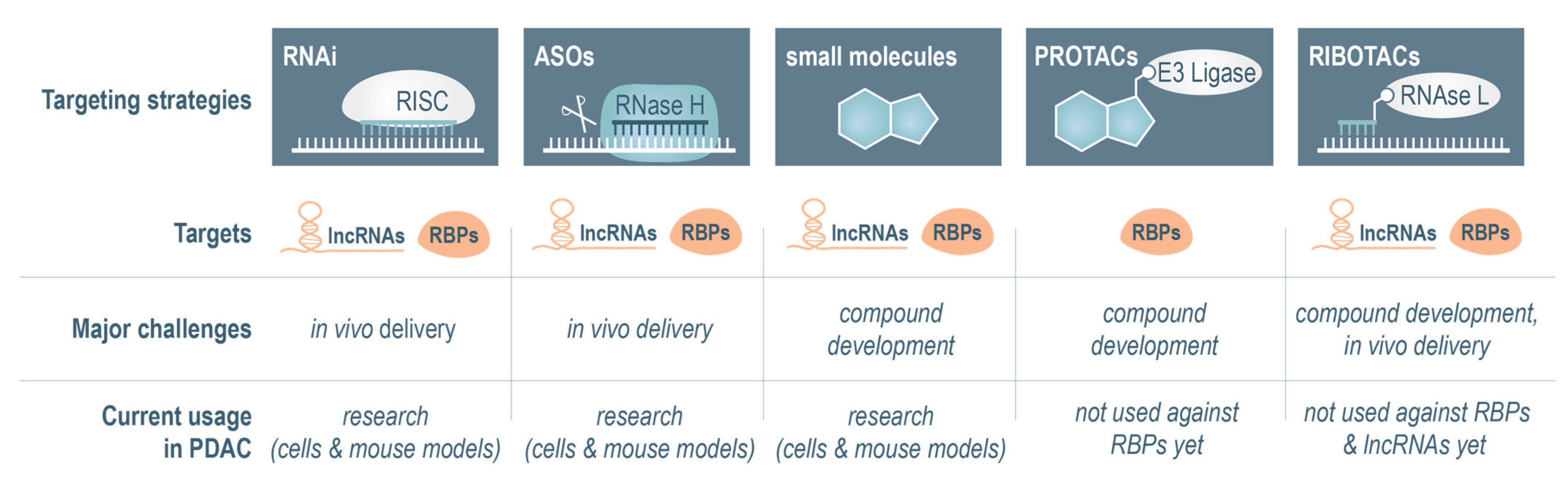
4. Strategies to Target lncRNAs and RBPs
4.1. RNA Interference-Based Targeting Approaches
4.2. Antisense Oligonucleotides to Target lncRNAs and RBPs
4.3. Small-Molecule Inhibitors
4.4. Targeting of lncRNAs and RBPs via Proximity-Inducing Bifunctional Molecules
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Beutel, A.K.; Ekizce, M.; Ettrich, T.J.; Seufferlein, T.; Lindenmayer, J.; Gout, J.; Kleger, A. Organoid-based precision medicine in pancreatic cancer. United Eur. Gastroenterol. J. 2024, 13, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P.S.; et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Anisetti, B.; Coston, T.W.; Ahmed, A.K.; Mahadevia, H.J.; Edgar, M.A.; Starr, J.S.; Babiker, H.M. An Excellent Response of Microsatellite Instability-High Pancreatic Adenocarcinoma to Pembrolizumab Treatment: The Role of Circulating Tumor DNA Testing. Cureus 2023, 15, e37239. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A., Jr.; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef]
- Strickler, J.H.; Satake, H.; George, T.J.; Yaeger, R.; Hollebecque, A.; Garrido-Laguna, I.; Schuler, M.; Burns, T.F.; Coveler, A.L.; Falchook, G.S.; et al. Sotorasib in KRAS p.G12C-Mutated Advanced Pancreatic Cancer. N. Engl. J. Med. 2023, 388, 33–43. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Raghavan, S.; Winter, P.S.; Navia, A.W.; Williams, H.L.; DenAdel, A.; Lowder, K.E.; Galvez-Reyes, J.; Kalekar, R.L.; Mulugeta, N.; Kapner, K.S.; et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 2021, 184, 6119–6137.e6126. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Dorn, A.; Huttelmaier, S.; Haemmerle, M.; Gutschner, T. Comprehensive Analysis of LincRNAs in Classical and Basal-Like Subtypes of Pancreatic Cancer. Cancers 2020, 12, 2077. [Google Scholar] [CrossRef]
- Henninger, J.E.; Young, R.A. An RNA-centric view of transcription and genome organization. Mol. Cell 2024, 84, 3627–3643. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Singh, A.; Liu, A.J.; Fuchs, S.Y.; Sharma, A.K.; Spiegelman, V.S. RNA Binding Proteins as Potential Therapeutic Targets in Colorectal Cancer. Cancers 2024, 16, 3502. [Google Scholar] [CrossRef]
- Weisse, J.; Rosemann, J.; Krauspe, V.; Kappler, M.; Eckert, A.W.; Haemmerle, M.; Gutschner, T. RNA-Binding Proteins as Regulators of Migration, Invasion and Metastasis in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2020, 21, 6835. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef]
- Mattick, J.S. Challenging the dogma: The hidden layer of non-protein-coding RNAs in complex organisms. Bioessays 2003, 25, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Melo, C.A.; Drost, J.; Wijchers, P.J.; van de Werken, H.; de Wit, E.; Oude Vrielink, J.A.; Elkon, R.; Melo, S.A.; Leveille, N.; Kalluri, R.; et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol. Cell 2013, 49, 524–535. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, J.; Li, Y.; Song, T.; Wu, Y.; Fang, S.; Bu, D.; Li, H.; Sun, L.; Pei, D.; et al. NONCODEV6: An updated database dedicated to long non-coding RNA annotation in both animals and plants. Nucleic Acids Res. 2021, 49, D165–D171. [Google Scholar] [CrossRef] [PubMed]
- Deveson, I.W.; Brunck, M.E.; Blackburn, J.; Tseng, E.; Hon, T.; Clark, T.A.; Clark, M.B.; Crawford, J.; Dinger, M.E.; Nielsen, L.K.; et al. Universal Alternative Splicing of Noncoding Exons. Cell Syst. 2018, 6, 245–255.e245. [Google Scholar] [CrossRef]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Gloss, B.S.; Dinger, M.E. The specificity of long noncoding RNA expression. Biochim. Biophys. Acta 2016, 1859, 16–22. [Google Scholar] [CrossRef]
- Quinn, J.J.; Zhang, Q.C.; Georgiev, P.; Ilik, I.A.; Akhtar, A.; Chang, H.Y. Rapid evolutionary turnover underlies conserved lncRNA-genome interactions. Genes. Dev. 2016, 30, 191–207. [Google Scholar] [CrossRef]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef]
- Bhan, A.; Mandal, S.S. LncRNA HOTAIR: A master regulator of chromatin dynamics and cancer. Biochim. Biophys. Acta 2015, 1856, 151–164. [Google Scholar] [CrossRef]
- Sahakyan, A.; Yang, Y.; Plath, K. The Role of Xist in X-Chromosome Dosage Compensation. Trends Cell Biol. 2018, 28, 999–1013. [Google Scholar] [CrossRef]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Romero-Barrios, N.; Legascue, M.F.; Benhamed, M.; Ariel, F.; Crespi, M. Splicing regulation by long noncoding RNAs. Nucleic Acids Res. 2018, 46, 2169–2184. [Google Scholar] [CrossRef]
- Toki, N.; Takahashi, H.; Sharma, H.; Valentine, M.N.Z.; Rahman, F.M.; Zucchelli, S.; Gustincich, S.; Carninci, P. SINEUP long non-coding RNA acts via PTBP1 and HNRNPK to promote translational initiation assemblies. Nucleic Acids Res. 2020, 48, 11626–11644. [Google Scholar] [CrossRef] [PubMed]
- Elguindy, M.M.; Mendell, J.T. NORAD-induced Pumilio phase separation is required for genome stability. Nature 2021, 595, 303–308. [Google Scholar] [CrossRef]
- Guo, C.J.; Ma, X.K.; Xing, Y.H.; Zheng, C.C.; Xu, Y.F.; Shan, L.; Zhang, J.; Wang, S.; Wang, Y.; Carmichael, G.G.; et al. Distinct Processing of lncRNAs Contributes to Non-conserved Functions in Stem Cells. Cell 2020, 181, 621–636.e622. [Google Scholar] [CrossRef]
- Liu, B.; Sun, L.; Liu, Q.; Gong, C.; Yao, Y.; Lv, X.; Lin, L.; Yao, H.; Su, F.; Li, D.; et al. A cytoplasmic NF-kappaB interacting long noncoding RNA blocks IkappaB phosphorylation and suppresses breast cancer metastasis. Cancer Cell 2015, 27, 370–381. [Google Scholar] [CrossRef]
- Lyu, Q.; Xu, S.; Lyu, Y.; Choi, M.; Christie, C.K.; Slivano, O.J.; Rahman, A.; Jin, Z.G.; Long, X.; Xu, Y.; et al. SENCR stabilizes vascular endothelial cell adherens junctions through interaction with CKAP4. Proc. Natl. Acad. Sci. USA 2019, 116, 546–555. [Google Scholar] [CrossRef]
- Yamazaki, T.; Souquere, S.; Chujo, T.; Kobelke, S.; Chong, Y.S.; Fox, A.H.; Bond, C.S.; Nakagawa, S.; Pierron, G.; Hirose, T. Functional Domains of NEAT1 Architectural lncRNA Induce Paraspeckle Assembly through Phase Separation. Mol. Cell 2018, 70, 1038–1053.e1037. [Google Scholar] [CrossRef]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Werner, M.S.; Ruthenburg, A.J. Nuclear Fractionation Reveals Thousands of Chromatin-Tethered Noncoding RNAs Adjacent to Active Genes. Cell Rep. 2015, 12, 1089–1098. [Google Scholar] [CrossRef]
- Arner, E.; Daub, C.O.; Vitting-Seerup, K.; Andersson, R.; Lilje, B.; Drablos, F.; Lennartsson, A.; Ronnerblad, M.; Hrydziuszko, O.; Vitezic, M.; et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 2015, 347, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.Y.; Marques, A.C. The activity of human enhancers is modulated by the splicing of their associated lncRNAs. PLoS Comput. Biol. 2022, 18, e1009722. [Google Scholar] [CrossRef]
- Vucicevic, D.; Corradin, O.; Ntini, E.; Scacheri, P.C.; Orom, U.A. Long ncRNA expression associates with tissue-specific enhancers. Cell Cycle 2015, 14, 253–260. [Google Scholar] [CrossRef]
- Anderson, K.M.; Anderson, D.M.; McAnally, J.R.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature 2016, 539, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhang, J.; Liu, Y.; Fan, X.; Ai, S.; Luo, Y.; Li, X.; Jin, H.; Luo, S.; Zheng, H.; et al. The lncRNA Hand2os1/Uph locus orchestrates heart development through regulation of precise expression of Hand2. Development 2019, 146, dev176198. [Google Scholar] [CrossRef]
- Xiang, J.F.; Yin, Q.F.; Chen, T.; Zhang, Y.; Zhang, X.O.; Wu, Z.; Zhang, S.; Wang, H.B.; Ge, J.; Lu, X.; et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014, 24, 513–531. [Google Scholar] [CrossRef]
