
Immunotherapeutic Strategies for the Treatment of Glioblastoma: Current Challenges and Future Perspectives

Abstract
Simple Summary
Abstract
1. Introduction
2. Standard of Care for GBM Patients
3. Therapeutic Challenges for GBM Therapies
3.1. Anatomical Location
3.2. Presence of the Blood–Brain Barrier
3.3. Tumor Heterogeneity and Plasticity
3.4. Infiltrative Nature
3.5. Systemic and Local Immunosuppression
- (i)
- Soluble molecules: Secreted by various cellular players of the GBM microenvironment, the TME contains various growth factors and cytokines, such as (i) tumor-promoting cytokines, including interleukin (IL)-1, and basic fibroblast growth factor (bFGF) and (ii) immunosuppressive chemical mediators, including TGF-β, IL-10, IL-6 and prostaglandin E-2 (PGE2) [95,96]. While IL-1 and bFGF promote tumorigenesis, TGF-β, IL-10, IL-6, and PGE2 generally shift the immune response from an inflammatory response to a pro-tumoral and wound-healing one. This alteration leads to a reduced ability of immune cells to efficiently eliminate tumor cells. Moreover, the GBM TME is characterized by high levels of CC Chemokine Ligand 2 (CCL2), a very potent chemoattractant essential for the recruitment of regulatory T cells (Tregs) and myeloid cells [97].
- (ii)
- Extracellular matrix (ECM): In GBM, ECM composition is altered due to an overexpression and increased secretion of laminin, collagen, and fibronectin, and this physically results in elevated overall density and tumor stiffness [98]. This contributes to limiting the ability of chemotherapeutic drugs to diffuse and penetrate the tumor, reducing their effectiveness. Moreover, high levels of fibronectin and hyaluronic acid, along with surrounding ECM degradation via metalloproteinases, increases the mobility and invasiveness of glioma cells [99].
- (iii)
- Vasculature: The GBM TME is characterized by abnormal vasculature, and the central areas of the tumor experience poor blood flow, leading to a decrease in oxygen delivery [100]. This hypoxic microenvironment increases the expression of hypoxia-inducible factor 1-α, promoting angiogenesis and tumor cell invasion [100]. HIF1-α upregulates immunomodulatory surface ligands such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed death-ligand 1 (PD-L1), inhibiting efficient anti-tumor immune responses [101].
- (iv)
- Healthy brain cells: In response to CNS injury, astrocytes normally secrete growth factors and cytokines to facilitate tissue repair in a process known as astrogliosis [102]. However, in GBM, this process is exploited to promote tumor growth. In particular, the TME promotes crosstalk between astrocytes and neighboring microglia, resulting in the activation of the JAK/STAT and PD-L1 pathways within astrocytes. This activation triggers an elevated production of anti-inflammatory cytokines like IL-10, TGF-β, and STAT3, thereby fostering an immunosuppressive milieu [103]. Moreover, neurons play a role in facilitating GBM tumor progression by upregulating neuroligin-3. This leads to the activation of the PI3K signaling pathway, promoting the proliferative activity of glioma cells [104].
- (v)
- Tumor-associated myeloid cells: Tumor-associated microglia and macrophages (TAMs) are the main infiltrating population in GBM, attracted towards the tumors in response to high concentrations of various chemoattractants secreted by glioma cells, including CCL2 [105,106,107]. Within the TME, they adopt immunosuppressive and tumor-supportive phenotypes [108]. Activation of the mTOR signaling pathway leads to increased STAT3 phosphorylation and suppression of the NF-κB pathway, resulting in the upregulation of anti-inflammatory cytokines such as IL-6, and IL-10 [109]. TAMs exhibit a decreased expression of surface MHC class II molecules and costimulatory molecules (CD40, CD80, and CD86), impairing antigen presentation and activation of T cells [110,111,112]. Myeloid-derived suppressor cells (MDSCs) suppress the immune system through multiple mechanisms. They express arginase, which reduces L-arginine levels necessary for TCR expression and function. They also secrete nitric oxide and reactive oxygen species, further inhibiting T cell activity. Additionally, MDSCs express PD-L1, promoting T cell exhaustion [113,114].
