
The Application of Long-Read Sequencing to Cancer



Abstract
Simple Summary
Abstract
1. Introduction
2. The Promise and Limits of NGS in Cancer Research
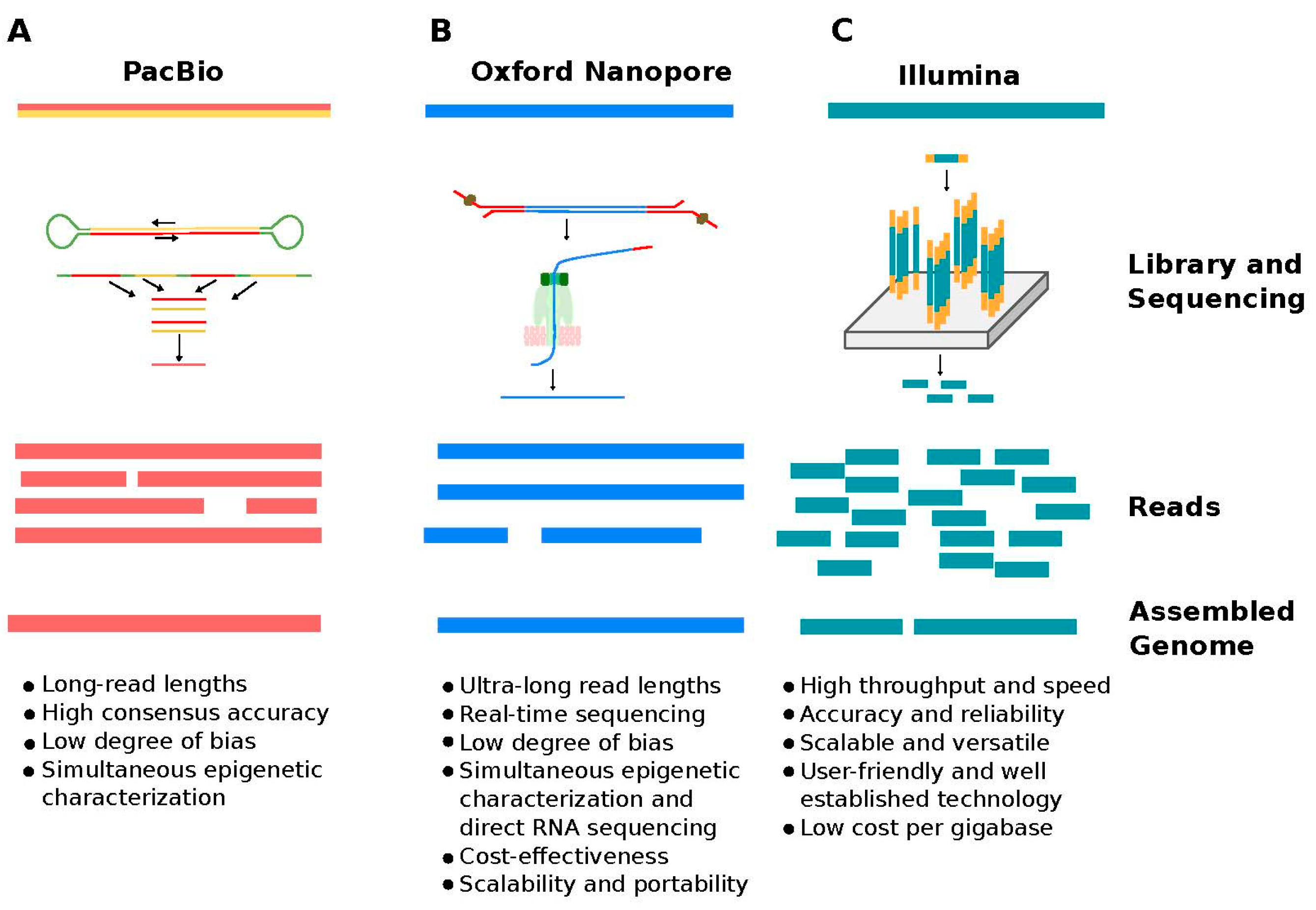
3. Long Read Sequencing
3.1. Technology Background
3.2. Advantages of TGS
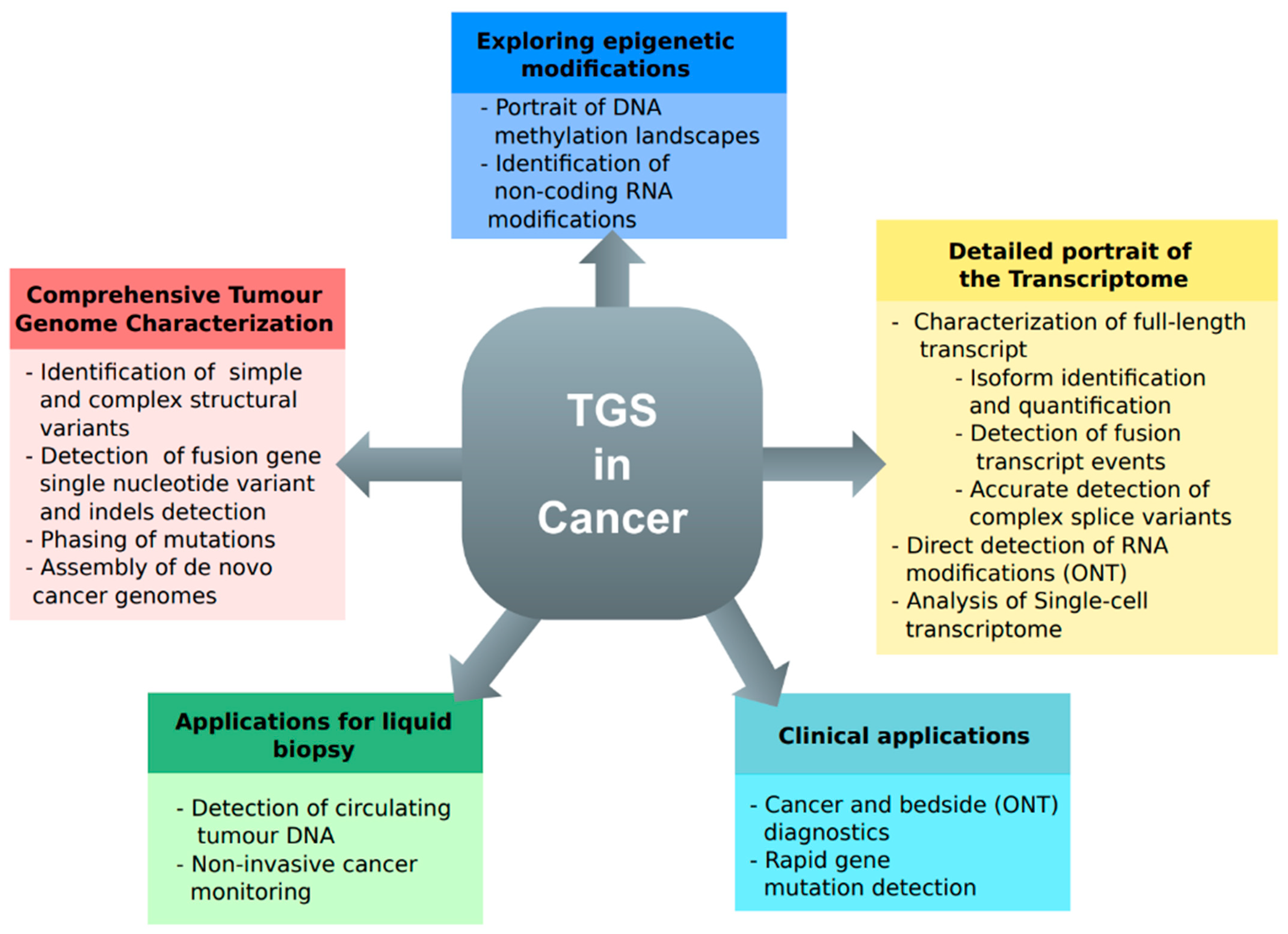
4. The Long-Read Approach in Cancer
4.1. Cancer Genomes with Long Reads
4.1.1. Identify and Phase Single Nucleotide Variants
4.1.2. Characterization of Structural Variants
4.1.3. Identification of Fusion Genes
4.1.4. Whole Genome of Single Cells
4.1.5. A Personalised Cancer Genome
4.2. Transcriptome Variation in Cancer Tissues
4.2.1. Full-Length Transcriptome of Cancer Cells
4.2.2. Post-Transcriptional RNA Modifications
4.2.3. Single-Cell Transcriptomics
4.2.4. Cancer Epigenomics in Long-Read Sequencing
4.3. Liquid Biopsy
4.4. Data Analysis of Cancer Genomes with Long Reads
5. The Challenge of Long Read Sequencing
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Brown, J.S.; Amend, S.R.; Austin, R.H.; Gatenby, R.A.; Hammarlund, E.U.; Pienta, K.J. Updating the Definition of Cancer. Mol. Cancer Res. 2023, 21, 1142–1147. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Ermini, L.; Taurone, S.; Greco, A.; Artico, M. Cancer Progression: A Single Cell Perspective. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 5721–5747. [Google Scholar]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A Window into Third-Generation Sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A.; et al. Telomere-to-Telomere Assembly of a Complete Human X Chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.; Holmes, N.; Rakyan, V.; Loose, M. BulkVis: A Graphical Viewer for Oxford Nanopore Bulk FAST5 Files. Bioinformatics 2019, 35, 2193–2198. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA Sequencing at 40: Past, Present and Future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef]