- Cho, S.W.; Xu, J.; Sun, R.; Mumbach, M.R.; Carter, A.C.; Chen, Y.G.; Yost, K.E.; Kim, J.; He, J.; Nevins, S.A.; et al. Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 2018, 173, 1398–1412.e1322. [Google Scholar] [CrossRef]
- Penny, G.D.; Kay, G.F.; Sheardown, S.A.; Rastan, S.; Brockdorff, N. Requirement for Xist in X chromosome inactivation. Nature 1996, 379, 131–137. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Hwang, T.; Gooding, A.R.; Goodrich, K.J.; Rinn, J.L.; Cech, T.R. RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells. Nat. Genet. 2020, 52, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Roden, C.; Gladfelter, A.S. RNA contributions to the form and function of biomolecular condensates. Nat. Rev. Mol. Cell Biol. 2021, 22, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, R.; Tano, K.; Mizuno, R.; Nakamura, Y.; Ijiri, K.; Rakwal, R.; Shibato, J.; Masuo, Y.; Mayeda, A.; Hirose, T.; et al. Identification of cis- and trans-acting factors involved in the localization of MALAT-1 noncoding RNA to nuclear speckles. RNA 2012, 18, 738–751. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Denzler, R.; Agarwal, V.; Stefano, J.; Bartel, D.P.; Stoffel, M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol. Cell 2014, 54, 766–776. [Google Scholar] [CrossRef]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef]
- Vitiello, M.; Evangelista, M.; Zhang, Y.; Salmena, L.; Pandolfi, P.P.; Poliseno, L. PTENP1 is a ceRNA for PTEN: It’s CRISPR clear. J. Hematol. Oncol. 2020, 13, 73. [Google Scholar] [CrossRef]
- Khurana, E.; Fu, Y.; Colonna, V.; Mu, X.J.; Kang, H.M.; Lappalainen, T.; Sboner, A.; Lochovsky, L.; Chen, J.; Harmanci, A.; et al. Integrative annotation of variants from 1092 humans: Application to cancer genomics. Science 2013, 342, 1235587. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef]
- Vancura, A.; Gutierrez, A.H.; Hennig, T.; Pulido-Quetglas, C.; Slack, F.J.; Johnson, R.; Haefliger, S. Is Evolutionary Conservation a Useful Predictor for Cancer Long Noncoding RNAs? Insights from the Cancer LncRNA Census 3. Noncoding RNA 2022, 8, 82. [Google Scholar] [CrossRef]
- Yang, M.; Lu, H.; Liu, J.; Wu, S.; Kim, P.; Zhou, X. lncRNAfunc: A knowledgebase of lncRNA function in human cancer. Nucleic Acids Res. 2022, 50, D1295–D1306. [Google Scholar] [CrossRef]
- Gutschner, T.; Richtig, G.; Haemmerle, M.; Pichler, M. From biomarkers to therapeutic targets-the promises and perils of long non-coding RNAs in cancer. Cancer Metastasis Rev. 2018, 37, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Hu, Z.; Feng, Y.; Hu, X.; Yuan, J.; Zhao, S.D.; Zhang, Y.; Yang, L.; Shan, W.; He, Q.; et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer Cell 2015, 28, 529–540. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Rabiee, N.; Kumar, A.P.; Sethi, G.; Zarrabi, A.; Wang, Y. Long noncoding RNAs (lncRNAs) in pancreatic cancer progression. Drug Discov. Today 2022, 27, 2181–2198. [Google Scholar] [CrossRef] [PubMed]
- Duguang, L.; Jin, H.; Xiaowei, Q.; Peng, X.; Xiaodong, W.; Zhennan, L.; Jianjun, Q.; Jie, Y. The involvement of lncRNAs in the development and progression of pancreatic cancer. Cancer Biol. Ther. 2017, 18, 927–936. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Fathi, M.; Zhai, T.; Taheri, M.; Dong, P. LncRNAs: Novel Biomarkers for Pancreatic Cancer. Biomolecules 2021, 11, 1665. [Google Scholar] [CrossRef]
- Huang, X.; Zhi, X.; Gao, Y.; Ta, N.; Jiang, H.; Zheng, J. LncRNAs in pancreatic cancer. Oncotarget 2016, 7, 57379–57390. [Google Scholar] [CrossRef]
- Xu, J.; Xu, J.; Liu, X.; Jiang, J. The role of lncRNA-mediated ceRNA regulatory networks in pancreatic cancer. Cell Death Discov. 2022, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Jutooru, I.; Chadalapaka, G.; Johnson, G.; Frank, J.; Burghardt, R.; Kim, S.; Safe, S. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 2013, 32, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Nong, K.; Zhu, H.; Wang, W.; Huang, X.; Yuan, Z.; Ai, K. H19 promotes pancreatic cancer metastasis by derepressing let-7′s suppression on its target HMGA2-mediated EMT. Tumour Biol. 2014, 35, 9163–9169. [Google Scholar] [CrossRef] [PubMed]
- Pang, E.J.; Yang, R.; Fu, X.B.; Liu, Y.F. Overexpression of long non-coding RNA MALAT1 is correlated with clinical progression and unfavorable prognosis in pancreatic cancer. Tumour Biol. 2015, 36, 2403–2407. [Google Scholar] [CrossRef]
- Li, X.; Zhou, S.; Fan, T.; Feng, X. lncRNA DGCR 5/miR-27a-3p/BNIP3 promotes cell apoptosis in pancreatic cancer by regulating the p38 MAPK pathway. Int. J. Mol. Med. 2020, 46, 729–739. [Google Scholar] [CrossRef]
- Ma, L.; Wang, F.; Du, C.; Zhang, Z.; Guo, H.; Xie, X.; Gao, H.; Zhuang, Y.; Kornmann, M.; Gao, H.; et al. Long non-coding RNA MEG3 functions as a tumour suppressor and has prognostic predictive value in human pancreatic cancer. Oncol. Rep. 2018, 39, 1132–1140. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, X.; Hu, H.; He, Y.; Lu, Z.; Wu, P.; Tian, L.; Xia, T.; Yin, J.; Yuan, H.; et al. Long non-coding RNA lnc-PCTST predicts prognosis through inhibiting progression of pancreatic cancer by downregulation of TACC-3. Int. J. Cancer 2018, 143, 3143–3154. [Google Scholar] [CrossRef]
- Liu, Y.; Feng, W.; Liu, W.; Kong, X.; Li, L.; He, J.; Wang, D.; Zhang, M.; Zhou, G.; Xu, W.; et al. Circulating lncRNA ABHD11-AS1 serves as a biomarker for early pancreatic cancer diagnosis. J. Cancer 2019, 10, 3746–3756. [Google Scholar] [CrossRef]
- Ou, Z.L.; Luo, Z.; Lu, Y.B. Long non-coding RNA HULC as a diagnostic and prognostic marker of pancreatic cancer. World J. Gastroenterol. 2019, 25, 6728–6742. [Google Scholar] [CrossRef]
- Permuth, J.B.; Chen, D.T.; Yoder, S.J.; Li, J.; Smith, A.T.; Choi, J.W.; Kim, J.; Balagurunathan, Y.; Jiang, K.; Coppola, D.; et al. Linc-ing Circulating Long Non-coding RNAs to the Diagnosis and Malignant Prediction of Intraductal Papillary Mucinous Neoplasms of the Pancreas. Sci. Rep. 2017, 7, 10484. [Google Scholar] [CrossRef] [PubMed]
- Mello, S.S.; Sinow, C.; Raj, N.; Mazur, P.K.; Bieging-Rolett, K.; Broz, D.K.; Imam, J.F.C.; Vogel, H.; Wood, L.D.; Sage, J.; et al. Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes. Dev. 2017, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Ingram, H.B.; Fox, A.H. Unveiling the intricacies of paraspeckle formation and function. Curr. Opin. Cell Biol. 2024, 90, 102399. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhang, Y.; Yang, J.; He, S.; Li, M.; Yan, S.; Chen, Y.; Qu, C.; Xu, L. NEAT1 regulates pancreatic cancer cell growth, invasion and migration though mircroRNA-335-5p/c-met axis. Am. J. Cancer Res. 2016, 6, 2361–2374. [Google Scholar]
- Feng, Y.; Gao, L.; Cui, G.; Cao, Y. LncRNA NEAT1 facilitates pancreatic cancer growth and metastasis through stabilizing ELF3 mRNA. Am. J. Cancer Res. 2020, 10, 237–248. [Google Scholar]
- Huang, B.; Liu, C.; Wu, Q.; Zhang, J.; Min, Q.; Sheng, T.; Wang, X.; Zou, Y. Long non-coding RNA NEAT1 facilitates pancreatic cancer progression through negative modulation of miR-506-3p. Biochem. Biophys. Res. Commun. 2017, 482, 828–834. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Ollinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef]
- Li, X.; Deng, S.J.; Zhu, S.; Jin, Y.; Cui, S.P.; Chen, J.Y.; Xiang, C.; Li, Q.Y.; He, C.; Zhao, S.F.; et al. Hypoxia-induced lncRNA-NUTF2P3-001 contributes to tumorigenesis of pancreatic cancer by derepressing the miR-3923/KRAS pathway. Oncotarget 2016, 7, 6000–6014. [Google Scholar] [CrossRef]