- (vi)
- Tumor-infiltrating lymphocytes (TILs): In GBM, TILs often exhibit dysfunction and exhaustion caused by factors released by glioma and microenvironmental cells, including TGF-β, IL-10, and CCL2, which recruit Tregs, MDSCs, and TAMs to the tumor site [115]. In response to TGF-β, CD4+ T cells upregulate FoxP3 and differentiate into Tregs. They account for 25% of TILs and are associated with a poor prognosis in GBM [116]. Through IL-10 and TGF-β signaling, Tregs promote the transition of other T cells into regulatory ones, exert an immunosuppressive function over natural killer (NK) and CD8+ T cells, help to generate MDSCs, and impair the antigen presentation capability of DCs [117]. TGF-β1 leads to a reduction in the expression of the activating receptor natural killer group 2 (NKG2D) on the surface of both CD8+ T cells and NK cells, thereby hindering their cytotoxic effects on GBM cells [118]. Moreover, Tregs highly express immune checkpoint molecules such as PD-1 and CTLA-4 that, via interaction with their respective receptors on the surface of T cells, suppress their effector functions [119]. Glioma cells further suppress lymphocyte activity through molecules such as FasL, PD-L1, PD-L2, CD70, and ganglioside [120,121,122]. The scarcity of TILs and accumulation of exhausted T cells in the tumor microenvironment contribute to immunotherapy resistance and relapse.
4. Immunotherapeutic Strategies for the Treatment of GBM
4.1. Immune Checkpoint Therapy
4.2. Vaccination Therapy
4.2.1. DNA/RNA Vaccines
4.2.2. Peptide Vaccines
4.2.3. Dendritic Cell Vaccines
4.3. Adoptive T Cell Therapy
4.3.1. TIL Therapy
4.3.2. CAR-T Cell Therapy
4.4. Virus-Based Therapy
4.4.1. Adenovirus (AdV)
4.4.2. Retrovirus
4.4.3. Herpes Simplex Virus (HSV)
4.4.4. Poliovirus
4.4.5. Respiratory Enteric Orphan Virus (Reovirus)
4.4.6. Measles Virus (MeV)
4.4.7. Newcastle Disease Virus (NDV)
4.4.8. H-1 Parvovirus (H-1PV)
4.4.9. Vaccinia Virus (VACV)
5. Combination Therapy
6. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 12 (Suppl. 2), iii1–iii105. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef] [PubMed]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Rev. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. Short-Course Radiation plus Temozolomide in Elderly Patients with Glioblastoma. N. Engl. J. Med. 2017, 376, 1027–1037. [Google Scholar] [CrossRef]
- Stupp, R.; Wong, E.T.; Kanner, A.A.; Steinberg, D.; Engelhard, H.; Heidecke, V.; Kirson, E.D.; Taillibert, S.; Liebermann, F.; Dbaly, V.; et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: A randomised phase III trial of a novel treatment modality. Eur. J. Cancer 2012, 48, 2192–2202. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Pacheco, P.; Stupp, R. Tumor treating fields: A novel treatment modality and its use in brain tumors. Neuro-Oncology 2016, 18, 1338–1349. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Lassman, A.B. NovoTTF: Where to go from here? Neuro-Oncology 2017, 19, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Taphoorn, M.J.B.; Dirven, L.; Kanner, A.A.; Lavy-Shahaf, G.; Weinberg, U.; Taillibert, S.; Toms, S.A.; Honnorat, J.; Chen, T.C.; Sroubek, J.; et al. Influence of Treatment with Tumor-Treating Fields on Health-Related Quality of Life of Patients with Newly Diagnosed Glioblastoma: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2018, 4, 495–504. [Google Scholar] [CrossRef]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma--are we there yet? Neuro-Oncology 2013, 15, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Audureau, E.; Chivet, A.; Ursu, R.; Corns, R.; Metellus, P.; Noel, G.; Zouaoui, S.; Guyotat, J.; Le Reste, P.J.; Faillot, T.; et al. Prognostic factors for survival in adult patients with recurrent glioblastoma: A decision-tree-based model. J. Neuro-Oncol. 2018, 136, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Le Rhun, E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat. Rev. 2020, 87, 102029. [Google Scholar] [CrossRef]