- Mardis, E.R. The Impact of Next-Generation Sequencing on Cancer Genomics: From Discovery to Clinic. Cold Spring Harb. Perspect. Med. 2019, 9, a036269. [Google Scholar] [CrossRef]
- Borden, E.S.; Buetow, K.H.; Wilson, M.A.; Hastings, K.T. Cancer Neoantigens: Challenges and Future Directions for Prediction, Prioritization, and Validation. Front. Oncol. 2022, 12, 836821. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef]
- Zhong, Y.; Xu, F.; Wu, J.; Schubert, J.; Li, M.M. Application of Next Generation Sequencing in Laboratory Medicine. Ann. Lab. Med. 2021, 41, 25–43. [Google Scholar] [CrossRef]
- Gilson, P.; Merlin, J.-L.; Harlé, A. Deciphering Tumour Heterogeneity: From Tissue to Liquid Biopsy. Cancers 2022, 14, 1384. [Google Scholar] [CrossRef]
- Choo, Z.-N.; Behr, J.M.; Deshpande, A.; Hadi, K.; Yao, X.; Tian, H.; Takai, K.; Zakusilo, G.; Rosiene, J.; Da Cruz Paula, A.; et al. Most Large Structural Variants in Cancer Genomes Can Be Detected without Long Reads. Nat. Genet. 2023, 55, 2139–2148. [Google Scholar] [CrossRef] [PubMed]
- Robasky, K.; Lewis, N.E.; Church, G.M. The Role of Replicates for Error Mitigation in next-Generation Sequencing. Nat. Rev. Genet. 2014, 15, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhou, Y.; Jia, E.; Pan, M.; Bai, Y.; Ge, Q. Bias in RNA-Seq Library Preparation: Current Challenges and Solutions. Biomed. Res. Int. 2021, 2021, 6647597. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Begik, O.; Lucas, M.C.; Ramirez, J.M.; Mason, C.E.; Wiener, D.; Schwartz, S.; Mattick, J.S.; Smith, M.A.; Novoa, E.M. Accurate Detection of m6A RNA Modifications in Native RNA Sequences. Nat. Commun. 2019, 10, 4079. [Google Scholar] [CrossRef] [PubMed]
- Genome Assembly. Available online: https://nanoporetech.com/applications/investigations/genome-assembly (accessed on 10 February 2024).