- Liu, P.; Yang, H.; Zhang, J.; Peng, X.; Lu, Z.; Tong, W.; Chen, J. The lncRNA MALAT1 acts as a competing endogenous RNA to regulate KRAS expression by sponging miR-217 in pancreatic ductal adenocarcinoma. Sci. Rep. 2017, 7, 5186. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.C.; Li, L.; Jiang, Z.C.; Liu, Z.C.; Wang, L.; Wang, H.J. The Functional Role of LncRNA UCA1 in Pancreatic Cancer: A mini-review. J. Cancer 2023, 14, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Feng, W.; Gu, S.; Wang, H.; Zhang, Y.; Chen, W.; Xu, W.; Lin, C.; Gong, A.; Xu, M. The UCA1/KRAS axis promotes human pancreatic ductal adenocarcinoma stem cell properties and tumor growth. Am. J. Cancer Res. 2019, 9, 496–510. [Google Scholar]
- Barcelo, C.; Etchin, J.; Mansour, M.R.; Sanda, T.; Ginesta, M.M.; Sanchez-Arevalo Lobo, V.J.; Real, F.X.; Capella, G.; Estanyol, J.M.; Jaumot, M.; et al. Ribonucleoprotein HNRNPA2B1 interacts with and regulates oncogenic KRAS in pancreatic ductal adenocarcinoma cells. Gastroenterology 2014, 147, 882–892.e888. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ding, Y.; He, Y.; Yu, Z.; Zhou, Y.; Gong, A.; Xu, M. LncRNA UCA1 promotes pancreatic cancer cell migration by regulating mitochondrial dynamics via the MAPK pathway. Arch. Biochem. Biophys. 2023, 748, 109783. [Google Scholar] [CrossRef]
- Liang, X.; Qi, M.; Wu, R.; Liu, A.; Chen, D.; Tang, L.; Chen, J.; Hu, X.; Li, W.; Zhan, L.; et al. Long non-coding RNA CUDR promotes malignant phenotypes in pancreatic ductal adenocarcinoma via activating AKT and ERK signaling pathways. Int. J. Oncol. 2018, 53, 2671–2682. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, W.; Yang, C.; Wu, W.; Wu, S.; Qin, X.; Li, X. Long non-coding RNA UCA1a(CUDR) promotes proliferation and tumorigenesis of bladder cancer. Int. J. Oncol. 2012, 41, 276–284. [Google Scholar] [CrossRef]
- Qiao, X.; Lv, S.X.; Qiao, Y.; Li, Q.P.; Ye, B.; Wang, C.C.; Miao, L. Long noncoding RNA ABHD11-AS1 predicts the prognosis of pancreatic cancer patients and serves as a promoter by activating the PI3K-AKT pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8630–8639. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, R.; Luo, G.; Ai, K. Long noncoding RNA SNHG1 promotes cell proliferation through PI3K/AKT signaling pathway in pancreatic ductal adenocarcinoma. J. Cancer 2018, 9, 2713–2722. [Google Scholar] [CrossRef]
- Cao, W.; Zhang, H.F.; Ding, X.L.; Zhu, S.Z.; Zhou, G.X. The progression of pancreatic cancer cells is promoted by a long non-coding RNA LUCAT1 by activating AKT phosphorylation. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 738–748. [Google Scholar] [CrossRef]
- Nai, Y.; Pan, C.; Hu, X.; Ma, Y. LncRNA LUCAT1 contributes to cell proliferation and migration in human pancreatic ductal adenocarcinoma via sponging miR-539. Cancer Med. 2020, 9, 757–767. [Google Scholar] [CrossRef]
- Meng, L.D.; Shi, G.D.; Ge, W.L.; Huang, X.M.; Chen, Q.; Yuan, H.; Wu, P.F.; Lu, Y.C.; Shen, P.; Zhang, Y.H.; et al. Linc01232 promotes the metastasis of pancreatic cancer by suppressing the ubiquitin-mediated degradation of HNRNPA2B1 and activating the A-Raf-induced MAPK/ERK signaling pathway. Cancer Lett. 2020, 494, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Shilo, A.; Ben Hur, V.; Denichenko, P.; Stein, I.; Pikarsky, E.; Rauch, J.; Kolch, W.; Zender, L.; Karni, R. Splicing factor hnRNP A2 activates the Ras-MAPK-ERK pathway by controlling A-Raf splicing in hepatocellular carcinoma development. RNA 2014, 20, 505–515. [Google Scholar] [CrossRef]
- Ishikawa, T.; Fukushige, S.; Saiki, Y.; Hirose, K.; Hiyoshi, T.; Ogawa, T.; Katori, Y.; Furukawa, T. Long Non-Coding RNAs Associated with Mitogen-Activated Protein Kinase in Human Pancreatic Cancer. Cancers 2023, 15, 303. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, Z.; Liu, X.; Xu, J.; Jiang, X.; Quan, G.; Jiang, J. LINC00941 promotes pancreatic cancer malignancy by interacting with ANXA2 and suppressing NEDD4L-mediated degradation of ANXA2. Cell Death Dis. 2022, 13, 718. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Cui, R.; Ye, L.; Wang, Y.; Wang, X.; Zhang, Q.; Wang, K.; Dong, C.; Le, W.; Chen, B. LINC00941 promotes glycolysis in pancreatic cancer by modulating the Hippo pathway. Mol. Ther. Nucleic Acids 2021, 26, 280–294. [Google Scholar] [CrossRef]
- Shen, W.; Tao, G.Q.; Zhang, Y.; Cai, B.; Sun, J.; Tian, Z.Q. TGF-beta in pancreatic cancer initiation and progression: Two sides of the same coin. Cell Biosci. 2017, 7, 39. [Google Scholar] [CrossRef]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef]
- McCarthy, A.J.; Chetty, R. Smad4/DPC4. J. Clin. Pathol. 2018, 71, 661–664. [Google Scholar] [CrossRef]
- Tascilar, M.; Skinner, H.G.; Rosty, C.; Sohn, T.; Wilentz, R.E.; Offerhaus, G.J.; Adsay, V.; Abrams, R.A.; Cameron, J.L.; Kern, S.E.; et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2001, 7, 4115–4121. [Google Scholar]
- Yamada, S.; Fujii, T.; Shimoyama, Y.; Kanda, M.; Nakayama, G.; Sugimoto, H.; Koike, M.; Nomoto, S.; Fujiwara, M.; Nakao, A.; et al. SMAD4 expression predicts local spread and treatment failure in resected pancreatic cancer. Pancreas 2015, 44, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wu, W.; Huang, C.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Qiu, Z. SMAD4 and its role in pancreatic cancer. Tumour Biol. 2015, 36, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Derderian, C.; Orunmuyi, A.T.; Olapade-Olaopa, E.O.; Ogunwobi, O.O. PVT1 Signaling Is a Mediator of Cancer Progression. Front. Oncol. 2019, 9, 502. [Google Scholar] [CrossRef]
- Li, R.; Wang, X.; Zhu, C.; Wang, K. lncRNA PVT1: A novel oncogene in multiple cancers. Cell Mol. Biol. Lett. 2022, 27, 84. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, W.; Zhang, J.; Ge, L.; Zhang, Y.; Jiang, X.; Peng, W.; Wang, D.; Gong, A.; Xu, M. Long non-coding RNA PVT1 promotes epithelial-mesenchymal transition via the TGF-beta/Smad pathway in pancreatic cancer cells. Oncol. Rep. 2018, 40, 1093–1102. [Google Scholar] [CrossRef]
- Li, Z.; Ma, Z.; Wang, S.; Yan, Q.; Zhuang, H.; Zhou, Z.; Liu, C.; Chen, Y.; Han, M.; Wu, Z.; et al. LINC00909 up-regulates pluripotency factors and promotes cancer stemness and metastasis in pancreatic ductal adenocarcinoma by targeting SMAD4. Biol. Direct 2024, 19, 24. [Google Scholar] [CrossRef]
- Chen, G.; Xu, L.; Ye, G.; Lin, J.; Meng, Z.; Shen, Y. Overexpression of a Long Non-Coding RNA BC037916 is Associated with Pancreatic Tumorigenesis and Poor Prognosis. Onco Targets Ther. 2020, 13, 13451–13463. [Google Scholar] [CrossRef]
- Zhou, B.; Guo, W.; Sun, C.; Zhang, B.; Zheng, F. Linc00462 promotes pancreatic cancer invasiveness through the miR-665/TGFBR1-TGFBR2/SMAD2/3 pathway. Cell Death Dis. 2018, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, G.Q.; Chen, H.; Zhao, Z.J.; Chen, H.Z.; Liu, H.; Wang, G.; Jia, Y.H.; Pan, S.H.; Kong, R.; et al. Plasma and tumor levels of Linc-pint are diagnostic and prognostic biomarkers for pancreatic cancer. Oncotarget 2016, 7, 71773–71781. [Google Scholar] [CrossRef]
- Lu, H.; Yang, D.; Zhang, L.; Lu, S.; Ye, J.; Li, M.; Hu, W. Linc-pint inhibits early stage pancreatic ductal adenocarcinoma growth through TGF-beta pathway activation. Oncol. Lett. 2019, 17, 4633–4639. [Google Scholar] [CrossRef]
- Dorn, A.; Glass, M.; Neu, C.T.; Heydel, B.; Huttelmaier, S.; Gutschner, T.; Haemmerle, M. LINC00261 Is Differentially Expressed in Pancreatic Cancer Subtypes and Regulates a Pro-Epithelial Cell Identity. Cancers 2020, 12, 1227. [Google Scholar] [CrossRef]
- Wu, B.Q.; Jiang, Y.; Zhu, F.; Sun, D.L.; He, X.Z. Long Noncoding RNA PVT1 Promotes EMT and Cell Proliferation and Migration Through Downregulating p21 in Pancreatic Cancer Cells. Technol. Cancer Res. Treat. 2017, 16, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, F.; Schwarzl, T.; Valcarcel, J.; Hentze, M.W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2021, 22, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Pereira, B.; Billaud, M.; Almeida, R. RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef]