- Vaz-Salgado, M.A.; Villamayor, M.; Albarrán, V.; Alía, V.; Sotoca, P.; Chamorro, J.; Rosero, D.; Barrill, A.M.; Martín, M.; Fernandez, E.; et al. Recurrent Glioblastoma: A Review of the Treatment Options. Cancers 2023, 15, 4279. [Google Scholar] [CrossRef] [PubMed]
- Taal, W.; Oosterkamp, H.M.; Walenkamp, A.M.; Dubbink, H.J.; Beerepoot, L.V.; Hanse, M.C.; Buter, J.; Honkoop, A.H.; Boerman, D.; de Vos, F.Y.; et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): A randomised controlled phase 2 trial. Lancet Oncol. 2014, 15, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Mulholland, P.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218. [Google Scholar] [CrossRef]
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro-Oncology 2016, 18, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Puduvalli, V.K.; Chamberlain, M.C.; van den Bent, M.J.; Carpentier, A.F.; Cher, L.M.; Mason, W.; Weller, M.; Hong, S.; Musib, L.; et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J. Clin. Oncol. 2010, 28, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bent, M.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.-S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F.; et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro-Oncology 2019, 22, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Duerinck, J.; Du Four, S.; Bouttens, F.; Andre, C.; Verschaeve, V.; Van Fraeyenhove, F.; Chaskis, C.; D’Haene, N.; Le Mercier, M.; Rogiers, A.; et al. Randomized phase II trial comparing axitinib with the combination of axitinib and lomustine in patients with recurrent glioblastoma. J. Neuro-Oncol. 2018, 136, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Bonosi, L.; Marrone, S.; Benigno, U.E.; Buscemi, F.; Musso, S.; Porzio, M.; Silven, M.P.; Torregrossa, F.; Grasso, G. Maximal Safe Resection in Glioblastoma Surgery: A Systematic Review of Advanced Intraoperative Image-Guided Techniques. Brain Sci. 2023, 13, 216. [Google Scholar] [CrossRef]
- Gerritsen, J.K.W.; Broekman, M.L.D.; De Vleeschouwer, S.; Schucht, P.; Jungk, C.; Krieg, S.M.; Nahed, B.V.; Berger, M.S.; Vincent, A. Decision making and surgical modality selection in glioblastoma patients: An international multicenter survey. J. Neuro-Oncol. 2022, 156, 465–482. [Google Scholar] [CrossRef]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.-J. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Tonn, J.C.; Mehdorn, H.M.; Nestler, U.; Franz, K.; Goetz, C.; Bink, A.; Pichlmeier, U.; Group, A.L.-G.S. Counterbalancing risks and gains from extended resections in malignant glioma surgery: A supplemental analysis from the randomized 5-aminolevulinic acid glioma resection study. Clinical article. J. Neurosurg. 2011, 114, 613–623. [Google Scholar] [CrossRef]
- Obermeier, B.; Verma, A.; Ransohoff, R.M. The blood-brain barrier. Handb. Clin. Neurol. 2016, 133, 39–59. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, P. Das sauerstufbudurfnis des organismus. In Eine Farbenanalytische Studies; Hirschwald: Berlin, Germany, 1885; p. 167. [Google Scholar]
- Ehrlich, F. Ueber das natürliche Isomere des Leucins. Berichte Dtsch. Chem. Ges. 1904, 37, 1809–1840. [Google Scholar] [CrossRef]
