
Synergistic Action of Benzyl Isothiocyanate and Sorafenib in a Nanoparticle Delivery System for Enhanced Triple-Negative Breast Cancer Treatment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Characterization of BITC–SOR-NPs
2.3. In Vitro Drug Release
2.4. Cell Culture
2.5. In Vitro Cytotoxicity Studies
2.6. Three-Dimensional Spheroid Formation
2.7. Colony Formation Assay
2.8. Wound-Healing Assay
2.9. Invasion Assay
2.10. Tube Formation Assay
2.11. Cell Cycle Analysis
2.12. Western Blot Assay
2.13. Statistics
3. Results
3.1. Synthesis of BITC and Sorafenib-Encapsulating Nanoparticles
3.2. Inhibition of TNBC Cell Viability by BITC and SOR
3.3. Inhibition of TNBC Cell Migration and Invasion by BITC and SOR
3.4. Inhibition of TNBC Tube Formation by BITC and SOR
3.5. Induction of Cell Cycle Arrest by BITC and SOR in TNBC Cells
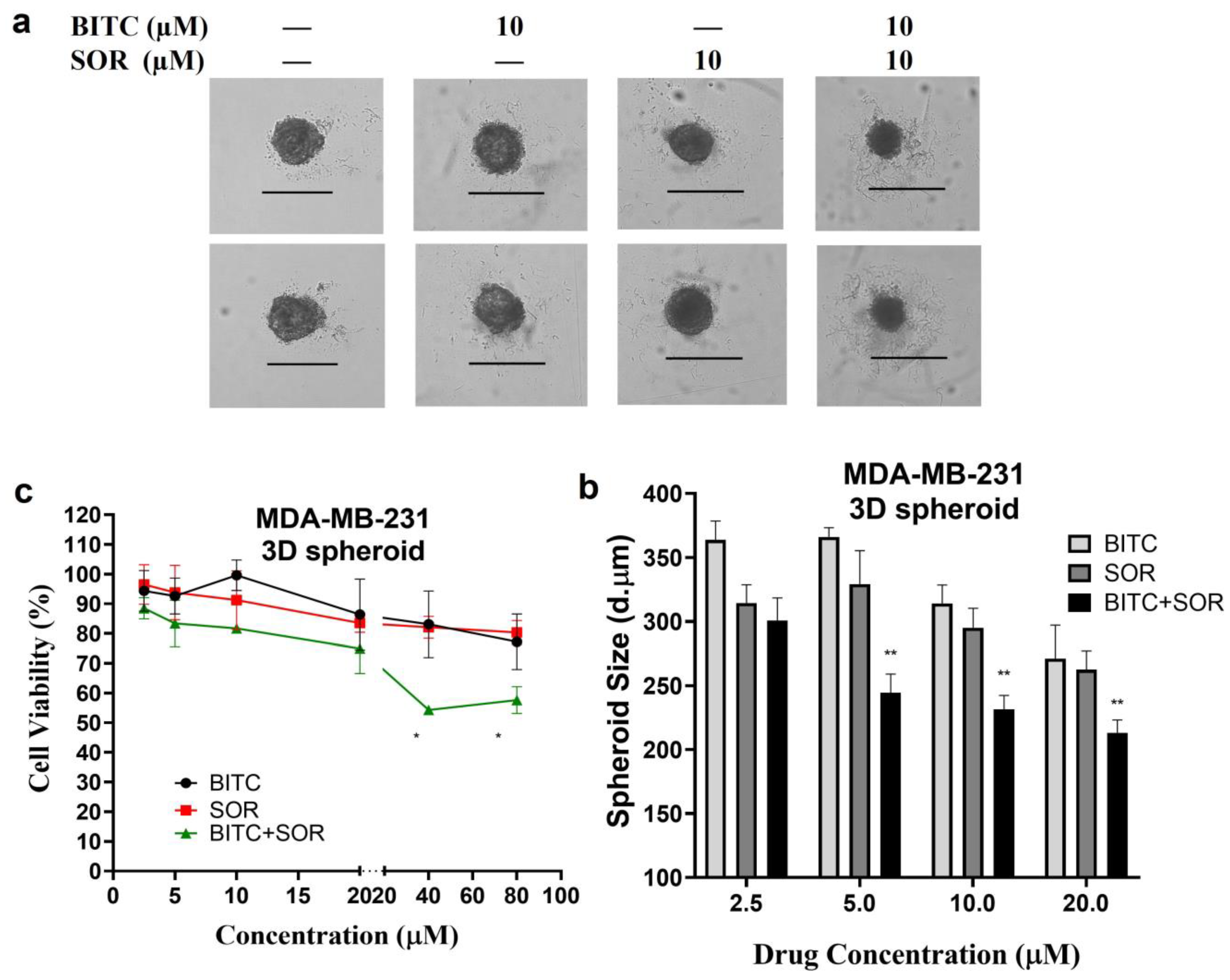
3.6. Inhibition of TNBC 3D Spheroids Growth by BITC and SOR
3.7. Induction of Cell Cycle Arrest via Inhibition of Cyclin B1, Chk1, and Cdc2 in TNBC Cells
3.8. Enhanced Anticancer Efficacy of BITC–SOR-NPs in TNBC Cells and 3D Spheroids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Jerusalem, G.; Collignon, J.; Schroeder, H.; Lousberg, L. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer Targets Ther. 2016, 8, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Zafrakas, M.; Papasozomenou, P.; Emmanouilides, C. Sorafenib in breast cancer treatment: A systematic review and overview of clinical trials. World J. Clin. Oncol. 2016, 7, 331. [Google Scholar] [CrossRef] [PubMed]
- Mangana, J.; Levesque, M.P.; Karpova, M.B.; Dummer, R. Sorafenib in melanoma. Expert. Opin. Pharmacother. 2012, 21, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Kacan, T.; Nayir, E.; Altun, A.; Kilickap, S.; Babacan, N.A.; Ataseven, H.; Kaya, T. Antitumor activity of sorafenib on colorectal cancer. J. Oncol. Sci. 2016, 2, 53–57. [Google Scholar] [CrossRef]