- Glass, M.; Michl, P.; Huttelmaier, A.S. RNA Binding Proteins as Drivers and Therapeutic Target Candidates in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 4190. [Google Scholar] [CrossRef] [PubMed]
- Grammatikakis, I.; Abdelmohsen, K.; Gorospe, M. Posttranslational control of HuR function. Wiley Interdiscip. Rev. RNA 2017, 8, e1372. [Google Scholar] [CrossRef]
- Finan, J.M.; Sutton, T.L.; Dixon, D.A.; Brody, J.R. Targeting the RNA-Binding Protein HuR in Cancer. Cancer Res. 2023, 83, 3507–3516. [Google Scholar] [CrossRef]
- Srikantan, S.; Gorospe, M. HuR function in disease. Front. Biosci. 2012, 17, 189–205. [Google Scholar] [CrossRef]
- Richards, N.G.; Rittenhouse, D.W.; Freydin, B.; Cozzitorto, J.A.; Grenda, D.; Rui, H.; Gonye, G.; Kennedy, E.P.; Yeo, C.J.; Brody, J.R.; et al. HuR status is a powerful marker for prognosis and response to gemcitabine-based chemotherapy for resected pancreatic ductal adenocarcinoma patients. Ann. Surg. 2010, 252, 499–505; discussion 505–506. [Google Scholar] [CrossRef]
- Costantino, C.L.; Witkiewicz, A.K.; Kuwano, Y.; Cozzitorto, J.A.; Kennedy, E.P.; Dasgupta, A.; Keen, J.C.; Yeo, C.J.; Gorospe, M.; Brody, J.R. The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR Up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Res. 2009, 69, 4567–4572. [Google Scholar] [CrossRef] [PubMed]
- Elebro, J.; Ben Dror, L.; Heby, M.; Nodin, B.; Jirstrom, K.; Eberhard, J. Prognostic effect of hENT1, dCK and HuR expression by morphological type in periampullary adenocarcinoma, including pancreatic cancer. Acta Oncol. 2016, 55, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Telonis, A.G.; Jing, Y.; Xia, N.L.; Biederman, L.; Jimbo, M.; Blanco, F.; Londin, E.; Brody, J.R.; Rigoutsos, I. GPRC5A is a potential oncogene in pancreatic ductal adenocarcinoma cells that is upregulated by gemcitabine with help from HuR. Cell Death Dis. 2016, 7, e2294. [Google Scholar] [CrossRef]
- Lal, S.; Burkhart, R.A.; Beeharry, N.; Bhattacharjee, V.; Londin, E.R.; Cozzitorto, J.A.; Romeo, C.; Jimbo, M.; Norris, Z.A.; Yeo, C.J.; et al. HuR posttranscriptionally regulates WEE1: Implications for the DNA damage response in pancreatic cancer cells. Cancer Res. 2014, 74, 1128–1140. [Google Scholar] [CrossRef]
- Jain, A.; McCoy, M.; Coats, C.; Brown, S.Z.; Addya, S.; Pelz, C.; Sears, R.C.; Yeo, C.J.; Brody, J.R. HuR Plays a Role in Double-Strand Break Repair in Pancreatic Cancer Cells and Regulates Functional BRCA1-Associated-Ring-Domain-1(BARD1) Isoforms. Cancers 2022, 14, 1848. [Google Scholar] [CrossRef]
- Burkhart, R.A.; Pineda, D.M.; Chand, S.N.; Romeo, C.; Londin, E.R.; Karoly, E.D.; Cozzitorto, J.A.; Rigoutsos, I.; Yeo, C.J.; Brody, J.R.; et al. HuR is a post-transcriptional regulator of core metabolic enzymes in pancreatic cancer. RNA Biol. 2013, 10, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.Z.; McCarthy, G.A.; Carroll, J.R.; Di Niro, R.; Pelz, C.; Jain, A.; Sutton, T.L.; Holly, H.D.; Nevler, A.; Schultz, C.W.; et al. The RNA-Binding Protein HuR Posttranscriptionally Regulates the Protumorigenic Activator YAP1 in Pancreatic Ductal Adenocarcinoma. Mol. Cell Biol. 2022, 42, e0001822. [Google Scholar] [CrossRef]
- Pineda, D.M.; Rittenhouse, D.W.; Valley, C.C.; Cozzitorto, J.A.; Burkhart, R.A.; Leiby, B.; Winter, J.M.; Weber, M.C.; Londin, E.R.; Rigoutsos, I.; et al. HuR’s post-transcriptional regulation of Death Receptor 5 in pancreatic cancer cells. Cancer Biol. Ther. 2012, 13, 946–955. [Google Scholar] [CrossRef]
- Dong, R.; Chen, P.; Polireddy, K.; Wu, X.; Wang, T.; Ramesh, R.; Dixon, D.A.; Xu, L.; Aube, J.; Chen, Q. An RNA-Binding Protein, Hu-antigen R, in Pancreatic Cancer Epithelial to Mesenchymal Transition, Metastasis, and Cancer Stem Cells. Mol. Cancer Ther. 2020, 19, 2267–2277. [Google Scholar] [CrossRef]
- McCarthy, G.A.; Di Niro, R.; Finan, J.M.; Jain, A.; Guo, Y.; Wyatt, C.R.; Guimaraes, A.R.; Waugh, T.A.; Keith, D.; Morgan, T.K.; et al. Deletion of the mRNA stability factor ELAVL1 (HuR) in pancreatic cancer cells disrupts the tumor microenvironment integrity. NAR Cancer 2023, 5, zcad016. [Google Scholar] [CrossRef]
- Bell, J.L.; Wachter, K.; Muhleck, B.; Pazaitis, N.; Kohn, M.; Lederer, M.; Huttelmaier, S. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): Post-transcriptional drivers of cancer progression? Cell Mol. Life Sci. 2013, 70, 2657–2675. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Liu, H.; Xu, S.; Yang, Z.; Zhang, F.; Wang, G.; Wang, Y.; Zhao, S.; Jiang, X. IGF2BPs as novel m(6)A readers: Diverse roles in regulating cancer cell biological functions, hypoxia adaptation, metabolism, and immunosuppressive tumor microenvironment. Genes. Dis. 2024, 11, 890–920. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Misiak, D.; Bley, N.; Muller, S.; Hagemann, S.; Busch, B.; Rausch, A.; Huttelmaier, S. IGF2BP1, a Conserved Regulator of RNA Turnover in Cancer. Front. Mol. Biosci. 2021, 8, 632219. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef]
- Muller, S.; Bley, N.; Glass, M.; Busch, B.; Rousseau, V.; Misiak, D.; Fuchs, T.; Lederer, M.; Huttelmaier, S. IGF2BP1 enhances an aggressive tumor cell phenotype by impairing miRNA-directed downregulation of oncogenic factors. Nucleic Acids Res. 2018, 46, 6285–6303. [Google Scholar] [CrossRef]
- Wan, B.S.; Cheng, M.; Zhang, L. Insulin-like growth factor 2 mRNA-binding protein 1 promotes cell proliferation via activation of AKT and is directly targeted by microRNA-494 in pancreatic cancer. World J. Gastroenterol. 2019, 25, 6063–6076. [Google Scholar] [CrossRef]
- Di Fusco, D.; Segreto, M.T.; Di Maggio, G.; Iannucci, A.; Maresca, C.; Di Grazia, A.; Colella, M.; Stolfi, C.; Monteleone, G.; Monteleone, I. Insulin-like Growth Factor II mRNA-Binding Protein 1 Regulates Pancreatic Cancer Cell Growth through the Surveillance of CDC25A mRNA. Cancers 2023, 15, 4983. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, H.; Yao, W.; Zhu, N.; Miao, R.; Liu, Z.; Song, X.; Xue, C.; Cai, C.; Cheng, M.; et al. RRP9 promotes gemcitabine resistance in pancreatic cancer via activating AKT signaling pathway. Cell Commun. Signal 2022, 20, 188. [Google Scholar] [CrossRef]
- Muller, S.; Bley, N.; Busch, B.; Glass, M.; Lederer, M.; Misiak, C.; Fuchs, T.; Wedler, A.; Haase, J.; Bertoldo, J.B.; et al. The oncofetal RNA-binding protein IGF2BP1 is a druggable, post-transcriptional super-enhancer of E2F-driven gene expression in cancer. Nucleic Acids Res. 2020, 48, 8576–8590. [Google Scholar] [CrossRef]
- Lin, C.; Wang, Y.; Dong, Y.; Lai, S.; Wang, L.; Weng, S.; Zhang, X. N6-methyladenosine-mediated SH3BP5-AS1 upregulation promotes GEM chemoresistance in pancreatic cancer by activating the Wnt signaling pathway. Biol. Direct 2022, 17, 33. [Google Scholar] [CrossRef]
- Cao, P.; Zhang, W.; Qiu, J.; Tang, Z.; Xue, X.; Feng, T. Gemcitabine Inhibits the Progression of Pancreatic Cancer by Restraining the WTAP/MYC Chain in an m6A-Dependent Manner. Cancer Res. Treat. 2024, 56, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wang, J.Z.; Chen, D.; He, Y.T.; Meng, N.; Chen, M.; Lu, R.X.; Chen, X.H.; Zhang, X.L.; Yan, G.R. An oncopeptide regulates m(6)A recognition by the m(6)A reader IGF2BP1 and tumorigenesis. Nat. Commun. 2020, 11, 1685. [Google Scholar] [CrossRef]
- Xu, X.; Yu, Y.; Zong, K.; Lv, P.; Gu, Y. Up-regulation of IGF2BP2 by multiple mechanisms in pancreatic cancer promotes cancer proliferation by activating the PI3K/Akt signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 497. [Google Scholar] [CrossRef] [PubMed]