- Goldmann, E.E. Die Aussere und Innere Skeretion des Gesunden Organismus im Lichte der “Vitalen Farbung”; Lauppsche: Tübingen, Germany, 1909. [Google Scholar]
- Reese, T.S.; Karnovsky, M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 1967, 34, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, P.; Cioni, C.; Toneatto, S.; Paccagnini, E. HIV-1 gp120 increases the permeability of rat brain endothelium cultures by a mechanism involving substance P. Aids 1998, 12, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Kustova, Y.; Grinberg, A.; Basile, A.S. Increased blood-brain barrier permeability in LP-BM5 infected mice is mediated by neuroexcitatory mechanisms. Brain Res. 1999, 839, 153–163. [Google Scholar] [CrossRef] [PubMed]
- St’astný, F.; Skultétyová, I.; Pliss, L.; Jezová, D. Quinolinic acid enhances permeability of rat brain microvessels to plasma albumin. Brain Res. Bull. 2000, 53, 415–420. [Google Scholar] [CrossRef]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef]
- Mo, F.; Pellerino, A.; Soffietti, R.; Ruda, R. Blood-Brain Barrier in Brain Tumors: Biology and Clinical Relevance. Int. J. Mol. Sci. 2021, 22, 12654. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.H.; Canney, M.; Carpentier, A.; Thanou, M.; Idbaih, A. Unveiling the enigma of the blood-brain barrier in glioblastoma: Current advances from preclinical and clinical studies. Curr. Opin. Oncol. 2023, 35, 522–528. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.M.; Taylor, J.W.; Wiencke, J.K.; Wrensch, M.R. Genetic and molecular epidemiology of adult diffuse glioma. Nat. Rev. Neurol. 2019, 15, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Reifenberger, G.; French, P.J.; Schweizer, L.; Weller, M.; Touat, M.; Niclou, S.P.; Euskirchen, P.; Haberler, C.; Hegi, M.E.; et al. EANO guideline on rational molecular testing of gliomas, glioneuronal, and neuronal tumors in adults for targeted therapy selection. Neuro-Oncology 2023, 25, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Yabo, Y.A.; Niclou, S.P.; Golebiewska, A. Cancer cell heterogeneity and plasticity: A paradigm shift in glioblastoma. Neuro-Oncology 2022, 24, 669–682. [Google Scholar] [CrossRef]
- White, K.; Connor, K.; Meylan, M.; Bougoüin, A.; Salvucci, M.; Bielle, F.; O’Farrell, A.C.; Sweeney, K.; Weng, L.; Bergers, G.; et al. Identification, validation and biological characterisation of novel glioblastoma tumour microenvironment subtypes: Implications for precision immunotherapy. Ann. Oncol. 2023, 34, 300–314. [Google Scholar] [CrossRef]
- Eisenbarth, D.; Wang, Y.A. Glioblastoma heterogeneity at single cell resolution. Oncogene 2023, 42, 2155–2165. [Google Scholar] [CrossRef]
- Larsson, I.; Dalmo, E.; Elgendy, R.; Niklasson, M.; Doroszko, M.; Segerman, A.; Jörnsten, R.; Westermark, B.; Nelander, S. Modeling glioblastoma heterogeneity as a dynamic network of cell states. Mol. Syst. Biol. 2021, 17, e10105. [Google Scholar] [CrossRef] [PubMed]
- So, J.S.; Kim, H.; Han, K.S. Mechanisms of Invasion in Glioblastoma: Extracellular Matrix, Ca(2+) Signaling, and Glutamate. Front. Cell Neurosci. 2021, 15, 663092. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Mittal, S.; Berens, M.E. Targeting adaptive glioblastoma: An overview of proliferation and invasion. Neuro-Oncology 2014, 16, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Claes, A.; Idema, A.J.; Wesseling, P. Diffuse glioma growth: A guerilla war. Acta Neuropathol. 2007, 114, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.; Friedl, P. Molecular mechanisms of cancer cell invasion and plasticity. