
Inhibition of TNBC Cell Growth by Paroxetine: Induction of Apoptosis and Blockage of Autophagy Flux

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Lines and Cell Cultures
2.3. Cell Viability Assay
2.4. Colony Formation Assay
2.5. Apoptosis Assays by Flow Cytometry
2.6. Analysis of Reactive Oxygen Species (ROS)
2.7. Analysis of Mitochondrial Membrane Potential ()
2.8. Analysis of Calcium Mobilization
2.9. Drug Synergy Studies and Data Analysis
2.10. Western Blot Analysis
2.11. RNA Sequencing (RNA-Seq)
2.12. Transient Transfection and Immunofluorescence Analysis
2.13. In Vivo Murine Experiments
2.14. Statistical Analysis
3. Results
3.1. Inhibition of TNBC Cell Line Growth In Vitro by PX
3.2. Induction of Apoptosis in TNBC Cell Lines by PX In Vitro
3.3. Induction of Apoptosis in TNBC Cell Lines by PX via Activation of the Mitochondria-Mediated Intrinsic Pathway
3.4. Induction of Cytoprotective Autophagy and Autophagic Flux Inhibition by PX in TNBC Cell Lines
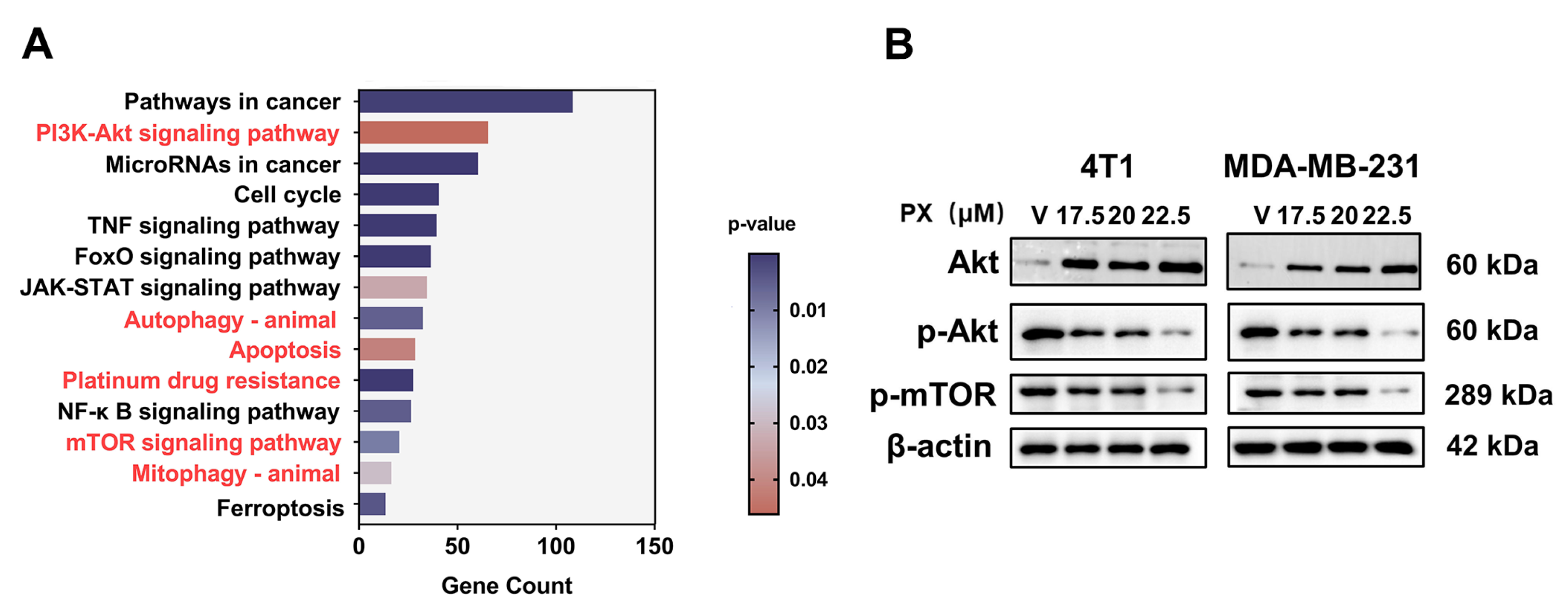
3.5. Inhibition of TNBC by PX via the PI3K-AKT-mTOR Pathway
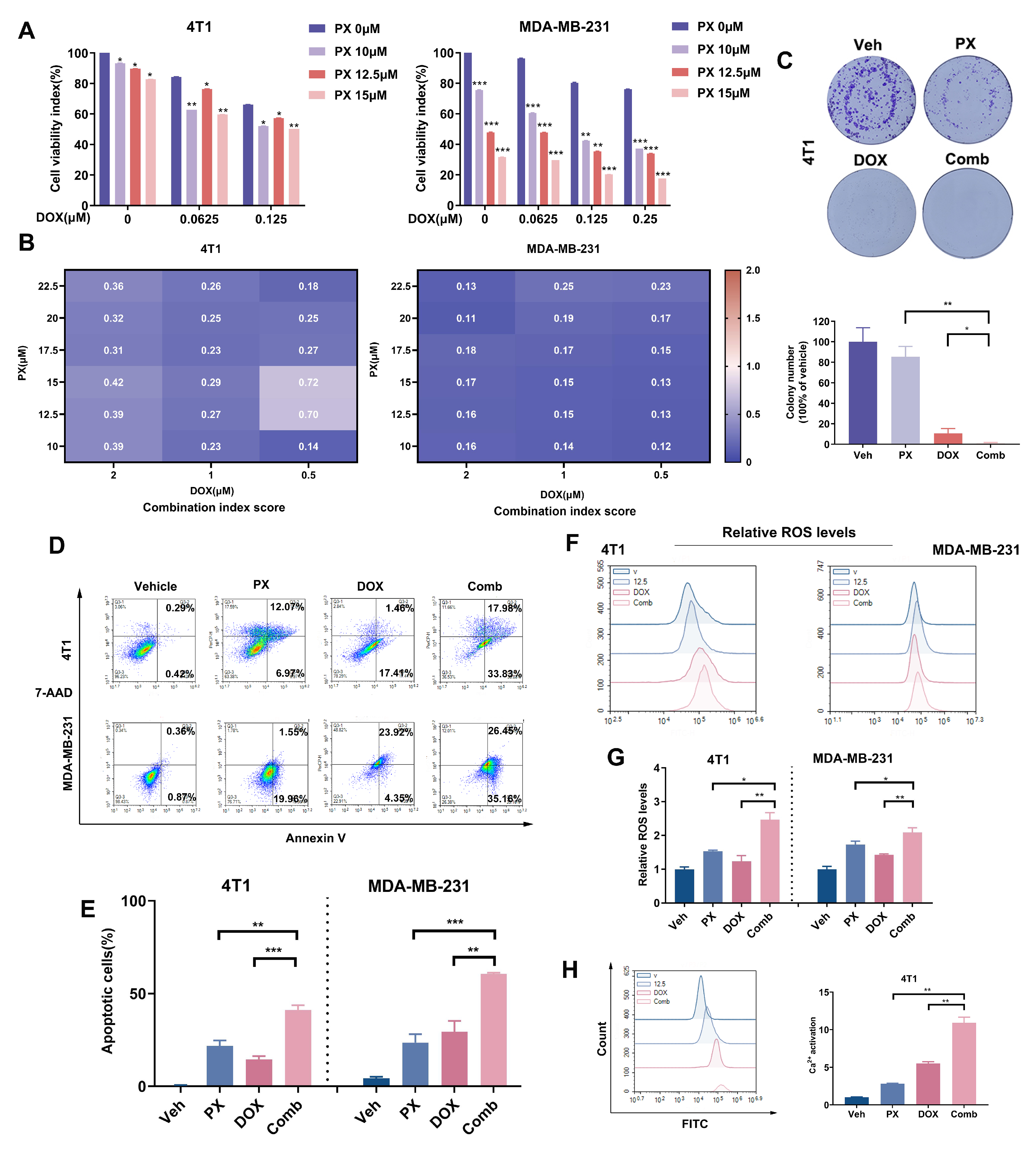
3.6. Potentiation of TNBC Cancer Cell Sensitivity to DOX Treatment by PX In Vitro
3.7. Potentiation of TNBC Tumor Sensitivity to Chemotherapy In Vivo by PX
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Atg | Autophagy-associated gene |
| Bcl-2 | B-cell lymphoma 2 |
| CQ | Chloroquine |
| CI | Combination index |
| DEGs | Differentially expressed genes |
| DMSO | Dimethyl sulfoxide |
