

The Engineered Drug 3′UTRMYC1-18 Degrades the c-MYC-STAT5A/B-PD-L1 Complex In Vivo to Inhibit Metastatic Triple-Negative Breast Cancer
 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines: Cell Cultures of MDA MB231, BT474, BT474 Clone 5, DAOY, 22Rv1, D283med, MCF7, T47D, U20S, RWPE1
2.2. Mice Animal Study according to ARRIVE
2.3. Extraction of RNA from Multiple Cancer Cell Lines
2.4. Amplification of the c-MYC 3′UTR by qPCR Primers
2.5. Sanger Sequencing of 3′UTR of c-MYC cDNA Amplicon
2.6. Design of Destabilized 3′UTR of c-MYC
2.7. Synthesis of Destabilized ARE 3′UTR of c-MYC as Gblock
2.8. Vector
2.9. Design of Gibson Assembly Primers
c-MYC and Vector Gibson Assembly Primers’ Design and Assembly
2.10. Transformation Recombination of Deficient E. coli (NEB Cat no C3019H)
2.11. Colony Picking and Miniprep
2.12. Colony PCR with c-MYC Gibson Primers
2.13. Gel Extraction
2.14. Sanger Sequencing of the Cloned Destabilized 3′UTR of c-MYC Amplicon
2.15. Sequence Alignment of Wildtype cDNA, RNA versus the Destabilized 3′UTR c-MYC cDNA and RNA Sequences
2.16. Transfection/Electroporation of Destabilized 3′UTR of c-MYC into MDA MB231 Cancer Cells and RWPE1 Normal Epithelial Cells
2.17. Cell Microscopy
2.18. Cellular Immunofluorescence
2.19. Antibodies and Reagents
2.20. Western Blot
2.21. Cell Viability
2.22. Phosphorylation Kinase Array
2.23. RNA Seq
2.24. Quantitative Reverse Transcript PCR
2.25. Generation and Structural Analysis of DNA Plasmid–IO-Nanocage Complexes
2.26. DHCA-Coated IO-Nanocage Synthesis