- Epigenetics and Methylation Analysis. Available online: https://nanoporetech.com/applications/investigations/epigenetics-and-methylation-analysis (accessed on 10 February 2024).
- Hon, T.; Mars, K.; Young, G.; Tsai, Y.-C.; Karalius, J.W.; Landolin, J.M.; Maurer, N.; Kudrna, D.; Hardigan, M.A.; Steiner, C.C.; et al. Highly Accurate Long-Read HiFi Sequencing Data for Five Complex Genomes. Sci. Data 2020, 7, 399. [Google Scholar] [CrossRef]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct Detection of DNA Methylation during Single-Molecule, Real-Time Sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef]
- Ni, P.; Nie, F.; Zhong, Z.; Xu, J.; Huang, N.; Zhang, J.; Zhao, H.; Zou, Y.; Huang, Y.; Li, J.; et al. DNA 5-Methylcytosine Detection and Methylation Phasing Using PacBio Circular Consensus Sequencing. Nat. Commun. 2023, 14, 4054. [Google Scholar] [CrossRef]
- Dohm, J.C.; Peters, P.; Stralis-Pavese, N.; Himmelbauer, H. Benchmarking of Long-Read Correction Methods. NAR Genom. Bioinform. 2020, 2, lqaa037. [Google Scholar] [CrossRef]
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the Performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef]
- Jain, M.; Tyson, J.R.; Loose, M.; Ip, C.L.C.; Eccles, D.A.; O’Grady, J.; Malla, S.; Leggett, R.M.; Wallerman, O.; Jansen, H.J.; et al. MinION Analysis and Reference Consortium: Phase 2 Data Release and Analysis of R9.0 Chemistry. F1000Research 2017, 6, 760. [Google Scholar] [CrossRef]
- Nanopore Sequencing Accuracy. Available online: https://nanoporetech.com/platform/accuracy (accessed on 15 March 2024).
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate Circular Consensus Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rosikiewicz, W.; Pan, Z.; Jillette, N.; Wang, P.; Taghbalout, A.; Foox, J.; Mason, C.; Carroll, M.; Cheng, A.; et al. DNA Methylation-Calling Tools for Oxford Nanopore Sequencing: A Survey and Human Epigenome-Wide Evaluation. Genome Biol. 2021, 22, 295. [Google Scholar] [CrossRef] [PubMed]
- Gershman, A.; Sauria, M.E.G.; Guitart, X.; Vollger, M.R.; Hook, P.W.; Hoyt, S.J.; Jain, M.; Shumate, A.; Razaghi, R.; Koren, S.; et al. Epigenetic Patterns in a Complete Human Genome. Science 2022, 376, eabj5089. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Miano, V.; Migliori, V.; Toolan-Kerr, P.; Sideri, T.; Enright, A.J.; et al. RNA Modifications Detection by Comparative Nanopore Direct RNA Sequencing. Nat. Commun. 2021, 12, 7198. [Google Scholar] [CrossRef] [PubMed]
- Djirackor, L.; Halldorsson, S.; Niehusmann, P.; Leske, H.; Capper, D.; Kuschel, L.P.; Pahnke, J.; Due-Tønnessen, B.J.; Langmoen, I.A.; Sandberg, C.J.; et al. Intraoperative DNA Methylation Classification of Brain Tumors Impacts Neurosurgical Strategy. Neurooncol Adv. 2021, 3, vdab149. [Google Scholar] [PubMed]
- Goenka, S.D.; Gorzynski, J.E.; Shafin, K.; Fisk, D.G.; Pesout, T.; Jensen, T.D.; Monlong, J.; Chang, P.-C.; Baid, G.; Bernstein, J.A.; et al. Accelerated Identification of Disease-Causing Variants with Ultra-Rapid Nanopore Genome Sequencing. Nat. Biotechnol. 2022, 40, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, B.; Chan, J.J.; Tabatabaeian, H.; Tong, Q.Y.; Chew, X.H.; Fan, X.; Driguez, P.; Chan, C.; Cheong, F.; et al. An Isoform-Resolution Transcriptomic Atlas of Colorectal Cancer from Long-Read Single-Cell Sequencing. bioRxiv 2023. [Google Scholar] [CrossRef]
- Penter, L.; Borji, M.; Nagler, A.; Lyu, H.; Lu, W.S.; Cieri, N.; Maurer, K.; Oliveira, G.; Al’Khafaji, A.M.; Garimella, K.V.; et al. Integrative Genotyping of Cancer and Immune Phenotypes by Long-Read Sequencing. Nat. Commun. 2024, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Dondi, A.; Lischetti, U.; Jacob, F.; Singer, F.; Borgsmüller, N.; Coelho, R.; Tumor Profiler Consortium; Heinzelmann-Schwarz, V.; Beisel, C.; Beerenwinkel, N. Detection of Isoforms and Genomic Alterations by High-Throughput Full-Length Single-Cell RNA Sequencing in Ovarian Cancer. Nat. Commun. 2023, 14, 7780. [Google Scholar] [CrossRef] [PubMed]
- Al’Khafaji, A.M.; Smith, J.T.; Garimella, K.V.; Babadi, M.; Popic, V.; Sade-Feldman, M.; Gatzen, M.; Sarkizova, S.; Schwartz, M.A.; Blaum, E.M.; et al. High-Throughput RNA Isoform Sequencing Using Programmed cDNA Concatenation. Nat. Biotechnol. 2023, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kinnex. Available online: https://www.pacb.com/technology/kinnex/ (accessed on 10 February 2024).