| DOX | Doxorubicin |
| ER | Endoplasmic reticulum |
| GSEA | Gene set enrichment analysis |
| HRP | Horseradish peroxidase |
| IAPs | Inhibitor of apoptosis proteins |
| IC50 | Half-maximal inhibitory concentration |
| i.p. | Intraperitoneal injection |
| i.v. | Intravenous injection |
| MTT | 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide |
| MOMP | Membrane permeabilization |
| PX | Paroxetine |
| PVDF | Polyvinylidene difluoride |
| Rh123 | Rhodamine123 |
| RNA-Seq | RNA sequencing |
| ROS | Reactive oxygen species |
| S.D. | Standard deviation |
| S.E.M. | Standard error of the mean |
| TNBC | Triple-negative breast cancer |
| 3-MA | 3-Methyladenine |
| Mitochondrial membrane potential. |
References
- Thorat, M.A.; Balasubramanian, R. Breast cancer prevention in high-risk women. Best Pract. Res. Clin. Obstet. Gynaecol. 2020, 65, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Vagia, E.; Mahalingam, D.; Cristofanilli, M. The landscape of targeted therapies in TNBC. Cancers 2020, 12, 916. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Deng, J.; Zhang, L.; Yuan, J.; Yang, H.; Li, Q. Tumor microenvironment characterization in triple-negative breast cancer identifies prognostic gene signature. Aging 2021, 13, 5485. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, Q.; Zhao, N. Identification of a prognostic chemoresistance-related gene signature associated with immune microenvironment in breast cancer. Bioengineered 2021, 12, 8419–8434. [Google Scholar] [CrossRef] [PubMed]
- Hofman, J.; Skarka, A.; Havrankova, J.; Wsol, V. Pharmacokinetic interactions of breast cancer chemotherapeutics with human doxorubicin reductases. Biochem. Pharmacol. 2015, 96, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Ramos-Medina, R.; Bernat, R.; García-Saenz, J.A.; del Monte-Millan, M.; Alvarez, E.; Cebollero, M.; Moreno, F.; Gonzalez-Haba, E.; Bueno, O.; et al. Activity of docetaxel, carboplatin, and doxorubicin in patient-derived triple-negative breast cancer xenografts. Sci. Rep. 2021, 11, 7064. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Paramanantham, A.; Jung, E.J.; Kim, H.J.; Jeong, B.K.; Jung, J.M.; Kim, G.S.; Hong, S.C.; Lee, W.S. Doxorubicin-resistant TNBC cells exhibit rapid growth with cancer stem cell-like properties and EMT phenotype, which can be transferred to parental cells through autocrine signaling. Int. J. Mol. Sci. 2021, 22, 12438. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Massie, M.J. Prevalence of depression in patients with cancer. JNCI Monogr. 2004, 2004, 57–71. [Google Scholar] [CrossRef]
- Purkayastha, D.; Venkateswaran, C.; Nayar, K.; Unnikrishnan, U. Prevalence of depression in breast cancer patients and its association with their quality of life: A cross-sectional observational study. Indian J. Palliat. Care 2017, 23, 268. [Google Scholar]
- National Cancer Institute. Side Effects of Cancer Treatment. 2023. Available online: https://www.cancer.org/cancer/managing-cancer/side-effects/emotional-mood-changes.html (accessed on 1 February 2020).
- Stearns, V.; Johnson, M.D.; Rae, J.M.; Morocho, A.; Novielli, A.; Bhargava, P.; Hayes, D.F.; Desta, Z.; Flockhart, D.A. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J. Natl. Cancer Inst. 2003, 95, 1758–1764. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Desta, Z.; Stearns, V.; Ward, B.; Ho, H.; Lee, K.H.; Skaar, T.; Storniolo, A.M.; Li, L.; Araba, A.; et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J. Natl. Cancer Inst. 2005, 97, 30–39. [Google Scholar] [CrossRef]
- Haque, R.; Shi, J.; Schottinger, J.E.; Ahmed, S.A.; Cheetham, T.C.; Chung, J.; Avila, C.; Kleinman, K.; Habel, L.A.; Fletcher, S.W.; et al. Tamoxifen and antidepressant drug interaction among a cohort of 16887 breast cancer survivors. J. Natl. Cancer Inst. 2016, 108, djv337. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.W.; Kim, E.J.; Nyiramana, M.M.; Shin, E.J.; Jin, H.; Ryu, J.H.; Kang, K.R.; Lee, G.W.; Kim, H.J.; Han, J.; et al. Paroxetine induces apoptosis of human breast cancer MCF-7 cells through Ca2+-and p38 MAP kinase-dependent ROS generation. Cancers 2019, 11, 64. [Google Scholar] [CrossRef]
- Jang, W.J.; Jung, S.K.; Vo, T.T.L.; Jeong, C.H. Anticancer activity of paroxetine in human colon cancer cells: Involvement of MET and ERBB3. J. Cell. Mol. Med. 2019, 23, 1106–1115. [Google Scholar] [CrossRef]
- Serafeim, A.; Holder, M.J.; Grafton, G.; Chamba, A.; Drayson, M.T.; Luong, Q.T.; Bunce, C.M.; Gregory, C.D.; Barnes, N.M.; Gordon, J. Selective serotonin reuptake inhibitors directly signal for apoptosis in biopsy-like Burkitt lymphoma cells. Blood, J. Am. Soc. Hematol. 2003, 101, 3212–3219. [Google Scholar] [CrossRef]
- Levkovitz, Y.; Gil-Ad, I.; Zeldich, E.; Dayag, M.; Weizman, A. Differential induction of apoptosis by antidepressants in glioma and neuroblastoma cell lines: Evidence for pc-Jun, cytochrome c, and caspase-3 involvement. J. Mol. Neurosci. 2005, 27, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Taler, M.; Gil-Ad, I.; Lomnitski, L.; Korov, I.; Baharav, E.; Bar, M.; Zolokov, A.; Weizman, A. Immunomodulatory effect of selective serotonin reuptake inhibitors (SSRIs) on human T lymphocyte function and gene expression. Eur. Neuropsychopharmacol. 2007, 17, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, J.; Yamada, T.; Egashira, N.; Ueda, M.; Zukeyama, N.; Ushio, S.; Masuda, S. Comparison of the anti-tumor effects of selective serotonin reuptake inhibitors as well as serotonin and norepinephrine reuptake inhibitors in human hepatocellular carcinoma cells. Biol. Pharm. Bull. 2015, 38, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.H.; Yuan, T.M.; Huang, C.J.; Hsu, D.T.; Chen, S.W.; Hsiao, N.W.; Lin, S.C.; Wu, S.W.; Lin, Y.M.J.; Chuang, S.M. DNA repair proteins as the targets for paroxetine to induce cytotoxicity in gastric cancer cell AGS. Am. J. Cancer Res. 2022, 12, 1465. [Google Scholar]
- Wang, T.; Zhang, H.; Han, Y.; Zheng, Q.; Liu, H.; Han, M.; Li, Z. Reversing T Cell Dysfunction to Boost Glioblastoma Immunotherapy by Paroxetine-Mediated GRK2 Inhibition and Blockade of Multiple Checkpoints through Biomimetic Nanoparticles. Adv. Sci. 2023, 10, 2204961. [Google Scholar] [CrossRef]
- Crudden, C.; Shibano, T.; Song, D.; Dragomir, M.P.; Cismas, S.; Serly, J.; Nedelcu, D.; Fuentes-Mattei, E.; Tica, A.; Calin, G.A.; et al. Inhibition of G Protein–Coupled Receptor Kinase 2 Promotes Unbiased Downregulation of IGF1 Receptor and Restrains Malignant Cell Growth. Cancer Res. 2021, 81, 501–514. [Google Scholar] [CrossRef]
- Xia, Y.; Xu, F.; Xiong, M.; Yang, H.; Lin, W.; Xie, Y.; Xi, H.; Xue, Q.; Ye, T.; Yu, L. Repurposing of antipsychotic trifluoperazine for treating brain metastasis, lung metastasis and bone metastasis of melanoma by disrupting autophagy flux. Pharmacol. Res. 2021, 163, 105295. [Google Scholar] [CrossRef] [PubMed]
- Lyublinskaya, O.; Ivanova, J.S.; Pugovkina, N.; Kozhukharova, I.; Kovaleva, Z.; Shatrova, A.; Aksenov, N.; Zenin, V.; Kaulin, Y.A.; Gamaley, I.; et al. Redox environment in stem and differentiated cells: A quantitative approach. Redox Biol. 2017, 12, 758–769. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, J.; Shi, W.; Liu, Y. Anticancer effects of 3,3′-diindolylmethane are associated with G1 arrest and mitochondria-dependent apoptosis in human nasopharyngeal carcinoma cells. Oncol. Lett. 2013, 5, 655–662. [Google Scholar] [CrossRef]
- Verma, N.; Vinik, Y.; Saroha, A.; Nair, N.U.; Ruppin, E.; Mills, G.; Karn, T.; Dubey, V.; Khera, L.; Raj, H.; et al. Synthetic lethal combination targeting BET uncovered intrinsic susceptibility of TNBC to ferroptosis. Sci. Adv. 2020, 6, eaba8968. [Google Scholar] [CrossRef]
- Xia, J.; Inagaki, Y.; Gao, J.; Qi, F.; Song, P.; Han, G.; Sawakami, T.; Gao, B.; Luo, C.; Kokudo, N.; et al. Combination of cinobufacini and doxorubicin increases apoptosis of hepatocellular carcinoma cells through the Fas-and mitochondria-mediated pathways. Am. J. Chin. Med. 2017, 45, 1537–1556. [Google Scholar] [CrossRef]
- Wu, M.; Huang, Q.; Liao, M.; Wu, X.; Xi, H.; Ma, H.; Li, S.; Zhang, Y.; Xia, Y. OSW-1 induces apoptosis and cyto-protective autophagy, and synergizes with chemotherapy on triple negative breast cancer metastasis. Cell. Oncol. 2022, 45, 1255–1275. [Google Scholar] [CrossRef]
- Wang, K.; Gong, Q.; Zhan, Y.; Chen, B.; Yin, T.; Lu, Y.; Zhang, Y.; Wang, H.; Ke, J.; Du, B.; et al. Blockage of autophagic flux and induction of mitochondria fragmentation by paroxetine hydrochloride in lung cancer cells promotes apoptosis via the ROS-MAPK pathway. Front. Cell Dev. Biol. 2020, 7, 397. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 2002, 9, 459–470. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; Seol, D.W. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef]
- Wang, H.; Xie, M.; Rizzi, G.; Li, X.; Tan, K.; Fussenegger, M. Identification of Sclareol As a Natural Neuroprotective Cav1. 3-Antagonist Using Synthetic Parkinson-Mimetic Gene Circuits and Computer-Aided Drug Discovery. Adv. Sci. 2022, 9, 2102855. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Ferrari, D.; Chami, M.; Szabadkai, G.; Magalhaes, P.J.; Virgilio, F.D.; Pozzan, T. Calcium and apoptosis: Facts and hypotheses. Oncogene 2003, 22, 8619–8627. [Google Scholar] [CrossRef]
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef]
- Ola, M.S.; Nawaz, M.; Ahsan, H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol. Cell. Biochem. 2011, 351, 41–58. [Google Scholar] [CrossRef]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Johnson, C.; Jarvis, W. Caspase-9 regulation: An update. Apoptosis 2004, 9, 423–427. [Google Scholar] [CrossRef]
- Zhang, Q.; Hu, X.m.; Zhao, W.j.; Ban, X.x.; Li, Y.; Huang, Y.x.; Wan, H.; He, Y.; Liao, L.s.; Shang, L.; et al. Targeting necroptosis: A novel therapeutic option for retinal degenerative diseases. Int. J. Biol. Sci. 2023, 19, 658. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Mei, C.; Yang, L.; Zheng, J.; Lu, H.; Xia, Y.; Hsu, S.; Liang, H.; Hong, L. Vitamin K2 promotes PI3K/AKT/HIF-1α-mediated glycolysis that leads to AMPK-dependent autophagic cell death in bladder cancer cells. Sci. Rep. 2020, 10, 7714. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin. Cancer Biol. 2022, 85, 69–94. [Google Scholar] [CrossRef]
- Schmitt, M.; Ceteci, F.; Gupta, J.; Pesic, M.; Böttger, T.W.; Nicolas, A.M.; Kennel, K.B.; Engel, E.; Schewe, M.; Callak Kirisözü, A.; et al. Colon tumour cell death causes mTOR dependence by paracrine P2X4 stimulation. Nature 2022, 612, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Xiaojun, X.; Peilong, C. Magnoflorine improves sensitivity to doxorubicin (DOX) of breast cancer cells via inducing apoptosis and autophagy through AKT/mTOR and p38 signaling pathways. Biomed. Pharmacother. 2020, 121, 109139. [Google Scholar] [CrossRef] [PubMed]
- Musa, F.; Alard, A.; David-West, G.; Curtin, J.P.; Blank, S.V.; Schneider, R.J. Dual mTORC1/2 inhibition as a novel strategy for the resensitization and treatment of platinum-resistant ovarian cancer. Mol. Cancer Ther. 2016, 15, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T.; Tsai, Y.H.; Tan, P.; Hsu, F.T.; Wang, H.M.D.; Lin, W.C.; Lin, F.H.; Wu, C.T. Fluoxetine inhibits STAT3-mediated survival and invasion of osteosarcoma cells. Anticancer. Res. 2023, 43, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, F.; Jay, T.M. Antidepressants promote and prevent cancers. Cancer Investig. 2020, 38, 572–598. [Google Scholar] [CrossRef] [PubMed]
- Dang, S.; Kumari, P. Anti-cancer potential of some commonly used drugs. Curr. Pharm. Des. 2021, 27, 4530–4538. [Google Scholar]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L. The concept of intrinsic versus extrinsic apoptosis. Biochem. J. 2022, 479, 357–384. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Breckenridge, D.G.; Germain, M.; Mathai, J.P.; Nguyen, M.; Shore, G.C. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 2003, 22, 8608–8618. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef] [PubMed]
- Frérart, F.; Sonveau, P.; Rath, G.; Smoos, A.; Meqor, A.; Charlier, N.; Jordan, B.F.; Saliez, J.; Noël, A.; Dessy, C.; et al. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar]
- Khan, M.A.; Jain, V.K.; Rizwanullah, M.; Ahmad, J.; Jain, K. PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: A review on drug discovery and future challenges. Drug Discov. Today 2019, 24, 2181–2191. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR pathway and its role in cancer therapeutics: Are we making headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef]