2.27. Complexation of DNA Plasmids with IO-Nanocages
2.28. Structural Analysis of DNA Plasmid-Loaded IO-Nanocages by TEM
2.29. Migration Assay
2.30. Tissue Immunofluorescence
2.31. PD-L1 Immunohistochemistry
2.32. Prussian Blue Staining and Detection of IO-Nanocage in Tissues
2.33. 4SU mRNA Labeling Pulse Chase Experiments
2.34. Targeted Sequencing to Detect Genomic Sites of Construct Integration
2.35. Quantification and Statistical Analysis
Cell Viability Measurement
2.36. Immunofluorescence
2.37. Transcript Quantification
2.38. Tumor and Animal Weight Measurement
2.39. Mouse Survival Quantification
2.40. IHC Image Capturing and Measurement
2.41. Phosphorylation Kinase Assay Quantification
2.42. Migration Assay Measurement
2.43. Prussian Blue Stain Quantification
2.44. Number of Experimental Replicates and Statistical Analysis
2.45. ImageJ
2.46. GraphPad Prism
3. Results
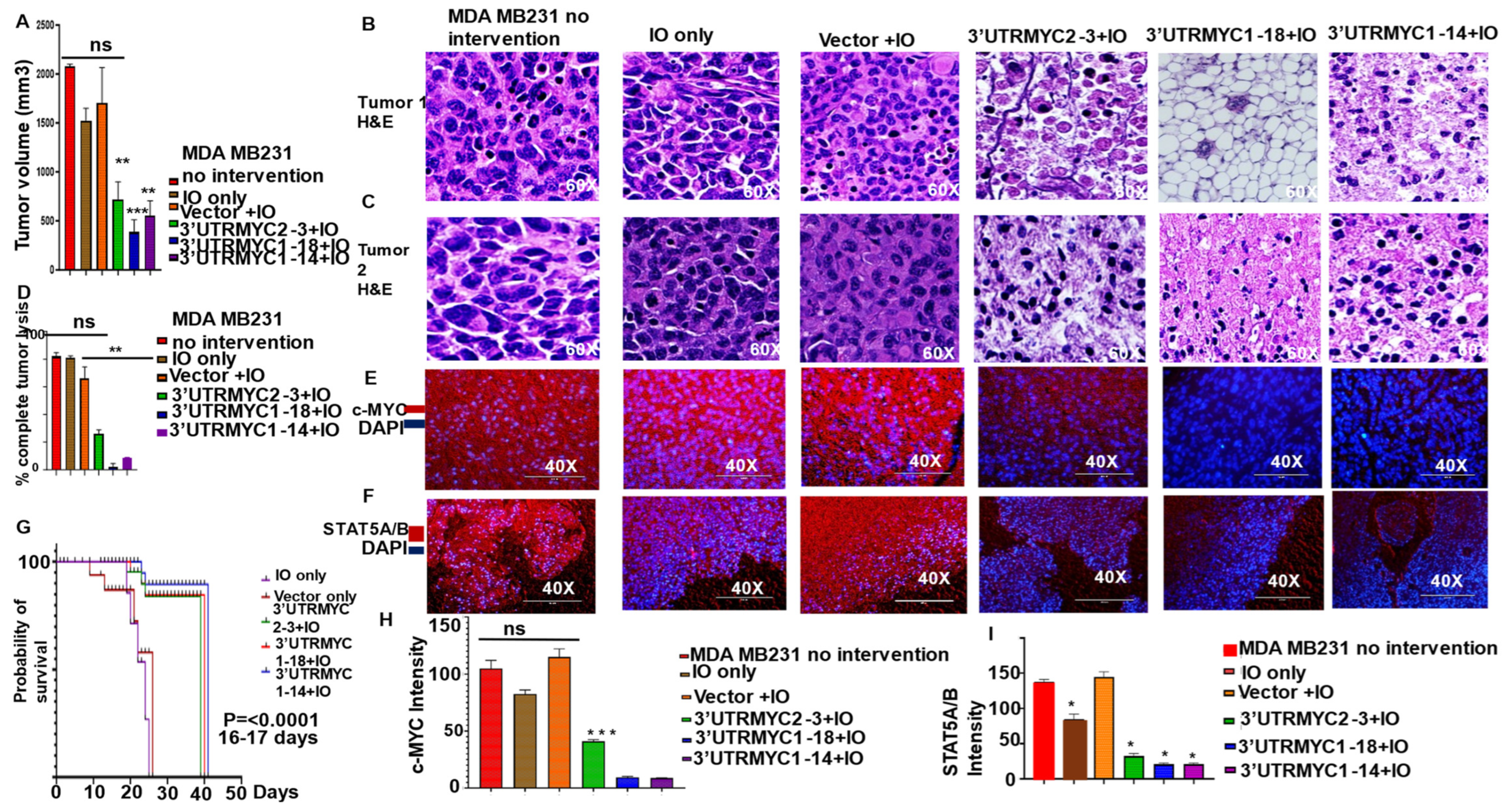
3.1. Engineered Destabilized 3′UTR of MYC Degrades c-MYC-STAT5A/5B-PD-L1 Complex to Inhibit Primary and Metastatic Tumors in c-MYC-Driven TNBC In Vivo with Significant Survival Outcome
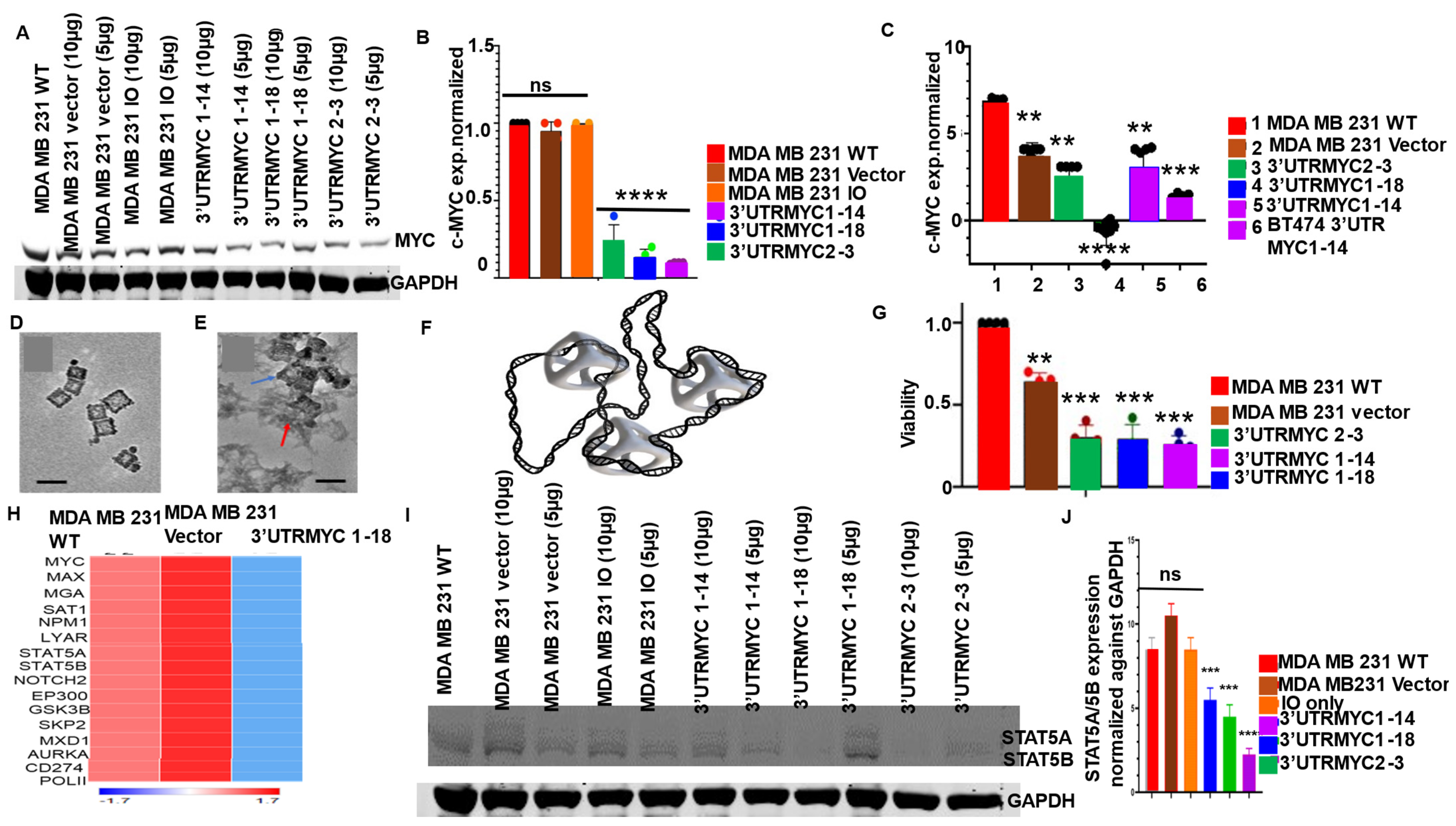
3.2. Engineered Destabilized 3′UTR of c-MYC Degrades c-MYC Transcript and Proteins in c-MYC-Driven TNBC and Medulloblastoma and Prostate Cancer with Specificity, and It Is Safe for Normal Healthy Epithelial Cells
3.3. Nonresponsive Metastatic TNBCs Are Driven by c-MYC-Independent STAT5A/5B and PD-L1 Expression
3.4. IO-Nanocages Delivered Destabilized c-MYC Constructs to Tumors, Leading to Effective Targeting of c-MYC in the Responsive TNBC