- Kane, R.C.; Farrell, A.T.; Madabushi, R.; Booth, B.; Chattopadhyay, S.; Sridhara, R.; Justice, R.; Pazdur, R. Sorafenib for the Treatment of Unresectable Hepatocellular Carcinoma. Oncologist 2009, 14, 95–100. [Google Scholar] [CrossRef]
- Guevremont, C.; Jeldres, C.; Perrotte, P.; Karakiewicz, P.I. Sorafenib in the Management of Metastatic Renal Cell Carcinoma. Curr. Oncol. 2009, 16, 27–32. [Google Scholar] [CrossRef][Green Version]
- Baselga, J.; Zamagni, C.; Gómez, P.; Bermejo, B.; Nagai, S.E.; Melichar, B.; Chan, A.; Mángel, L.; Bergh, J.; Costa, F.; et al. RESILIENCE: Phase III Randomized, Double-Blind Trial Comparing Sorafenib with Capecitabine Versus Placebo With Capecitabine in Locally Advanced or Metastatic HER2-Negative Breast Cancer. Clin. Breast Cancer 2017, 17, 585–594.e584. [Google Scholar] [CrossRef] [PubMed]
- Bronte, G.; Andreis, D.; Bravaccini, S.; Maltoni, R.; Cecconetto, L.; Schirone, A.; Farolfi, A.; Fedeli, A.; Serra, P.; Donati, C.; et al. Sorafenib for the treatment of breast cancer. Expert Opin. Pharmacother. 2017, 18, 621–630. [Google Scholar] [CrossRef]
- Sun, W.; Powell, M.; O’Dwyer, P.J.; Catalano, P.; Ansari, R.H.; Benson, A.B. Phase II Study of Sorafenib in Combination With Docetaxel and Cisplatin in the Treatment of Metastatic or Advanced Gastric and Gastroesophageal Junction Adenocarcinoma: ECOG 5203. J. Clin. Oncol. 2010, 28, 2947–2951. [Google Scholar] [CrossRef]
- Ohio State University Comprehensive Cancer. Sorafenib Tosylate, Cisplatin, and Docetaxel in Treating Patients with Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck. Available online: https://ClinicalTrials.gov/show/NCT02035527 (accessed on 3 November 2023).
- Gradishar, W.J.; Kaklamani, V.; Sahoo, T.P.; Lokanatha, D.; Raina, V.; Bondarde, S.; Jain, M.; Ro, S.K.; Lokker, N.A.; Schwartzberg, L. A double-blind, randomised, placebo-controlled, phase 2b study evaluating sorafenib in combination with paclitaxel as a first-line therapy in patients with HER2-negative advanced breast cancer. Eur. J. Cancer 2013, 49, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.; Kaklamani, V.; Prasad Sahoo, T.; Lokanatha, D.; Raina, V.; Bondarde, S.; Jain, M. A Double-Blind, Randomized, Placebo-Controlled, Phase 2b Study Evaluating the Efficacy and Safety of Sorafenib (SOR) in Combination with Paclitaxel (PAC) as a First-Line Therapy in Patients (pts) with Locally Recurrent or Metastatic Breast Cancer (BC). Cancer Res. 2009, 69, 44. [Google Scholar] [CrossRef]
- Decker, T.; Overkamp, F.; Rösel, S.; Nusch, A.; Göhler, T.; Indorf, M.; Sahlmann, J.; Trarbach, T. A randomized phase II study of paclitaxel alone versus paclitaxel plus sorafenib in second- and third-line treatment of patients with HER2-negative metastatic breast cancer (PASO). BMC Cancer 2017, 17, 499. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Thompson, D.S.; Bismayer, J.A.; Gian, V.G.; Merritt, W.M.; Whorf, R.C.; Finney, L.H.; Dudley, B.S. Paclitaxel/carboplatin with or without sorafenib in the first-line treatment of patients with stage III/IV epithelial ovarian cancer: A randomized phase II study of the Sarah Cannon Research Institute. Cancer Med. 2015, 4, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.S.; Dudley, B.S.; Bismayer, J.A.; Gian, V.G.; Merritt, W.M.; Whorf, R.C.; Burris, H.A.; Hainsworth, J.D. Paclitaxel/carboplatin with or without sorafenib in the first-line treatment of patients with stage III/IV epithelial ovarian cancer: A randomized phase II study of the Sarah Cannon Research Institute. J. Clin. Oncol. 2013, 31, 5513. [Google Scholar] [CrossRef]
- Richly, H.; Henning, B.; Kupsch, P.; Passarge, K.; Grubert, M.; Hilger, R.; Christensen, O.; Brendel, E.; Schwartz, B.; Ludwig, M. Results of a Phase I trial of sorafenib (BAY 43-9006) in combination with doxorubicin in patients with refractory solid tumors. Ann. Oncol. 2006, 17, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Kupsch, P.; Henning, B.F.; Passarge, K.; Richly, H.; Wiesemann, K.; Hilger, R.A.; Scheulen, M.E.; Christensen, O.; Brendel, E.; Schwartz, B.; et al. Results of a phase I trial of sorafenib (BAY 43-9006) in combination with oxaliplatin in patients with refractory solid tumors, including colorectal cancer. Clin. Color. Cancer 2005, 5, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Sun Yat-sen University. Sorafenib or Lenvatinib Plus HAIC of 130 mg/m² Oxaliplatin, and 5-fu vs Sorafenib or Lenvatinib Plus HAIC of 85 mg/m² Oxaliplatin, and 5-fu for Unresectable Advanced HCC: A Randomised Phase 3 Trial. Available online: https://ClinicalTrials.gov/show/NCT04687163 (accessed on 1 December 2023).
- Siu, L.; Takimoto, C.; Awada, A.; Moore, M.; Piccart, M.; Poulin-Costello, M.; Lathia, C.; Petrenciuc, O. A phase I/II trial of BAY 43–9006 and gemcitabine in advanced solid tumors and in advanced pancreatic cancer. J. Clin. Oncol. 2004, 22, 3059. [Google Scholar] [CrossRef]
- Kindler, H.L.; Wroblewski, K.; Wallace, J.A.; Hall, M.J.; Locker, G.; Nattam, S.; Agamah, E.; Stadler, W.M.; Vokes, E.E. Gemcitabine plus sorafenib in patients with advanced pancreatic cancer: A phase II trial of the University of Chicago Phase II Consortium. Investig. New Drugs 2012, 30, 382–386. [Google Scholar] [CrossRef]
- George, T.J.; Ivey, A.M.; Ali, A.; Lee, J.-H.; Wang, Y.; Daily, K.C.; Ramnaraign, B.H.; Tan, S.A.; Terracina, K.P.; Read, T.E.; et al. Activity of Sorafenib Plus Capecitabine in Previously Treated Metastatic Colorectal Cancer. Oncologist 2021, 26, 362.e724. [Google Scholar] [CrossRef]
- Cardin, D.B.; Goff, L.; Li, C.I.; Shyr, Y.; Winkler, C.; Devore, R.; Schlabach, L.; Holloway, M.; McClanahan, P.; Meyer, K.; et al. Phase II trial of sorafenib and erlotinib in advanced pancreatic cancer. Cancer Med. 2014, 3, 572–579. [Google Scholar] [CrossRef]
- M.D. Anderson Cancer Center. Vemurafenib with Sorafenib Tosylate or Crizotinib in Treating Patients With Advanced Malignancies with BRAF Mutations. Available online: https://ClinicalTrials.gov/show/NCT01531361 (accessed on 13 January 2024).
- HonorHealth Research Institute Phase II Trial of Vemurafenib and Sorafenib in Pancreatic Cancer. Available online: https://ClinicalTrials.gov/show/NCT05068752 (accessed on 1 December 2023).
- BeiGene. Phase 3 Study of Tislelizumab Versus Sorafenib in Participants With Unresectable HCC. Available online: https://ClinicalTrials.gov/show/NCT03412773 (accessed on 1 December 2023).
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef]
- Keating, G.M.; Santoro, A. Sorafenib. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef]
- Conaway, C.; Yang, Y.; Chung, F. Isothiocyanates as cancer chemopreventive agents: Their biological activities and metabolism in rodents and humans. Curr. Drug Metab. 2002, 3, 233–255. [Google Scholar] [CrossRef]
- Hecht, S.S. Inhibition of Carcinogenesis by Isothiocyanates. Drug Metab. Rev. 2000, 32, 395–411. [Google Scholar] [CrossRef]
- Wang, Q.; Bao, Y. Nanodelivery of natural isothiocyanates as a cancer therapeutic. Free Radic. Biol. Med. 2021, 167, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Miyoshi, N.; Osawa, T.; Uchida, K.; Kawakami, M.; Yoshihiro, A.; Ohigashi, H.; Kawai, K. Involvement of the mitochondrial death pathway in chemopreventive benzyl isothiocyanate-induced apoptosis. J. Biol. Chem. 2002, 277, 8492–8499. [Google Scholar] [CrossRef]
- Lui, V.W.; Wentzel, A.L.; Xiao, D.; Lew, K.L.; Singh, S.V.; Grandis, J.R. Requirement of a carbon spacer in benzyl isothiocyanate-mediated cytotoxicity and MAPK activation in head and neck squamous cell carcinoma. Carcinogenesis 2003, 24, 1705–1712. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Singh, S.V. Cell cycle arrest, apoptosis induction and inhibition of nuclear factor kappa B activation in anti-proliferative activity of benzyl isothiocyanate against human pancreatic cancer cells. Carcinogenesis 2004, 25, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Uchida, K.; Osawa, T.; Nakamura, Y. A link between benzyl isothiocyanate-induced cell cycle arrest and apoptosis: Involvement of mitogen-activated protein kinases in the Bcl-2 phosphorylation. Cancer Res. 2004, 64, 2134–2142. [Google Scholar] [CrossRef] [PubMed]