- Costa, R.L.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Bentires-Alj, M. Tackling resistance to PI3K inhibition by targeting the epigenome. Cancer Cell 2017, 31, 616–618. [Google Scholar] [CrossRef]
- Perry, J.M.; Tao, F.; Roy, A.; Lin, T.; He, X.C.; Chen, S.; Lu, X.; Nemechek, J.; Ruan, L.; Yu, X.; et al. Overcoming Wnt–β-catenin dependent anticancer therapy resistance in leukaemia stem cells. Nat. Cell Biol. 2020, 22, 689–700. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Kosić, M.; Nešić, Z.; Glumac, S.; Vasić, M.; Pajović, V.; Savić, B.; Japundžić-Žigon, N. Paroxetine mitigates cardiac remodelling by doxorubicin and increases survival. Biomed. Pharmacother. 2022, 145, 112411. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27. [Google Scholar] [CrossRef]
- Pollack, M.H.; Zaninelli, R.; Goddard, A.; McCafferty, J.P.; Bellew, K.M.; Burnham, D.B.; Iyengar, M.K. Paroxetine in the treatment of generalized anxiety disorder: Results of a placebo-controlled, flexible-dosage trial. J. Clin. Psychiatry 2001, 62, 350–357. [Google Scholar] [CrossRef]
- Bourin, M.; Chue, P.; Guillon, Y. Paroxetine: A review. CNS Drug Rev. 2001, 7, 25–47. [Google Scholar] [CrossRef]
- Cassano, G.B.; Puca, F.; Scapicchio, P.L.; Trabucchi, M.; Italian Study Group on Depression in Elderly Patients. Paroxetine and fluoxetine effects on mood and cognitive functions in depressed nondemented elderly patients. J. Clin. Psychiatry 2002, 63, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, R.; Gascon, A.; Calvo, M.; Caramella, C.; Conte, U.; Dominguez-Gil, A.; Pedraz, J. Influence of route of administration and dosage form in the pharmacokinetics and bioavailability of salbutamol. Eur. J. Drug Metab. Pharmacokinet. 1997, 22, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.L. Strategies to Modify the Drug Release from Pharmaceutical Systems; Woodhead Publishing: Sawston, UK, 2015. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Q.; Wu, M.; Pu, Y.; Zhou, J.; Zhang, Y.; Li, R.; Xia, Y.; Zhang, Y.; Ma, Y. Inhibition of TNBC Cell Growth by Paroxetine: Induction of Apoptosis and Blockage of Autophagy Flux. Cancers 2024, 16, 885. https://doi.org/10.3390/cancers16050885
Huang Q, Wu M, Pu Y, Zhou J, Zhang Y, Li R, Xia Y, Zhang Y, Ma Y. Inhibition of TNBC Cell Growth by Paroxetine: Induction of Apoptosis and Blockage of Autophagy Flux. Cancers. 2024; 16(5):885. https://doi.org/10.3390/cancers16050885
Chicago/Turabian StyleHuang, Qianrui, Mengling Wu, Yamin Pu, Junyou Zhou, Yiqian Zhang, Ru Li, Yong Xia, Yiwen Zhang, and Yimei Ma. 2024. "Inhibition of TNBC Cell Growth by Paroxetine: Induction of Apoptosis and Blockage of Autophagy Flux" Cancers 16, no. 5: 885. https://doi.org/10.3390/cancers16050885
APA StyleHuang, Q., Wu, M., Pu, Y., Zhou, J., Zhang, Y., Li, R., Xia, Y., Zhang, Y., & Ma, Y. (2024). Inhibition of TNBC Cell Growth by Paroxetine: Induction of Apoptosis and Blockage of Autophagy Flux. Cancers, 16(5), 885. https://doi.org/10.3390/cancers16050885