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The Cancer Genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- International Cancer Genome Consortium; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. International Network of Cancer Genome Projects. Nature 2010, 464, 993–998. [Google Scholar]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef]
- Suzuki, A.; Suzuki, M.; Mizushima-Sugano, J.; Frith, M.C.; Makalowski, W.; Kohno, T.; Sugano, S.; Tsuchihara, K.; Suzuki, Y. Sequencing and Phasing Cancer Mutations in Lung Cancers Using a Long-Read Portable Sequencer. DNA Res. 2017, 24, 585–596. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Miyake, S.; Oka, M.; Kanai, A.; Kawai, Y.; Nagasawa, S.; Shiraishi, Y.; Tokunaga, K.; Kohno, T.; Seki, M.; et al. Phasing Analysis of Lung Cancer Genomes Using a Long Read Sequencer. Nat. Commun. 2022, 13, 3464. [Google Scholar] [CrossRef]
- Yoo, B.; Cheung, W.; Pushel, I.; Lansdon, L.; Bi, C.; Guest, E.; Pastinen, T.; Farooqi, M. Abstract 243: Long-Read Sequencing of Pediatric Cancer Genomes Identifies Multiple Clinically Relevant Variants. Cancer Res. 2023, 83, 243. [Google Scholar] [CrossRef]
- Weinhold, N.; Jacobsen, A.; Schultz, N.; Sander, C.; Lee, W. Genome-Wide Analysis of Noncoding Regulatory Mutations in Cancer. Nat. Genet. 2014, 46, 1160–1165. [Google Scholar] [CrossRef]
- Lee, S.; Chang, T.-C.; Schreiner, P.; Fan, Y.; Agarwal, N.; Owens, C.; Dummer, R.; Kirkwood, J.M.; Barnhill, R.L.; Theodorescu, D.; et al. Targeted Long-Read Bisulfite Sequencing Identifies Differences in the TERT Promoter Methylation Profiles between TERT Wild-Type and TERT Mutant Cancer Cells. Cancers 2022, 14, 18. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 174, 1034–1035. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Razavi, P.; Johnson, J.L.; Shao, H.; Shah, H.; Antoine, A.; Ladewig, E.; Gorelick, A.; Lin, T.-Y.; Toska, E.; et al. Double PIK3CA Mutations in Cis Increase Oncogenicity and Sensitivity to PI3Kα Inhibitors. Science 2019, 366, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural Variation in the Human Genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef]
- Mills, R.E.; Walter, K.; Stewart, C.; Handsaker, R.E.; Chen, K.; Alkan, C.; Abyzov, A.; Yoon, S.C.; Ye, K.; Cheetham, R.K.; et al. Mapping Copy Number Variation by Population-Scale Genome Sequencing. Nature 2011, 470, 59–65. [Google Scholar] [CrossRef] [PubMed]
- ICGC/TCGA. Pan-Cancer Analysis of Whole Genomes Consortium Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Weischenfeldt, J.; Dubash, T.; Drainas, A.P.; Mardin, B.R.; Chen, Y.; Stütz, A.M.; Waszak, S.M.; Bosco, G.; Halvorsen, A.R.; Raeder, B.; et al. Pan-Cancer Analysis of Somatic Copy-Number Alterations Implicates IRS4 and IGF2 in Enhancer Hijacking. Nat. Genet. 2017, 49, 65–74. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Gao, P.; Ding, Y.-Y.; Chen, C.-H.; Chen, G.; Chen, C.; Kim, H.; Tasian, S.K.; Hunger, S.P.; Tan, K. Diverse Noncoding Mutations Contribute to Deregulation of Cis-Regulatory Landscape in Pediatric Cancers. Sci. Adv. 2020, 6, eaba3064. [Google Scholar] [CrossRef]
- Herz, H.-M. Enhancer Deregulation in Cancer and Other Diseases. Bioessays 2016, 38, 1003–1015. [Google Scholar] [CrossRef]
- Wu, S.; Turner, K.M.; Nguyen, N.; Raviram, R.; Erb, M.; Santini, J.; Luebeck, J.; Rajkumar, U.; Diao, Y.; Li, B.; et al. Circular ecDNA Promotes Accessible Chromatin and High Oncogene Expression. Nature 2019, 575, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.L.; Workman, R.E.; Fan, Y.; Eshleman, J.R.; Timp, W. Nanopore Sequencing Detects Structural Variants in Cancer. Cancer Biol. Ther. 2016, 17, 246–253. [Google Scholar] [CrossRef]
- Nattestad, M.; Goodwin, S.; Ng, K.; Baslan, T.; Sedlazeck, F.J.; Rescheneder, P.; Garvin, T.; Fang, H.; Gurtowski, J.; Hutton, E.; et al. Complex Rearrangements and Oncogene Amplifications Revealed by Long-Read DNA and RNA Sequencing of a Breast Cancer Cell Line. Genome Res. 2018, 28, 1126–1135. [Google Scholar] [CrossRef]
- Aganezov, S.; Goodwin, S.; Sherman, R.M.; Sedlazeck, F.J.; Arun, G.; Bhatia, S.; Lee, I.; Kirsche, M.; Wappel, R.; Kramer, M.; et al. Comprehensive Analysis of Structural Variants in Breast Cancer Genomes Using Single-Molecule Sequencing. Genome Res. 2020, 30, 1258–1273. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate Detection of Complex Structural Variations Using Single-Molecule Sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef]