- Dahlem, C.; Barghash, A.; Puchas, P.; Haybaeck, J.; Kessler, S.M. The Insulin-Like Growth Factor 2 mRNA Binding Protein IMP2/IGF2BP2 is Overexpressed and Correlates with Poor Survival in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 3204. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.X.; Liu, F.; Jiang, J.H.; Yuan, H.; Zhang, Z.; Yang, L.; Mo, Y.Y. N6-methyladenosine modified LINC00901 promotes pancreatic cancer progression through IGF2BP2/MYC axis. Genes. Dis. 2023, 10, 554–567. [Google Scholar] [CrossRef]
- Yao, Y.; Luo, L.; Xiang, G.; Xiong, J.; Ke, N.; Tan, C.; Chen, Y.; Liu, X. The expression of m(6)A regulators correlated with the immune microenvironment plays an important role in the prognosis of pancreatic ductal adenocarcinoma. Gland. Surg. 2022, 11, 147–165. [Google Scholar] [CrossRef]
- Deng, H.; Yao, H.; Zhou, S.; He, C.; Huang, Y.; Li, Y.; Chen, H.; Shu, J. Pancancer analysis uncovers an immunological role and prognostic value of the m6A reader IGF2BP2 in pancreatic cancer. Mol. Cell Probes 2024, 73, 101948. [Google Scholar] [CrossRef]
- Huang, S.; Wu, Z.; Cheng, Y.; Wei, W.; Hao, L. Insulin-like growth factor 2 mRNA binding protein 2 promotes aerobic glycolysis and cell proliferation in pancreatic ductal adenocarcinoma via stabilizing GLUT1 mRNA. Acta Biochim. Biophys. Sin. 2019, 51, 743–752. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J.; Chen, W.; Xu, Y.; Shen, Y.; Xu, X. Targeting IGF2BP3 in Cancer. Int. J. Mol. Sci. 2023, 24, 9423. [Google Scholar] [CrossRef]
- Mueller-Pillasch, F.; Lacher, U.; Wallrapp, C.; Micha, A.; Zimmerhackl, F.; Hameister, H.; Varga, G.; Friess, H.; Buchler, M.; Beger, H.G.; et al. Cloning of a gene highly overexpressed in cancer coding for a novel KH-domain containing protein. Oncogene 1997, 14, 2729–2733. [Google Scholar] [CrossRef]
- Wang, B.J.; Wang, L.; Yang, S.Y.; Liu, Z.J. Expression and clinical significance of IMP3 in microdissected premalignant and malignant pancreatic lesions. Clin. Transl. Oncol. 2015, 17, 215–222. [Google Scholar] [CrossRef]
- Senoo, J.; Mikata, R.; Kishimoto, T.; Hayashi, M.; Kusakabe, Y.; Yasui, S.; Yamato, M.; Ohyama, H.; Sugiyama, H.; Tsuyuguchi, T.; et al. Immunohistochemical analysis of IMP3 and p53 expression in endoscopic ultrasound-guided fine needle aspiration and resected specimens of pancreatic diseases. Pancreatology 2018, 18, 176–183. [Google Scholar] [CrossRef]
- Johnson, B.; Khalil, M.; Blansfield, J.; Lin, F.; Zhu, S.; Kirchner, H.L.; Weir, A.B., 3rd. Investigating the prognostic value of KOC (K homology domain containing protein overexpressed in cancer) overexpression after curative intent resection of pancreatic ductal adenocarcinoma. J. Gastrointest. Oncol. 2016, 7, E113–E117. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, X.; Fang, Y.; Huo, Z.; Lu, X.; Zhan, X.; Deng, X.; Peng, C.; Shen, B. Integrated expression profiles analysis reveals novel predictive biomarker in pancreatic ductal adenocarcinoma. Oncotarget 2017, 8, 52571–52583. [Google Scholar] [CrossRef] [PubMed]
- Yantiss, R.K.; Cosar, E.; Fischer, A.H. Use of IMP3 in identification of carcinoma in fine needle aspiration biopsies of pancreas. Acta Cytol. 2008, 52, 133–138. [Google Scholar] [CrossRef]
- Wachter, D.L.; Schlabrakowski, A.; Hoegel, J.; Kristiansen, G.; Hartmann, A.; Riener, M.O. Diagnostic value of immunohistochemical IMP3 expression in core needle biopsies of pancreatic ductal adenocarcinoma. Am. J. Surg. Pathol. 2011, 35, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Mikata, R.; Yasui, S.; Kishimoto, T.; Kouchi, Y.; Shingyoji, A.; Senoo, J.; Takahashi, K.; Nagashima, H.; Kusakabe, Y.; Ohyama, H.; et al. Diagnostic value of IMP3 and p53 immunohistochemical staining in EUS-guided fine-needle aspiration for solid pancreatic tumors. Sci. Rep. 2021, 11, 17257. [Google Scholar] [CrossRef]
- Ennajdaoui, H.; Howard, J.M.; Sterne-Weiler, T.; Jahanbani, F.; Coyne, D.J.; Uren, P.J.; Dargyte, M.; Katzman, S.; Draper, J.M.; Wallace, A.; et al. IGF2BP3 Modulates the Interaction of Invasion-Associated Transcripts with RISC. Cell Rep. 2016, 15, 1876–1883. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, X.; Lin, C.; Huang, Y.; Zhong, Y.; Guo, H.; Zheng, Z.; Weng, S. METTL3-IGF2BP3-axis mediates the proliferation and migration of pancreatic cancer by regulating spermine synthase m6A modification. Front. Oncol. 2022, 12, 962204. [Google Scholar] [CrossRef]
- Pasiliao, C.C.; Chang, C.W.; Sutherland, B.W.; Valdez, S.M.; Schaeffer, D.; Yapp, D.T.; Ng, S.S. The involvement of insulin-like growth factor 2 binding protein 3 (IMP3) in pancreatic cancer cell migration, invasion, and adhesion. BMC Cancer 2015, 15, 266. [Google Scholar] [CrossRef] [PubMed]
- Taniuchi, K.; Furihata, M.; Hanazaki, K.; Saito, M.; Saibara, T. IGF2BP3-mediated translation in cell protrusions promotes cell invasiveness and metastasis of pancreatic cancer. Oncotarget 2014, 5, 6832–6845. [Google Scholar] [CrossRef] [PubMed]
- Taniuchi, K.; Furihata, M.; Saibara, T. KIF20A-mediated RNA granule transport system promotes the invasiveness of pancreatic cancer cells. Neoplasia 2014, 16, 1082–1093. [Google Scholar] [CrossRef]
- Fu, W.; Lei, X.; Lu, Q.; Zhang, J.; Guo, J.; Zhao, J.; Tong, X.; Hu, X. UBE2K regulated by IGF2BP3 promotes cell proliferation and stemness in pancreatic ductal adenocarcinoma. Int. J. Oncol. 2023, 62, 52. [Google Scholar] [CrossRef] [PubMed]
- Dowdle, M.E.; Kanzler, C.R.; Harder, C.R.K.; Moffet, S.; Walker, M.N.; Sheets, M.D. Bicaudal-C Post-transcriptional regulator of cell fates and functions. Front. Cell Dev. Biol. 2022, 10, 981696. [Google Scholar] [CrossRef]
- Mahone, M.; Saffman, E.E.; Lasko, P.F. Localized Bicaudal-C RNA encodes a protein containing a KH domain, the RNA binding motif of FMR1. EMBO J. 1995, 14, 2043–2055. [Google Scholar] [CrossRef]
- Mohler, J.; Wieschaus, E.F. Dominant maternal-effect mutations of Drosophila melanogaster causing the production of double-abdomen embryos. Genetics 1986, 112, 803–822. [Google Scholar] [CrossRef]
- Saffman, E.E.; Styhler, S.; Rother, K.; Li, W.; Richard, S.; Lasko, P. Premature translation of oskar in oocytes lacking the RNA-binding protein bicaudal-C. Mol. Cell Biol. 1998, 18, 4855–4862. [Google Scholar] [CrossRef]
- Meng, F.; Hua, S.; Chen, X.; Meng, N.; Lan, T. Lymph node metastasis related gene BICC1 promotes tumor progression by promoting EMT and immune infiltration in pancreatic cancer. BMC Med. Genom. 2023, 16, 263. [Google Scholar] [CrossRef]
- Huang, C.; Li, H.; Xu, Y.; Xu, C.; Sun, H.; Li, Z.; Ge, Y.; Wang, H.; Zhao, T.; Gao, S.; et al. BICC1 drives pancreatic cancer progression by inducing VEGF-independent angiogenesis. Signal Transduct. Target. Ther. 2023, 8, 271. [Google Scholar] [CrossRef]
- Sun, H.; Li, H.; Guan, Y.; Yuan, Y.; Xu, C.; Fu, D.; Xie, P.; Li, J.; Zhao, T.; Wang, X.; et al. BICC1 drives pancreatic cancer stemness and chemoresistance by facilitating tryptophan metabolism. Sci. Adv. 2024, 10, eadj8650. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Zhang, X.; Luo, H.; Xu, L.; Lu, X.; Lu, J. Adrenaline promotes epithelial-to-mesenchymal transition via HuR-TGFbeta regulatory axis in pancreatic cancer cells and the implication in cancer prognosis. Biochem. Biophys. Res. Commun. 2017, 493, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Lei, X.; Guo, J.; Fu, W.; Sun, W.; Lu, Q.; Su, W.; Xu, Q.; Tu, K. The Emerging Role of RNA N6-Methyladenosine Modification in Pancreatic Cancer. Front. Oncol. 2022, 12, 927640. [Google Scholar] [CrossRef]