3.5. IO-Nanocage Package Delivered Destabilized c-MYC Constructs, Effectively Targeted c-MYC-STAT5A/5B-PD-L1 in the Lungs, and Inhibited Distant Organ Lung and Liver Metastasis
3.6. The Engineered Destabilized c-MYC Degrades the Endogenous MYC by Overwriting Its mRNA Message through EEF2 Upregulation and Increased XRN1 and CNOT1 Expression
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vennstrom, B.; Sheiness, D. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J. Virol. 1982, 42, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Dhanaskeran, R.; Deutzmann, A. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 1, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.R.; Soucek, L. The long journey to bring a Myc inhibitor to the clinic. J. Cell Biol. 2021, 220, e202103090. [Google Scholar] [CrossRef]
- Horiuchi, D.; Kusdra, L. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med. 2012, 4, 679–696. [Google Scholar]
- Zimmerli, D.; Brambillasca, C.S. MYC promotes immune-suppression in triple-negative breast cancer via inhibition of interferon signaling. Nat. Commun. 2022, 13, 6579. [Google Scholar]
- Demma, M.J.; Mapelli, C. Omomyc Reveals New Mechanisms to Inhibit the MYC Oncogene. Mol. Cell. Biol. 2019, 39, e00248-19. [Google Scholar] [PubMed]
- Phase 1/2 Study to Evaluate Safety, PK and Efficacy of the MYC-Inhibitor OMO-103 in Solid Tumors (MYCure). Available online: https://clinicaltrials.gov/ct2/show/NCT04808362 (accessed on 1 January 2021).