- Fujimoto, A.; Wong, J.H.; Yoshii, Y.; Akiyama, S.; Tanaka, A.; Yagi, H.; Shigemizu, D.; Nakagawa, H.; Mizokami, M.; Shimada, M. Whole-Genome Sequencing with Long Reads Reveals Complex Structure and Origin of Structural Variation in Human Genetic Variations and Somatic Mutations in Cancer. Genome Med. 2021, 13, 65. [Google Scholar] [CrossRef]
- Xu, L.; Wang, X.; Lu, X.; Liang, F.; Liu, Z.; Zhang, H.; Li, X.; Tian, S.; Wang, L.; Wang, Z. Long-Read Sequencing Identifies Novel Structural Variations in Colorectal Cancer. PLoS Genet. 2023, 19, e1010514. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K.; Symer, D.E.; Mahmoud, M.; Jiang, B.; Goodwin, S.; Wangsa, D.; Li, Z.; Xiao, W.; Dunn, J.D.; Ried, T.; et al. Intratumoral Heterogeneity and Clonal Evolution Induced by HPV Integration. Cancer Discov. 2023, 13, 910–927. [Google Scholar] [CrossRef]
- Talsania, K.; Shen, T.-W.; Chen, X.; Jaeger, E.; Li, Z.; Chen, Z.; Chen, W.; Tran, B.; Kusko, R.; Wang, L.; et al. Structural Variant Analysis of a Cancer Reference Cell Line Sample Using Multiple Sequencing Technologies. Genome Biol. 2022, 23, 255. [Google Scholar] [CrossRef]
- Li, J.; Lu, H.; Ng, P.K.-S.; Pantazi, A.; Ip, C.K.M.; Jeong, K.J.; Amador, B.; Tran, R.; Tsang, Y.H.; Yang, L.; et al. A Functional Genomic Approach to Actionable Gene Fusions for Precision Oncology. Sci. Adv. 2022, 8, eabm2382. [Google Scholar] [CrossRef]
- Kohno, T.; Nakaoku, T.; Tsuta, K.; Tsuchihara, K.; Matsumoto, S.; Yoh, K.; Goto, K. Beyond ALK-RET, ROS1 and Other Oncogene Fusions in Lung Cancer. Transl. Lung Cancer Res. 2015, 4, 156–164. [Google Scholar] [PubMed]
- Potter, N.E.; Ermini, L.; Papaemmanuil, E.; Cazzaniga, G.; Vijayaraghavan, G.; Titley, I.; Ford, A.; Campbell, P.; Kearney, L.; Greaves, M. Single-Cell Mutational Profiling and Clonal Phylogeny in Cancer. Genome Res. 2013, 23, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG Gene Fusion in Prostate Cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.N.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-Year Follow-up of Patients Receiving Imatinib for Chronic Myeloid Leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Lee, J.; Robinson, H.; Le, L.P.; Iafrate, A.J.; Nardi, V. A Nanopore Sequencing-Based Assay for Rapid Detection of Gene Fusions. J. Mol. Diagn. 2019, 21, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Iafrate, A.J.; Nardi, V. Nanopore Flongle Sequencing as a Rapid, Single-Specimen Clinical Test for Fusion Detection. J. Mol. Diagn. 2021, 23, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Sala-Torra, O.; Reddy, S.; Hung, L.-H.; Beppu, L.; Wu, D.; Radich, J.; Yeung, K.Y.; Yeung, C.C.S. Rapid Detection of Myeloid Neoplasm Fusions Using Single-Molecule Long-Read Sequencing. PLoS Glob. Public Health 2023, 3, e0002267. [Google Scholar] [CrossRef]
- Hård, J.; Mold, J.E.; Eisfeldt, J.; Tellgren-Roth, C.; Häggqvist, S.; Bunikis, I.; Contreras-Lopez, O.; Chin, C.-S.; Nordlund, J.; Rubin, C.-J.; et al. Long-Read Whole-Genome Analysis of Human Single Cells. Nat. Commun. 2023, 14, 5164. [Google Scholar] [CrossRef]
- Xiao, C.; Chen, Z.; Chen, W.; Padilla, C.; Colgan, M.; Wu, W.; Fang, L.-T.; Liu, T.; Yang, Y.; Schneider, V.; et al. Personalized Genome Assembly for Accurate Cancer Somatic Mutation Discovery Using Tumor-Normal Paired Reference Samples. Genome Biol. 2022, 23, 237. [Google Scholar] [CrossRef]
- Huang, K.K.; Huang, J.; Wu, J.K.L.; Lee, M.; Tay, S.T.; Kumar, V.; Ramnarayanan, K.; Padmanabhan, N.; Xu, C.; Tan, A.L.K.; et al. Long-Read Transcriptome Sequencing Reveals Abundant Promoter Diversity in Distinct Molecular Subtypes of Gastric Cancer. Genome Biol. 2021, 22, 44. [Google Scholar] [CrossRef]
- Sun, Q.; Han, Y.; He, J.; Wang, J.; Ma, X.; Ning, Q.; Zhao, Q.; Jin, Q.; Yang, L.; Li, S.; et al. Long-Read Sequencing Reveals the Landscape of Aberrant Alternative Splicing and Novel Therapeutic Target in Colorectal Cancer. Genome Med. 2023, 15, 76. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, J.; Mor, G.; Sklar, J. A Neoplastic Gene Fusion Mimics Trans-Splicing of RNAs in Normal Human Cells. Science 2008, 321, 1357–1361. [Google Scholar] [CrossRef]
- Weirather, J.L.; Afshar, P.T.; Clark, T.A.; Tseng, E.; Powers, L.S.; Underwood, J.G.; Zabner, J.; Korlach, J.; Wong, W.H.; Au, K.F. Characterization of Fusion Genes and the Significantly Expressed Fusion Isoforms in Breast Cancer by Hybrid Sequencing. Nucleic Acids Res. 2015, 43, e116. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-W.; Chen, Y.-M.; Zhao, Q.-Q.; Zhao, X.; Wu, Y.-R.; Chen, D.-Z.; Liao, L.-D.; Chen, Y.; Yang, Q.; Xu, L.-Y.; et al. Long Read Single-Molecule Real-Time Sequencing Elucidates Transcriptome-Wide Heterogeneity and Complexity in Esophageal Squamous Cells. Front. Genet. 2019, 10, 915. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Li, J.; Long, M.; Wu, J.; Zhang, Z.; Xie, F.; Zhao, J.; Yang, H.; Song, Q.; Lian, S.; et al. Detection of Structural Variations and Fusion Genes in Breast Cancer Samples Using Third-Generation Sequencing. Front. Cell Dev. Biol. 2022, 10, 854640. [Google Scholar] [CrossRef] [PubMed]
- Mock, A.; Braun, M.; Scholl, C.; Fröhling, S.; Erkut, C. Transcriptome Profiling for Precision Cancer Medicine Using Shallow Nanopore cDNA Sequencing. Sci. Rep. 2023, 13, 2378. [Google Scholar] [CrossRef]
- Volden, R.; Kronenberg, Z.; Gillmor, A.; Verhey, T.; Monument, M.; Senger, D.; Dhillon, H.; Underwood, J.; Tseng, E.; Baker, D.; et al. Abstract LB078: Pbfusion: Detecting Gene-Fusion and Other Transcriptional Abnormalities Using PacBio HiFi Data. Cancer Res. 2023, 83, LB078. [Google Scholar] [CrossRef]
- Dorney, R.; Dhungel, B.P.; Rasko, J.E.J.; Hebbard, L.; Schmitz, U. Recent Advances in Cancer Fusion Transcript Detection. Brief. Bioinform. 2023, 24, bbac519. [Google Scholar] [CrossRef]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly Parallel Direct RNA Sequencing on an Array of Nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef]
- Krusnauskas, R.; Stakaitis, R.; Steponaitis, G.; Almstrup, K.; Vaitkiene, P. Identification and Comparison of m6A Modifications in Glioblastoma Non-Coding RNAs with MeRIP-Seq and Nanopore dRNA-Seq. Epigenetics 2023, 18, 2163365. [Google Scholar] [CrossRef]
- Van de Sande, B.; Lee, J.S.; Mutasa-Gottgens, E.; Naughton, B.; Bacon, W.; Manning, J.; Wang, Y.; Pollard, J.; Mendez, M.; Hill, J.; et al. Applications of Single-Cell RNA Sequencing in Drug Discovery and Development. Nat. Rev. Drug Discov. 2023, 22, 496–520. [Google Scholar] [CrossRef]
- Levitin, H.M.; Yuan, J.; Sims, P.A. Single-Cell Transcriptomic Analysis of Tumor Heterogeneity. Trends Cancer Res. 2018, 4, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Jabbari, J.S.; Thijssen, R.; Gouil, Q.; Amarasinghe, S.L.; Voogd, O.; Kariyawasam, H.; Du, M.R.M.; Schuster, J.; Wang, C.; et al. Comprehensive Characterization of Single-Cell Full-Length Isoforms in Human and Mouse with Long-Read Sequencing. Genome Biol. 2021, 22, 310. [Google Scholar] [CrossRef] [PubMed]
- Volden, R.; Palmer, T.; Byrne, A.; Cole, C.; Schmitz, R.J.; Green, R.E.; Vollmers, C. Improving Nanopore Read Accuracy with the R2C2 Method Enables the Sequencing of Highly Multiplexed Full-Length Single-Cell cDNA. Proc. Natl. Acad. Sci. USA 2018, 115, 9726–9731. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.; Machado, M.; Ferrão, J.; Sebastião Rodrigues, A.; Vieira, L. Whole Human Genome 5′-mC Methylation Analysis Using Long Read Nanopore Sequencing. Epigenetics 2022, 17, 1961–1975. [Google Scholar] [CrossRef] [PubMed]
- Ewing, A.D.; Smits, N.; Sanchez-Luque, F.J.; Faivre, J.; Brennan, P.M.; Richardson, S.R.; Cheetham, S.W.; Faulkner, G.J. Nanopore Sequencing Enables Comprehensive Transposable Element Epigenomic Profiling. Mol. Cell 2020, 80, 915–928.e5. [Google Scholar] [CrossRef]
- Euskirchen, P.; Bielle, F.; Labreche, K.; Kloosterman, W.P.; Rosenberg, S.; Daniau, M.; Schmitt, C.; Masliah-Planchon, J.; Bourdeaut, F.; Dehais, C.; et al. Same-Day Genomic and Epigenomic Diagnosis of Brain Tumors Using Real-Time Nanopore Sequencing. Acta Neuropathol. 2017, 134, 691–703. [Google Scholar] [CrossRef]
- Choy, L.Y.L.; Peng, W.; Jiang, P.; Cheng, S.H.; Yu, S.C.Y.; Shang, H.; Olivia Tse, O.Y.; Wong, J.; Wong, V.W.S.; Wong, G.L.H.; et al. Single-Molecule Sequencing Enables Long Cell-Free DNA Detection and Direct Methylation Analysis for Cancer Patients. Clin. Chem. 2022, 68, 1151–1163. [Google Scholar] [CrossRef]
- Athanasopoulou, K.; Boti, M.A.; Adamopoulos, P.G.; Skourou, P.C.; Scorilas, A. Third-Generation Sequencing: The Spearhead towards the Radical Transformation of Modern Genomics. Life 2021, 12, 30. [Google Scholar] [CrossRef]
- Gouil, Q.; Keniry, A. Latest Techniques to Study DNA Methylation. Essays Biochem. 2019, 63, 639–648. [Google Scholar]
- Cheng, S.H.; Jiang, P.; Sun, K.; Cheng, Y.K.Y.; Chan, K.C.A.; Leung, T.Y.; Chiu, R.W.K.; Lo, Y.M.D. Noninvasive Prenatal Testing by Nanopore Sequencing of Maternal Plasma DNA: Feasibility Assessment. Clin. Chem. 2015, 61, 1305–1306. [Google Scholar] [CrossRef] [PubMed]