- Visanji, J.M.; Duthie, S.J.; Pirie, L.; Thompson, D.G.; Padfield, P.J. Dietary isothiocyanates inhibit Caco-2 cell proliferation and induce G2/M phase cell cycle arrest, DNA damage, and G2/M checkpoint activation. J. Nutr. 2004, 134, 3121–3126. [Google Scholar] [CrossRef] [PubMed]
- Tseng, E.; Ramsay, E.A.S.; Morris, M.E. Dietary organic isothiocyanates are cytotoxic in human breast cancer MCF-7 and mammary epithelial MCF-12A cell lines. Exp. Biol. Med. 2004, 229, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Jakubíková, J.; Sedlák, J.; Bacon, J.; Goldson, A.; Bao, Y. Effects of MEK1 and PI3K inhibitors on allyl-, benzyl-and phenylethyl-isothiocyanate-induced G2/M arrest and cell death in Caco-2 cells. Int. J. Oncol. 2005, 27, 1449–1458. [Google Scholar] [CrossRef]
- Cheng, N.; Diao, H.; Lin, Z.; Gao, J.; Zhao, Y.; Zhang, W.; Wang, Q.; Lin, J.; Zhang, D.; Jin, Y.; et al. Benzyl Isothiocyanate Induces Apoptosis and Inhibits Tumor Growth in Canine Mammary Carcinoma via Downregulation of the Cyclin B1/Cdk1 Pathway. Front. Vet. Sci. 2020, 7, 580530. [Google Scholar] [CrossRef] [PubMed]
- Di Pasqua, A.J.; Hong, C.; Wu, M.Y.; McCracken, E.; Wang, X.T.; Mi, L.X.; Chung, F.L. Sensitization of Non-small Cell Lung Cancer Cells to Cisplatin by Naturally Occurring Isothiocyanates. Chem. Res. Toxicol. 2010, 23, 1307–1309. [Google Scholar] [CrossRef]
- Wolf, M.A.; Claudio, P.P. Benzyl Isothiocyanate Inhibits HNSCC Cell Migration and Invasion, and Sensitizes HNSCC Cells to Cisplatin. Nutr. Cancer 2014, 66, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Chiang, N.N.; Lu, Y.H.; Huang, Y.S.; Yang, J.S.; Tsai, S.C.; Lu, C.C.; Chen, F.A. Benzyl isothiocyanate (BITC) triggers mitochondria-mediated apoptotic machinery in human cisplatin-resistant oral cancer CAR cells. Biomedicine 2018, 8, 13–22. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, Y.J.; Choi, Y.J.; Lee, J.W.; Lee, S.; Chung, H.W. Enhancement of cisplatin cytotoxicity by benzyl isothiocyanate in HL-60 cells. Food Chem. Toxicol. 2012, 50, 2397–2406. [Google Scholar] [CrossRef]
- Zhang, R.; Loganathan, S.; Humphreys, I.; Srivastava, S.K. Benzyl Isothiocyanate-Induced DNA Damage Causes G2/M Cell Cycle Arrest and Apoptosis in Human Pancreatic Cancer Cells1. J. Nutr. 2006, 136, 2728–2734. [Google Scholar] [CrossRef]
- Na, G.; He, C.; Zhang, S.; Tian, S.; Bao, Y.; Shan, Y. Dietary Isothiocyanates: Novel Insights into the Potential for Cancer Prevention and Therapy. Int. J. Mol. Sci. 2023, 24, 1962. [Google Scholar] [CrossRef]
- Ji, Y.; Kuo, Y.; Morris, M.E. Pharmacokinetics of Dietary Phenethyl Isothiocyanate in Rats. Pharm. Res. 2005, 22, 1658–1666. [Google Scholar] [CrossRef]
- Chuang, W.-T.; Liu, Y.-T.; Huang, C.-S.; Lo, C.-W.; Yao, H.-T.; Chen, H.-W.; Lii, C.-K. Benzyl Isothiocyanate and Phenethyl Isothiocyanate Inhibit Adipogenesis and Hepatosteatosis in Mice with Obesity Induced by a High-Fat Diet. J. Agric. Food Chem. 2019, 67, 7136–7146. [Google Scholar] [CrossRef]
- Jain, L.; Woo, S.; Gardner, E.R.; Dahut, W.L.; Kohn, E.C.; Kummar, S.; Mould, D.R.; Giaccone, G.; Yarchoan, R.; Venitz, J.; et al. Population pharmacokinetic analysis of sorafenib in patients with solid tumours. Br. J. Clin. Pharmacol. 2011, 72, 294–305. [Google Scholar] [CrossRef]
- Boudou-Rouquette, P.; Ropert, S.; Mir, O.; Coriat, R.; Billemont, B.; Tod, M.; Cabanes, L.; Franck, N.; Blanchet, B.; Goldwasser, F. Variability of Sorafenib Toxicity and Exposure over Time: A Pharmacokinetic/Pharmacodynamic Analysis. Oncologist 2012, 17, 1204–1212. [Google Scholar] [CrossRef]
- Yoon, M.S.; Lee, Y.J.; Shin, H.J.; Park, C.-W.; Han, S.-B.; Jung, J.-K.; Kim, J.-S.; Shin, D.H. Recent Advances and Challenges in Controlling the Spatiotemporal Release of Combinatorial Anticancer Drugs from Nanoparticles. Pharmaceutics 2020, 12, 1156. [Google Scholar] [CrossRef]
- Alshaker, H.; Wang, Q.; Böhler, T.; Mills, R.; Winkler, M.; Arafat, T.; Kawano, Y.; Pchejetski, D. Combination of RAD001 (everolimus) and docetaxel reduces prostate and breast cancer cell VEGF production and tumour vascularisation independently of sphingosine-kinase-1. Sci. Rep. 2017, 7, 3493. [Google Scholar] [CrossRef]
- Alshaker, H.; Wang, Q.; Srivats, S.; Chao, Y.; Cooper, C.; Pchejetski, D. New FTY720-docetaxel nanoparticle therapy overcomes FTY720-induced lymphopenia and inhibits metastatic breast tumour growth. Breast Cancer Res. Treat. 2017, 165, 531–543. [Google Scholar] [CrossRef]
- Wu, I.Y.; Bala, S.; Škalko-Basnet, N.; Di Cagno, M.P. Interpreting non-linear drug diffusion data: Utilizing Korsmeyer-Peppas model to study drug release from liposomes. Eur. J. Pharm. Sci. 2019, 138, 105026. [Google Scholar] [CrossRef]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis Analyzer for ImageJ—A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef]
- Juhász, Á.; Ungor, D.; Berta, K.; Seres, L.; Csapó, E. Spreadsheet-based nonlinear analysis of in vitro release properties of a model drug from colloidal carriers. J. Mol. Liq. 2021, 328, 115405. [Google Scholar] [CrossRef]
- Xiao, D.; Johnson, C.S.; Trump, D.L.; Singh, S.V. Proteasome-mediated degradation of cell division cycle 25C and cyclin-dependent kinase 1 in phenethyl isothiocyanate-induced G2-M-phase cell cycle arrest in PC-3 human prostate cancer cells. Mol. Cancer Ther. 2004, 3, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, G.; Nicolas, V.; Matsusaki, M.; Akashi, M.; Couvreur, P.; Mura, S. Multicellular spheroid based on a triple co-culture: A novel 3D model to mimic pancreatic tumor complexity. Acta Biomater. 2018, 78, 296–307. [Google Scholar] [CrossRef]
- Nath, S.; Devi, G.R. Three-dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef]
- Brancato, V.; Gioiella, F.; Imparato, G.; Guarnieri, D.; Urciuolo, F.; Netti, P.A. 3D breast cancer microtissue reveals the role of tumor microenvironment on the transport and efficacy of free-doxorubicin in vitro. Acta Biomater. 2018, 75, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Wu, L.W.; Huang, A.C.; Yu, C.C.; Lien, J.C.; Huang, Y.P.; Yang, J.S.; Yang, J.H.; Hsiao, Y.P.; Wood, W.G.; et al. Benzyl Isothiocyanate (BITC) Induces G2/M Phase Arrest and Apoptosis in Human Melanoma A375.S2 Cells through Reactive Oxygen Species (ROS) and both Mitochondria-Dependent and Death Receptor-Mediated Multiple Signaling Pathways. J. Agric. Food Chem. 2012, 60, 665–675. [Google Scholar] [CrossRef]
- Shang, H.S.; Shih, Y.L.; Lu, T.J.; Lee, C.H.; Hsueh, S.C.; Chou, Y.C.; Lu, H.F.; Liao, N.C.; Chung, J.G. Benzyl Isothiocyanate (BITC) Induces Apoptosis of GBM 8401 Human Brain Glioblastoma Multiforms Cells via Activation of Caspase-8/Bid and the Reactive Oxygen Species-Dependent Mitochondrial Pathway. Environ. Toxicol 2016, 31, 1751–1760. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Mao, W.F.; Shao, M.H.; Gao, P.T.; Ma, J.; Li, H.J.; Li, G.L.; Han, B.H.; Yuan, C.G. The important roles of RET, VEGFR2 and the RAF/MEK/ERK pathway in cancer treatment with sorafenib. Acta Pharmacol. Sin. 2012, 33, 1311–1318. [Google Scholar] [CrossRef]
- Huynh, H.; Ong, R.; Goh, K.Y.; Lee, L.Y.; Puehler, F.; Scholz, A.; Politz, O.; Mumberg, D.; Ziegelbauer, K. Sorafenib/MEK inhibitor combination inhibits tumor growth and the Wnt/-catenin pathway in xenograft models of hepatocellular carcinoma. Int. J. Oncol. 2019, 54, 1123–1133. [Google Scholar] [CrossRef]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotech. 2012, 30, 679–691. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Kastan, M.B. Cell cycle control and cancer. Science 1994, 266, 1821–1828. [Google Scholar] [CrossRef]
- Royston, K.J.; Paul, B.; Nozell, S.; Rajbhandari, R.; Tollefsbol, T.O. Withaferin A and sulforaphane regulate breast cancer cell cycle progression through epigenetic mechanisms. Exp. Cell Res. 2018, 368, 67–74. [Google Scholar] [CrossRef]
- Paul, B.; Li, Y.; Tollefsbol, T.O. The effects of combinatorial genistein and sulforaphane in breast tumor inhibition: Role in epigenetic regulation. Int. J. Mol. Sci. 2018, 19, 1754. [Google Scholar] [CrossRef]
- Ghosh, S.; Javia, A.; Shetty, S.; Bardoliwala, D.; Maiti, K.; Banerjee, S.; Khopade, A.; Misra, A.; Sawant, K.; Bhowmick, S. Triple negative breast cancer and non-small cell lung cancer: Clinical challenges and nano-formulation approaches. J. Control Release 2021, 337, 27–58. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L.J. Three-Dimensional Cell Culture Systems and Their Applications in Drug Discovery and Cell-Based Biosensors. Assay. Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Cheng, N.; Wang, W.; Bao, Y. Synergistic Action of Benzyl Isothiocyanate and Sorafenib in a Nanoparticle Delivery System for Enhanced Triple-Negative Breast Cancer Treatment. Cancers 2024, 16, 1695. https://doi.org/10.3390/cancers16091695
Wang Q, Cheng N, Wang W, Bao Y. Synergistic Action of Benzyl Isothiocyanate and Sorafenib in a Nanoparticle Delivery System for Enhanced Triple-Negative Breast Cancer Treatment. Cancers. 2024; 16(9):1695. https://doi.org/10.3390/cancers16091695
Chicago/Turabian StyleWang, Qi, Nan Cheng, Wei Wang, and Yongping Bao. 2024. "Synergistic Action of Benzyl Isothiocyanate and Sorafenib in a Nanoparticle Delivery System for Enhanced Triple-Negative Breast Cancer Treatment" Cancers 16, no. 9: 1695. https://doi.org/10.3390/cancers16091695
APA StyleWang, Q., Cheng, N., Wang, W., & Bao, Y. (2024). Synergistic Action of Benzyl Isothiocyanate and Sorafenib in a Nanoparticle Delivery System for Enhanced Triple-Negative Breast Cancer Treatment. Cancers, 16(9), 1695. https://doi.org/10.3390/cancers16091695