- Awah, C.U.; Dong, F. Engineered destabilized AU rich elements on the 3UTR of HER2 degrades HER2, inhibits proliferation, and induces apoptosis in HER2 positive trastuzumab resistant breast cancer cells. Cancer Res. 2022, 82, 1788. [Google Scholar] [CrossRef]
- Awah, C.U.; Dong, F. Engineered destabilized ARE on the 3UTR of MYC degrades oncogenic c-MYC transcript and protein and induces apoptosis in multiple cancer type. Cancer Res. 2022, 82 (Suppl. S12), 1809. [Google Scholar] [CrossRef]
- Geisberg, J.V.; Moqtaderi, Z. Global analysis of mRNA isoform half-lives reveals stabilizing and destabilizing elements in yeast. Cell 2014, 13, 812–824. [Google Scholar]
- Awah, C.U.; Glemaud, Y. Destabilized 3′UTR ARE therapeutically degrades ERBB2 in drug-resistant ERBB2+ cancer models. Front. Genet. 2023, 14, 1184600. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.L.; Jensen, T.H. Nonsense mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Schonenberg, D.R.; Maquat, L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012, 13, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Rampersaud, S.; Fang, J. The Effect of Cage Shape on Nanoparticle-Based Drug Carriers: Anticancer Drug Release and Efficacy via Receptor Blockade Using Dextran-Coated Iron Oxide Nanocages. Nano Lett. 2016, 16, 7357–7363. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, T. Facile surface functionalization of hydrophobic magnetic nanoparticles. J. Am. Chem. Soc. 2014, 136, 12552–12555. [Google Scholar] [CrossRef]
- Raghubir, M.; Rahman, C.N. Osteosarcoma growth suppression by riluzole delivery via iron oxide nanocage in nude mice. Oncol. Rep. 2020, 43, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Selivanovitch, E.; LaFrance, B. Molecular exclusion limits for diffusion across a porous capsid. Nat. Commun. 2021, 12, 2903. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.H.; Yu, T. Galvanic Replacement Reactions in Metal Oxide Nanocrystals. Science 2013, 340, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by Glycogen synthase kinase -3 controls c-MYC proteolysis and subnuclear localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef]
- Alarcon-Vargas, D.; Ronia, Z. c-Jun-NH2 kinase (JNK) contributes to the regulation of c-Myc protein stability. J. Biol. Chem. 2004, 279, 5008–5016. [Google Scholar] [CrossRef]
- Chiariello, M.; Marinissen, M.J. Regulation of c-myc expression by PDGF through Rho GTPases. Nat. Cell Biol. 2001, 3, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Nesta, A.V.; Tafur, D. Hotspots of human mutation. Trends Genet. 2021, 37, 717–729. [Google Scholar] [CrossRef]
- Liang, Q.M.; Yu, F.Q. c-Myc regulates PD-L1 expression in esophageal squamous cell carcinoma. Am. J. Transl. Res. 2020, 2, 379–388. [Google Scholar]
- Casey, S.C.; Baylot, V. The MYC oncogene is a global regulator of immune response. Blood 2018, 13, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Prinz, S.; Unser, S. Signal transducer and activator of transcription STAT5 is recruited to c-Myc super-enhancer. BMC Mol. Biol. 2016, 17, s1867. [Google Scholar]
- Huang, X.; Kong, N. Landscape of mRNA nanomedicine. Nat. Med. 2022, 28, 2273–2287. [Google Scholar] [CrossRef]
- Garibaldi, A.; Carranza, F. Isolation of newly transcribed RNA using metabolic label 4-thiouridine. Methods Mol. Biol. 2017, 1648, 169–176. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awah, C.U.; Mun, J.S.; Paragodaarachchi, A.; Boylu, B.; Ochu, C.; Matsui, H.; Ogunwobi, O.O. The Engineered Drug 3′UTRMYC1-18 Degrades the c-MYC-STAT5A/B-PD-L1 Complex In Vivo to Inhibit Metastatic Triple-Negative Breast Cancer. Cancers 2024, 16, 2663. https://doi.org/10.3390/cancers16152663
Awah CU, Mun JS, Paragodaarachchi A, Boylu B, Ochu C, Matsui H, Ogunwobi OO. The Engineered Drug 3′UTRMYC1-18 Degrades the c-MYC-STAT5A/B-PD-L1 Complex In Vivo to Inhibit Metastatic Triple-Negative Breast Cancer. Cancers. 2024; 16(15):2663. https://doi.org/10.3390/cancers16152663
Chicago/Turabian StyleAwah, Chidiebere U., Joo Sun Mun, Aloka Paragodaarachchi, Baris Boylu, Chika Ochu, Hiroshi Matsui, and Olorunseun O. Ogunwobi. 2024. "The Engineered Drug 3′UTRMYC1-18 Degrades the c-MYC-STAT5A/B-PD-L1 Complex In Vivo to Inhibit Metastatic Triple-Negative Breast Cancer" Cancers 16, no. 15: 2663. https://doi.org/10.3390/cancers16152663
APA StyleAwah, C. U., Mun, J. S., Paragodaarachchi, A., Boylu, B., Ochu, C., Matsui, H., & Ogunwobi, O. O. (2024). The Engineered Drug 3′UTRMYC1-18 Degrades the c-MYC-STAT5A/B-PD-L1 Complex In Vivo to Inhibit Metastatic Triple-Negative Breast Cancer. Cancers, 16(15), 2663. https://doi.org/10.3390/cancers16152663