- Martignano, F.; Munagala, U.; Crucitta, S.; Mingrino, A.; Semeraro, R.; Del Re, M.; Petrini, I.; Magi, A.; Conticello, S.G. Nanopore Sequencing from Liquid Biopsy: Analysis of Copy Number Variations from Cell-Free DNA of Lung Cancer Patients. Mol. Cancer 2021, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Marcozzi, A.; Jager, M.; Elferink, M.; Straver, R.; van Ginkel, J.H.; Peltenburg, B.; Chen, L.-T.; Renkens, I.; van Kuik, J.; Terhaard, C.; et al. Accurate Detection of Circulating Tumor DNA Using Nanopore Consensus Sequencing. NPJ Genom. Med. 2021, 6, 106. [Google Scholar] [CrossRef] [PubMed]
- Katsman, E.; Orlanski, S.; Martignano, F.; Fox-Fisher, I.; Shemer, R.; Dor, Y.; Zick, A.; Eden, A.; Petrini, I.; Conticello, S.G.; et al. Detecting Cell-of-Origin and Cancer-Specific Methylation Features of Cell-Free DNA from Nanopore Sequencing. Genome Biol. 2022, 23, 158. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, Y.; Tantyo, N.A.; Evander, N.; Hentschel, A.E.; Wever, B.M.; Ramaker, J.; Bootsma, S.; Fransen, M.F.; Lenos, K.J.; Vermeulen, L.; et al. Real-Time Analysis of the Cancer Genome and Fragmentome from Plasma and Urine Cell-Free DNA Using Nanopore Sequencing. EMBO Mol. Med. 2023, 15, e17282. [Google Scholar] [CrossRef] [PubMed]
- Pagès-Gallego, M.; de Ridder, J. Comprehensive Benchmark and Architectural Analysis of Deep Learning Models for Nanopore Sequencing Basecalling. Genome Biol. 2023, 24, 71. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of Neural Network Basecalling Tools for Oxford Nanopore Sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef] [PubMed]
- Perešíni, P.; Boža, V.; Brejová, B.; Vinař, T. Nanopore Base Calling on the Edge. Bioinformatics 2021, 37, 4661–4667. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Jain, C.; Rhie, A.; Hansen, N.F.; Koren, S.; Phillippy, A.M. Long-Read Mapping to Repetitive Reference Sequences Using Winnowmap2. Nat. Methods 2022, 19, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Ekim, B.; Sahlin, K.; Medvedev, P.; Berger, B.; Chikhi, R. Efficient Mapping of Accurate Long Reads in Minimizer Space with Mapquik. Genome Res. 2023, 33, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, S.; Su, J.; Leung, A.W.-S.; Lam, T.-W.; Luo, R. Symphonizing Pileup and Full-Alignment for Deep Learning-Based Long-Read Variant Calling. Nat. Comput. Sci. 2022, 2, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.K.; Hendra, C.; Pratanwanich, P.N.; Göke, J. Beyond Sequencing: Machine Learning Algorithms Extract Biology Hidden in Nanopore Signal Data. Trends Genet. 2022, 38, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Kokot, M.; Gudyś, A.; Li, H.; Deorowicz, S. CoLoRd: Compressing Long Reads. Nat. Methods 2022, 19, 441–444. [Google Scholar] [CrossRef]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-Read Human Genome Sequencing and Its Applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Koren, S.; Bao, Z.; Guarracino, A.; Ou, S.; Goodwin, S.; Jenike, K.M.; Lucas, J.; McNulty, B.; Park, J.; Rautianinen, M.; et al. Gapless Assembly of Complete Human and Plant Chromosomes Using Only Nanopore Sequencing. bioRxiv 2024. [Google Scholar] [CrossRef]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-Resolved de Novo Assembly Using Phased Assembly Graphs with Hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Choudhury, O.; Chakrabarty, A.; Emrich, S.J. HECIL: A Hybrid Error Correction Algorithm for Long Reads with Iterative Learning. Sci. Rep. 2018, 8, 9936. [Google Scholar] [CrossRef]
- Zhu, W.; Liao, X. LCAT: An Isoform-Sensitive Error Correction for Transcriptome Sequencing Long Reads. Front. Genet. 2023, 14, 1166975. [Google Scholar] [CrossRef]
- Fukasawa, Y.; Ermini, L.; Wang, H.; Carty, K.; Cheung, M.-S. LongQC: A Quality Control Tool for Third Generation Sequencing Long Read Data. G3 2020, 10, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Ritchie, M.E.; Gouil, Q. Long-Read-Tools.org: An Interactive Catalogue of Analysis Methods for Long-Read Sequencing Data. Gigascience 2021, 10, giab003. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Sirén, J.; Novak, A.M.; Hickey, G.; Eizenga, J.M.; Dawson, E.T.; Jones, W.; Garg, S.; Markello, C.; Lin, M.F.; et al. Variation Graph Toolkit Improves Read Mapping by Representing Genetic Variation in the Reference. Nat. Biotechnol. 2018, 36, 875–879. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Feature | TGS | NGS |
|---|---|---|
| Throughput | Lower throughput (fewer reads but more runs) | Higher throughput (billions of reads per run) |
| Advantage | More flexibility, enables rapid sequencing of many runs | Cost-effective for sequencing many samples in a run |
| Disadvantage | Higher cost per gigabase sequenced | Fewer runs compared to TGS platforms |
| Read Length | Longer reads (10 kb–1 Mb+) | Shorter reads (150 bp–300 bp) |
| Advantage | Enables sequencing of entire transcripts and long-range variant detection | Suitable for most applications requiring moderate read lengths |
| Disadvantage | Lower accuracy as longer stretches can be more prone to errors. | Read lengths limit applications requiring long-range information |
| Error Rate | Higher error rate compared to NGS | Lower error rate compared to TGS |
| Advantage | Error rate is rapidly improving and approaching the NGS rate. | Provides high-accuracy data for most applications |
| Disadvantage | Lower accuracy compared to NGS | May require higher sequencing depth for some applications |
| Cost | Generally higher cost per gigabase | Generally lower cost per gigabase |
| Advantage | The cost has been steadily decreasing | Cost-effective for large-scale sequencing |
| Disadvantage | Costs can still be significant depending on project requirements | Costs can still be significant depending on project requirements |
| Data Analysis | Real-time analysis, portability and appropriate for whole genome assembly | Established analysis pipelines and bioinformatics tools readily available |
| Advantage | Reduced bias due to minimal amplification | Streamlined data analysis with well-established tools |
| Disadvantage | Can be computationally demanding | Data analysis can be complex for some applications |
| Applications | Ideal for large genome sequencing, de novo assembly, long-range variant detection, full-length transcriptomics, direct detection of DNA/RNA modifications, metagenomics | Wide range of applications including targeted sequencing, variant analysis, gene expression studies, and microbiome analysis |
| Advantage | Full-length transcript sequencing and accurate assembly | Versatile platform for various research areas |
| Disadvantage | Less suitable for targeted sequencing and high-depth variant analysis | May not be suitable for complex variant detection or de novo assembly |
| Sample requirements | More stringent quality and quantity requirements than NGS | Established lab workflows and less stringent requirements |
| Advantage | More stringent requirements produce long-read data | Standardised workflow and less stringent sample requirements. Degraded (FFPE) or low-input (surgical biopsy) samples can be sequenced. |
| Disadvantage | Many sample types cannot be easily sequenced due to reduced quality or small quantity. | Although more samples can be sequenced data suffers from disadvantages inherent in short-read data |
| Feature | TGS | NGS |
|---|---|---|
| Basecalling Complexity | More complex due to indirect signal interpretation and longer reads | Less complex due to direct imaging and shorter reads |
| Computational Analysis | More powerful computing resources are required for assembly and variant calling | Generally less computationally demanding |
| Data-File Size | Larger files per gigabase sequenced due to longer reads | Smaller files per gigabase sequenced due to shorter reads |
| Data-Analysis Challenges | Requires specialised algorithms to handle longer reads and higher error rates | Requires robust algorithms for high-throughput data processing |
| Genome Assembly | Easier for complex or repetitive genomes due to long reads More challenging due to higher error rates and potential for chimeric reads (merged from different fragments) | More challenging for complex genomes due to shorter reads Easier due to lower error rates and shorter reads providing more overlap |
| Variant Detection | More powerful for detecting large insertions/deletions and structural variants | Well-suited for detecting single nucleotide variants |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ermini, L.; Driguez, P. The Application of Long-Read Sequencing to Cancer. Cancers 2024, 16, 1275. https://doi.org/10.3390/cancers16071275
Ermini L, Driguez P. The Application of Long-Read Sequencing to Cancer. Cancers. 2024; 16(7):1275. https://doi.org/10.3390/cancers16071275
Chicago/Turabian StyleErmini, Luca, and Patrick Driguez. 2024. "The Application of Long-Read Sequencing to Cancer" Cancers 16, no. 7: 1275. https://doi.org/10.3390/cancers16071275
APA StyleErmini, L., & Driguez, P. (2024). The Application of Long-Read Sequencing to Cancer. Cancers, 16(7), 1275. https://doi.org/10.3390/cancers16071275