- Subramaniam, D.; Ramalingam, S.; Linehan, D.C.; Dieckgraefe, B.K.; Postier, R.G.; Houchen, C.W.; Jensen, R.A.; Anant, S. RNA binding protein CUGBP2/CELF2 mediates curcumin-induced mitotic catastrophe of pancreatic cancer cells. PLoS ONE 2011, 6, e16958. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Useros, J.; Georgiev-Hristov, T.; Fernandez-Acenero, M.J.; Borrero-Palacios, A.; Indacochea, A.; Guerrero, S.; Li, W.; Cebrian, A.; Gomez Del Pulgar, T.; Puime-Otin, A.; et al. UNR/CDSE1 expression as prognosis biomarker in resectable pancreatic ductal adenocarcinoma patients: A proof-of-concept. PLoS ONE 2017, 12, e0182044. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, L.; Du, K. Exosomal HSPB1, interacting with FUS protein, suppresses hypoxia-induced ferroptosis in pancreatic cancer by stabilizing Nrf2 mRNA and repressing P450. J. Cell Mol. Med. 2024, 28, e18209. [Google Scholar] [CrossRef]
- Yan-Sanders, Y.; Hammons, G.J.; Lyn-Cook, B.D. Increased expression of heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNP) in pancreatic tissue from smokers and pancreatic tumor cells. Cancer Lett. 2002, 183, 215–220. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Cai, L.; Zhu, J.; Chen, M.; Chen, J.; Li, Z.H.; Liu, X.D.; Wang, S.G.; Bie, P.; Jiang, P.; et al. Fyn requires HnRNPA2B1 and Sam68 to synergistically regulate apoptosis in pancreatic cancer. Carcinogenesis 2011, 32, 1419–1426. [Google Scholar] [CrossRef]
- Yang, N.; Liu, L.; Liu, X.; Chen, Y.; Lu, J.; Wang, Z. hnRNPC Promotes Malignancy in Pancreatic Cancer through Stabilization of IQGAP3. Biomed. Res. Int. 2022, 2022, 6319685. [Google Scholar] [CrossRef]
- Antal, C.E.; Oh, T.G.; Aigner, S.; Luo, E.C.; Yee, B.A.; Campos, T.; Tiriac, H.; Rothamel, K.L.; Cheng, Z.; Jiao, H.; et al. A super-enhancer-regulated RNA-binding protein cascade drives pancreatic cancer. Nat. Commun. 2023, 14, 5195. [Google Scholar] [CrossRef]
- Qiao, L.; Xie, N.; Bai, Y.; Li, Y.; Shi, Y.; Wang, J.; Liu, N. Identification of Upregulated HNRNPs Associated with Poor Prognosis in Pancreatic Cancer. Biomed. Res. Int. 2019, 2019, 5134050. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Guo, S.; Ouyang, Y.; Yin, L.; Liu, S.; Zhao, Z.; Yang, J.; Huang, W.; Qin, H.; et al. Lin28B facilitates the progression and metastasis of pancreatic ductal adenocarcinoma. Oncotarget 2017, 8, 60414–60428. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, D.; Zhou, M.; Chen, H.; Wang, H.; Min, J.; Chen, J.; Wu, S.; Ni, X.; Zhang, Y.; et al. The KRAS/Lin28B axis maintains stemness of pancreatic cancer cells via the let-7i/TET3 pathway. Mol. Oncol. 2021, 15, 262–278. [Google Scholar] [CrossRef]
- Shu, Z.; Fan, M.; Tu, B.; Tang, Z.; Wang, H.; Li, H.; Li, H.; Yuan, M.; Bai, J.; Huo, S.; et al. The Lin28b/Wnt5a axis drives pancreas cancer through crosstalk between cancer associated fibroblasts and tumor epithelium. Nat. Commun. 2023, 14, 6885. [Google Scholar] [CrossRef]
- Panzeri, V.; Manni, I.; Capone, A.; Naro, C.; Sacconi, A.; Di Agostino, S.; de Latouliere, L.; Montori, A.; Pilozzi, E.; Piaggio, G.; et al. The RNA-binding protein MEX3A is a prognostic factor and regulator of resistance to gemcitabine in pancreatic ductal adenocarcinoma. Mol. Oncol. 2021, 15, 579–595. [Google Scholar] [CrossRef]
- Sheng, W.; Dong, M.; Chen, C.; Wang, Z.; Li, Y.; Wang, K.; Li, Y.; Zhou, J. Cooperation of Musashi-2, Numb, MDM2, and P53 in drug resistance and malignant biology of pancreatic cancer. FASEB J. 2017, 31, 2429–2438. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Cui, J.; Quan, M.; Xie, D.; Jia, Z.; Wei, D.; Wang, L.; Gao, Y.; Ma, Q.; Xie, K. The Novel KLF4/MSI2 Signaling Pathway Regulates Growth and Metastasis of Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 687–696. [Google Scholar] [CrossRef]
- Yang, H.; Hu, J.; Chen, J.; Chen, Z.; Jiao, F.; Cui, J.; Quan, M.; Wang, L. RNA-binding protein Musashi2 regulates Hippo signaling via SAV1 and MOB1 in pancreatic cancer. Med. Oncol. 2020, 37, 84. [Google Scholar] [CrossRef]
- Sheng, W.; Dong, M.; Chen, C.; Li, Y.; Liu, Q.; Dong, Q. Musashi2 promotes the development and progression of pancreatic cancer by down-regulating Numb protein. Oncotarget 2017, 8, 14359–14373. [Google Scholar] [CrossRef]
- Chu, L.; Hu, Y.; Jiang, Y.H.; Xu, C.; Liu, W.C.; Lu, Z.F. Effects of RNA binding protein QKI on pancreatic cancer ductal epithelial cells and surrounding activation fibroblasts. J. Cell Biochem. 2019, 120, 11551–11561. [Google Scholar] [CrossRef]
- Maurin, M.; Ranjouri, M.; Megino-Luque, C.; Newberg, J.Y.; Du, D.; Martin, K.; Miner, R.E., 3rd; Prater, M.S.; Wee, D.K.B.; Centeno, B.; et al. RBFOX2 deregulation promotes pancreatic cancer progression and metastasis through alternative splicing. Nat. Commun. 2023, 14, 8444. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Xiao, W.; Chen, X.; Li, X.; Deng, K.; Liu, H.; Ma, J.; Wang, Z.; Hu, Y.; Hou, J. RBM10 regulates human TERT gene splicing and inhibits pancreatic cancer progression. Am. J. Cancer Res. 2021, 11, 157–170. [Google Scholar] [PubMed]
- Iwata, T.; Kishikawa, T.; Seimiya, T.; Notoya, G.; Suzuki, T.; Shibata, C.; Miyakawa, Y.; Odawara, N.; Funato, K.; Tanaka, E.; et al. Satellite double-stranded RNA induces mesenchymal transition in pancreatic cancer by regulating alternative splicing. J. Biol. Chem. 2024, 300, 105742. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, M.; Tan, J. Knockdown of ZFR suppresses cell proliferation and invasion of human pancreatic cancer. Biol. Res. 2016, 49, 26. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Tani, H. Recent Advances and Prospects in RNA Drug Development. Int. J. Mol. Sci. 2024, 25, 12284. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef]
- Neil, E.E.; Bisaccia, E.K. Nusinersen: A Novel Antisense Oligonucleotide for the Treatment of Spinal Muscular Atrophy. J. Pediatr. Pharmacol. Ther. 2019, 24, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Ammala, C.; Drury, W.J., 3rd; Knerr, L.; Ahlstedt, I.; Stillemark-Billton, P.; Wennberg-Huldt, C.; Andersson, E.M.; Valeur, E.; Jansson-Lofmark, R.; Janzen, D.; et al. Targeted delivery of antisense oligonucleotides to pancreatic beta-cells. Sci. Adv. 2018, 4, eaat3386. [Google Scholar] [CrossRef]
- Childs-Disney, J.L.; Yang, X.; Gibaut, Q.M.R.; Tong, Y.; Batey, R.T.; Disney, M.D. Targeting RNA structures with small molecules. Nat. Rev. Drug Discov. 2022, 21, 736–762. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K.; Thaxton, M.L.; Scherer, G.M.; Sorrentino, J.P.; Garg, N.K.; Rao, D.S. Small molecule inhibition of RNA binding proteins in haematologic cancer. RNA Biol. 2024, 21, 276–289. [Google Scholar] [CrossRef]
- Li, Q.; Kang, C. Targeting RNA-binding proteins with small molecules: Perspectives, pitfalls and bifunctional molecules. FEBS Lett. 2023, 597, 2031–2047. [Google Scholar] [CrossRef]
- Nguyen, L.D.; Chau, R.K.; Krichevsky, A.M. Small Molecule Drugs Targeting Non-Coding RNAs as Treatments for Alzheimer’s Disease and Related Dementias. Genes 2021, 12, 2005. [Google Scholar] [CrossRef]
- Zhao, R.; Fu, J.; Zhu, L.; Chen, Y.; Liu, B. Designing strategies of small-molecule compounds for modulating non-coding RNAs in cancer therapy. J. Hematol. Oncol. 2022, 15, 14. [Google Scholar] [CrossRef] [PubMed]
- Bertoldo, J.B.; Muller, S.; Huttelmaier, S. RNA-binding proteins in cancer drug discovery. Drug Discov. Today 2023, 28, 103580. [Google Scholar] [CrossRef]
- Bekes, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Gama-Brambila, R.A.; Chen, J.; Zhou, J.; Tascher, G.; Munch, C.; Cheng, X. A PROTAC targets splicing factor 3B1. Cell Chem. Biol. 2021, 28, 1616–1627.e1618. [Google Scholar] [CrossRef]
- Kaur, T.; Menon, A.; Garner, A.L. Synthesis of 7-benzylguanosine cap-analogue conjugates for eIF4E targeted degradation. Eur. J. Med. Chem. 2019, 166, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Ghidini, A.; Clery, A.; Halloy, F.; Allain, F.H.T.; Hall, J. RNA-PROTACs: Degraders of RNA-Binding Proteins. Angew. Chem. Int. Ed. Engl. 2021, 60, 3163–3169. [Google Scholar] [CrossRef]
- Costales, M.G.; Matsumoto, Y.; Velagapudi, S.P.; Disney, M.D. Small Molecule Targeted Recruitment of a Nuclease to RNA. J. Am. Chem. Soc. 2018, 140, 6741–6744. [Google Scholar] [CrossRef] [PubMed]
- Costales, M.G.; Aikawa, H.; Li, Y.; Childs-Disney, J.L.; Abegg, D.; Hoch, D.G.; Pradeep Velagapudi, S.; Nakai, Y.; Khan, T.; Wang, K.W.; et al. Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 2406–2411. [Google Scholar] [CrossRef]
- Meyer, S.M.; Tanaka, T.; Zanon, P.R.A.; Baisden, J.T.; Abegg, D.; Yang, X.; Akahori, Y.; Alshakarchi, Z.; Cameron, M.D.; Adibekian, A.; et al. DNA-Encoded Library Screening to Inform Design of a Ribonuclease Targeting Chimera (RiboTAC). J. Am. Chem. Soc. 2022, 144, 21096–21102. [Google Scholar] [CrossRef]
- Tong, Y.; Lee, Y.; Liu, X.; Childs-Disney, J.L.; Suresh, B.M.; Benhamou, R.I.; Yang, C.; Li, W.; Costales, M.G.; Haniff, H.S.; et al. Programming inactive RNA-binding small molecules into bioactive degraders. Nature 2023, 618, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Cirino, N.M.; Li, G.; Xiao, W.; Torrence, P.F.; Silverman, R.H. Targeting RNA decay with 2′,5′ oligoadenylate-antisense in respiratory syncytial virus-infected cells. Proc. Natl. Acad. Sci. USA 1997, 94, 1937–1942. [Google Scholar] [CrossRef]
- Fang, Y.; Wu, Q.; Wang, F.; Liu, Y.; Zhang, H.; Yang, C.; Zhu, Z. Aptamer-RIBOTAC Strategy Enabling Tumor-Specific Targeted Degradation of MicroRNA for Precise Cancer Therapy. In Small Methods; Wiley: New York, NY, USA, 2024; p. e2400349. [Google Scholar] [CrossRef]
- Su, X.; Ma, W.; Feng, D.; Cheng, B.; Wang, Q.; Guo, Z.; Zhou, D.; Tang, X. Efficient Inhibition of SARS-CoV-2 Using Chimeric Antisense Oligonucleotides through RNase L Activation. Angew. Chem. Int. Ed. Engl. 2021, 60, 21662–21667. [Google Scholar] [CrossRef]
- Torrence, P.F.; Maitra, R.K.; Lesiak, K.; Khamnei, S.; Zhou, A.; Silverman, R.H. Targeting RNA for degradation with a (2′-5′)oligoadenylate-antisense chimera. Proc. Natl. Acad. Sci. USA 1993, 90, 1300–1304. [Google Scholar] [CrossRef]
- Lu, J.; Yu, L.; Xie, N.; Wu, Y.; Li, B. METTL14 Facilitates the Metastasis of Pancreatic Carcinoma by Stabilizing LINC00941 in an m6A-IGF2BP2-Dependent Manner. J. Cancer 2023, 14, 1117–1131. [Google Scholar] [CrossRef]
- Zhai, S.; Xu, Z.; Xie, J.; Zhang, J.; Wang, X.; Peng, C.; Li, H.; Chen, H.; Shen, B.; Deng, X. Epigenetic silencing of LncRNA LINC00261 promotes c-myc-mediated aerobic glycolysis by regulating miR-222-3p/HIPK2/ERK axis and sequestering IGF2BP1. Oncogene 2021, 40, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, A.; Kureshi, N.; Wittig, A.; Sterner, M.; Huber, R.; Palma, N.A.; King, T.; Schiess, R. Biomarker Discovery for Early Detection of Pancreatic Ductal Adenocarcinoma (PDAC) Using Multiplex Proteomics Technology. J. Proteome Res. 2025, 24, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Khomiak, A.; Brunner, M.; Kordes, M.; Lindblad, S.; Miksch, R.C.; Ohlund, D.; Regel, I. Recent Discoveries of Diagnostic, Prognostic and Predictive Biomarkers for Pancreatic Cancer. Cancers 2020, 12, 3234. [Google Scholar] [CrossRef] [PubMed]
- Bin, W.; Yuan, C.; Qie, Y.; Dang, S. Long non-coding RNAs and pancreatic cancer: A multifaceted view. Biomed. Pharmacother. 2023, 167, 115601. [Google Scholar] [CrossRef]
- von Widdern, J.C.; Rosendahl, J.; Ammer-Herrmenau, C. Chronic and Idiopathic Pancreatitis-A Personalized Treatment Approach. United Eur. Gastroenterol. J. 2025, 13, 116–124. [Google Scholar] [CrossRef]
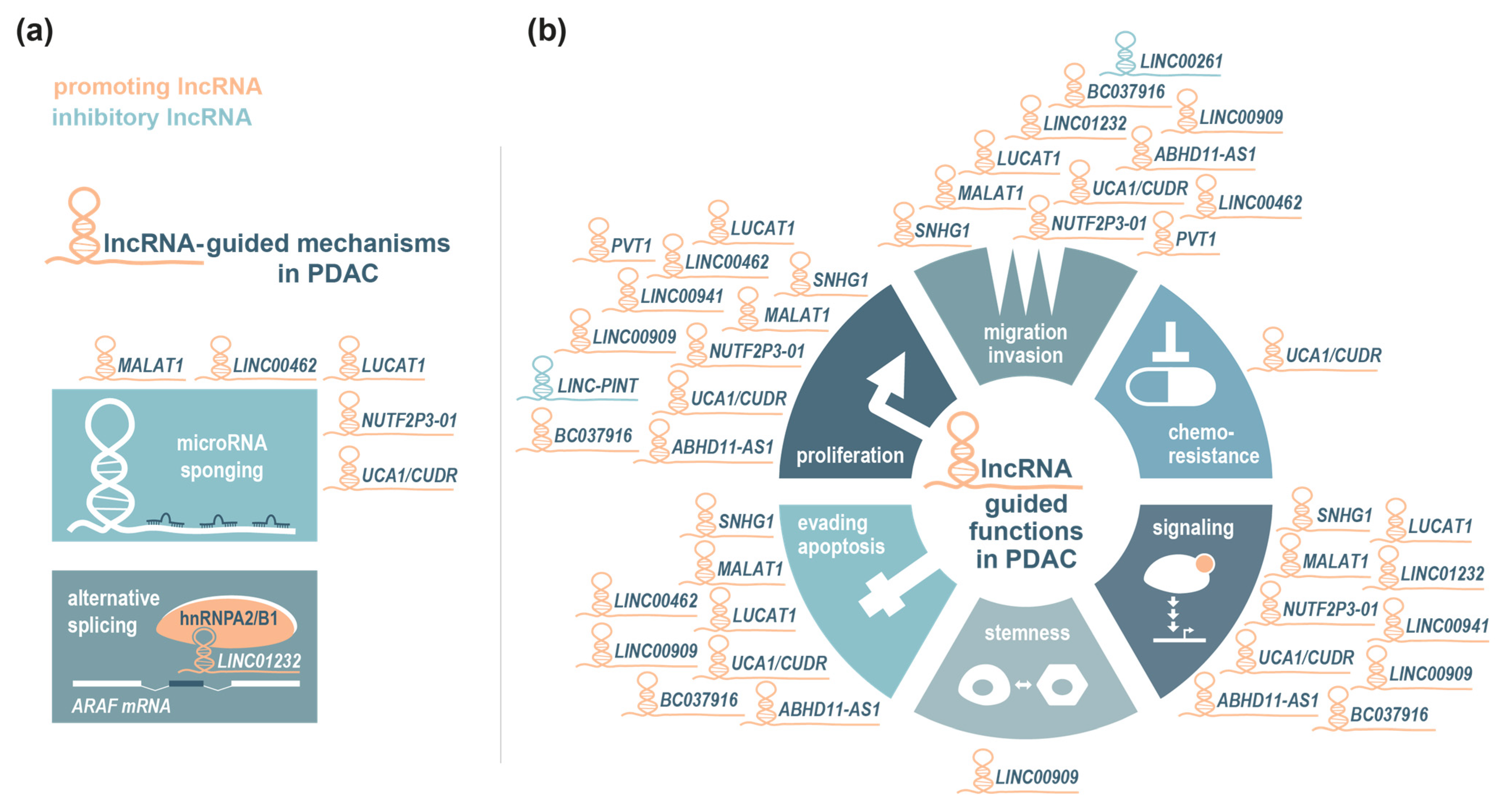


{kind=link}
{kind=link}
{kind=link}
| Function | Mode of Action | Description | Examples |
|---|---|---|---|
| Enhancer action | cis/trans | A total of 30–60% of lncRNAs are transcribed from enhancer regions, and enhancer activity is modulated by the active transcription and splicing of enhancer lncRNAs [44,45,46]. These transcripts may potentiate enhancer activity by recruiting proteins that participate in the formation of chromatin loops between distal enhancers and their promoters or direct the recruitment of transcriptional activators to gene-proximal enhancers [47,48,49]. Similar mechanisms can also result in the repression of enhancer activity when lncRNA loci compete for enhancers with protein-coding genes [50]. | Hand2os1 [47,48] CCAT1-L [49] PVT1 [50] |
| Chromatin architecture modulation | cis/trans | The process of X chromosome inactivation is dependent on the cis-acting lncRNA XIST, which spreads along and coats the inactive chromosome, ensuring dosage compensation [31,51]. Additionally, association with chromatin remodelling complexes is a common feature among lncRNAs [52]. The polycomp repressive complex 2 (PRC2) in particular is functionally dependent on RNAs and has been shown to interact with ~20% of human lncRNAs [52,53]. | HOTAIR [30,54] XIST [31,51] |
| Biomolecular condensate formation | trans | Intracellular condensates are formed from RNA and protein interactions through liquid–liquid phase separation. Many of these compartments depend on architectural lncRNAs for their structures, compositions and functions [55]. | NORAD [36] NEAT1 [40] MALAT1 [56] |
| miRNA sponge | trans | The target sites of miRNAs are found in mRNAs but are also present in many types of ncRNAs including lncRNAs. This has led to the hypothesis that all transcripts sharing binding sites for a particular miRNA may regulate each other by competing against each other [57]. An extensive number of publications have focused on the action of lncRNAs as competing endogenous RNAs (ceRNAs), but the hypothesis has also received criticism relating to the stoichiometries needed to support ceRNA regulatory networks [58]. | LINCMD1 [59], PTENP1 [60,61] |
| LncRNA | Expression in PDAC | Clinical Association | Function and Mechanism | References |
|---|---|---|---|---|
| NUTF2P3-01 | upregulated | high expression correlates with tumour size; poor tumour differentiation; the tumour, node and metastasis (TNM) stage; lymphatic invasion; distant metastasis; and shorter overall survival (OS) | promotes proliferation, invasion, xenograft tumour growth and hepatic metastasis and acts as a ceRNA to sequester miR-3923 and increase KRAS expression | [90] |
| MALAT1 | upregulated | high expression is associated with shorter OS | promotes proliferation, migration, invasion, and xenograft growth; reduces apoptosis; and acts as a ceRNA to sequester miR-217 and increase KRAS expression | [91] |
| UCA1/CUDR | upregulated | high expression correlates with an advanced T and N stage and is associated with poor OS | promotes proliferation, migration, drug resistance and xenograft tumour growth; reduces apoptosis; positively regulates MAPK/ERK and AKT/FAK signalling; enhances the interaction between KRAS and hnRNPA2/B1; and acts as a ceRNA to sequester miR-590-3p and increase KRAS expression | [93,95,96] |
| ABHD11-AS1 | upregulated | expression correlates with poor OS and disease-free survival (DFS), an advanced TNM stage, increased distant metastasis and poor tumour differentiation | promotes proliferation and migration, reduces apoptosis and positively regulates PI3K/AKT signalling and EMT | [98] |
| SNHG1 | upregulated | expression correlates with an advanced TNM stage and a larger tumour size | promotes proliferation, invasion and xenograft tumour growth; reduces apoptosis; and positively regulates PI3K/AKT signalling | [99] |
| LUCAT1 | upregulated | increased expression linked to a larger tumour size and lymphatic invasion | promotes proliferation, invasion and xenograft tumour growth; reduces apoptosis; and positively regulates AKT and p38-MAPK signalling and the sponge of miR-539 | [100,101] |
| LINC01232 | upregulated | expression is positively correlated with nerve invasion, and high LINC01232 levels are associated with worse outcomes | promotes migration and invasion and regulates the alternative splicing of ARAF by stabilizing hnRNPA2/B1, thereby activating MAPK/ERK signalling | [102] |
| LINC00941 | upregulated | high expression is associated with a poor prognosis | promotes proliferation, is a downstream target of MAPK/ETS-1 signalling and increases E2F7 | [104] |
| PVT1 | upregulated | high expression is associated with an advanced clinical stage and lymph node metastasis (LNM) | promotes cell adhesion, viability, migration and invasion; enhances EMT via TGF-β/SMAD signalling; and downregulates SMAD4 and p21 | [115,122] |
| LINC00909 | upregulated | high expression is associated with poor OS and DFS and correlates with an advanced TNM stage, a larger tumour size, poor differentiation and LNM | promotes cell viability, colony formation, migration and xenograft tumour growth and metastasis; inhibits apoptosis; upregulates the expression of pluripotency factors; promotes the pancreatic cancer stem cell phenotype; activates the MAPK/JNK pathway; and destabilizes the SMAD4 mRNA | [116] |
| BC037916 | upregulated | increased expression correlates with a clinical stage and advanced T and N stages and is associated with poor OS | promotes proliferation, invasion and xenograft tumour growth; reduces apoptosis; positively regulates EMT via SMAD2/3 signalling; and upregulates JAK/STAT signalling | [117] |
| LINC00462 | upregulated | expression is associated with a larger tumour size, poor tumour differentiation, a TNM stage and distant metastasis | promotes proliferation, migration, invasion, xenograft growth and invasion; reduces apoptosis and cellular adhesion; induces EMT through the activation of the TGF-β pathway via the upregulation of TGFBR1/2; and acts as a ceRNA to sequester miR-665 | [118] |
| LINC-PINT | downregulated | Low plasma levels correlate with tumour recurrence and are associated with tumour size; a low LINC-PINT level in tumour tissues correlates with a poor prognosis after pancreatectomy | reduces cell proliferation and increases TGF-β1 expression in a pancreatic cancer cell line | [119,120] |
| LINC00261 | downregulated | expression is subtype-dependent and shows an inverse correlation with tumour grade and stage, and high expression is associated with better OS | inhibits cell migration and invasion; TGF-β stimulation decreases LINC00261 levels, and its genetic ablation leads to the downregulation of the E-cadherin mRNA and protein and the induction of an EMT programme | [121] |
| RBP | Roles in PDAC | References |
|---|---|---|
| ELAVL1 (HuR) | upregulated in PDAC; associated with a poor prognosis; promotes proliferation, the cell cycle, migration and invasion; inhibits apoptosis; and might mediate gemcitabine sensitivity induced by promoting DCK expression; stabilized mRNAs in PDAC: BARD1, DCK, DR5/TNFRSF10B, GPRC5A, IDH1, PRPS2, PDGFA, SNAI1, WEE1 and YAP1 | [128,131,132,133,134,135,136,137,138,139,140,141,182] |
| IGF2BP1 | upregulated in PDAC; associated with a poor prognosis; promotes proliferation, cell cycle progression, EMT, metastasis and gemcitabine resistance; is an m6A reader; and stabilized RNAs in PDAC: SH3BP5-AS, E2F1 and CDC25A | [142,143,144,146,147,148,149,150,183] |
| IGF2BP2 | upregulated in PDAC; associated with a poor prognosis; promotes proliferation and glucose metabolism; associated with an immune-suppressive microenvironment; is an m6A reader; and stabilized mRNAs in PDAC: SLC2A1 and CD274 | [142,143,154,155,156,157,158,159,183] |
| IGF2BP3 | upregulated in PDAC; associated with a poor prognosis; frequently used as a biomarker (needle biopsy); promotes proliferation, adhesion, migration and invasion; associated with an immune suppressive microenvironment; is an m6A reader; and stabilized mRNAs in PDAC: ARF6, ARHGEF4, CD44, SMS and UBE2K | [142,143,157,161,162,164,165,166,167,168,169,170,171,172,173,174,183] |
| BICC1 | upregulated in PDAC; expression associated with EMT and immune infiltration; and supports angiogenesis and chemoresistance by promoting LCN2 and CXCL1 expression | [176,177,178,179,180,181] |
| CELF2 (CUGBP2) | enhances stability but represses the translation of VEGF and COX2 mRNAs and induced by curcumin | [184] |
| CSDE1 (UNR) | biomarker associated with a favourable prognosis and the immunogenic subtype | [185] |
| FUS | upregulated in PDAC, stabilizes the NRF2 mRNA and suppresses oxidative stress and ferroptosis | [186] |
| hnRNPA2/B1 | upregulated in early PDAC stages; inhibits apoptosis by suppressing BCL-X(S) splicing; regulates splicing of ARAF; and interacts with KRAS | [93,102,187,188] |
| hnRNPC | controls the mRNA stability of IQGAP3 to promote cell proliferation, migration, invasion and metastasis | [189] |
| hnRNPF | part of a druggable super-enhancer network that promotes PDAC cell and tumour growth and stabilizes the PRMT1 mRNA to promote MYC expression | [190] |
| hnRNPL | upregulated in PDAC, associated with a poor prognosis and promotes migration and EMT | [191] |
| LIN28B | upregulated in PDAC, associated with metastasis and a poor prognosis; promotes proliferation, stemness, migration and EMT; targets in PDAC (via de-regulating let-7): MYC, HMGA2, KRAS, TET3, WNT5A and PCSK9 | [192,193,194] |
| MEX3A | upregulated in PDAC, expression correlates with disease progression in human and mice; promotes cell cycle and gemcitabine resistance and stabilized mRNAs in PDAC: CDK6 | [195] |
| MSI2 | upregulated in PDAC; associated with metastasis and a poor prognosis; promotes proliferation, metastasis and chemoresistance; transcriptionally suppressed by KLF4; and controls HIPPO signalling by suppressing SAV1 and MOB1 translation; mRNA targets in PDAC: NUMB, MOB1, SAV1 | [196,197,198,199] |
| QKI | upregulated in PDAC tumour cells and fibroblasts and promotes proliferation, EMT and metastasis | [200] |
| RBFOX2 | prevents PDAC progression and metastasis and controls the splicing of ABI1 mRNA to inhibit migration | [201] |
| RBM10 | downregulated in PDAC; the RBM10 mutation in advanced-stage tumours is associated with favourable survival; regulates the splicing of hTERT | [202,203] |
| STRBP | Binds to the HSATII RNA to prevent EMT and invasion and controls isoform switching of CLSTN1 | [204] |
| ZFR | upregulated in PDAC and promotes proliferation, cell cycle progression and invasion | [205] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preckwinkel, P.; Mir, K.U.I.; Otto, F.W.; Elrewany, H.; Sinz, A.; Hüttelmaier, S.; Bley, N.; Gutschner, T. Long Non-Coding RNAs and RNA-Binding Proteins in Pancreatic Cancer Development and Progression. Cancers 2025, 17, 1601. https://doi.org/10.3390/cancers17101601
Preckwinkel P, Mir KUI, Otto FW, Elrewany H, Sinz A, Hüttelmaier S, Bley N, Gutschner T. Long Non-Coding RNAs and RNA-Binding Proteins in Pancreatic Cancer Development and Progression. Cancers. 2025; 17(10):1601. https://doi.org/10.3390/cancers17101601
Chicago/Turabian StylePreckwinkel, Pit, Khursheed Ul Islam Mir, Florian W. Otto, Hend Elrewany, Andrea Sinz, Stefan Hüttelmaier, Nadine Bley, and Tony Gutschner. 2025. "Long Non-Coding RNAs and RNA-Binding Proteins in Pancreatic Cancer Development and Progression" Cancers 17, no. 10: 1601. https://doi.org/10.3390/cancers17101601
APA StylePreckwinkel, P., Mir, K. U. I., Otto, F. W., Elrewany, H., Sinz, A., Hüttelmaier, S., Bley, N., & Gutschner, T. (2025). Long Non-Coding RNAs and RNA-Binding Proteins in Pancreatic Cancer Development and Progression. Cancers, 17(10), 1601. https://doi.org/10.3390/cancers17101601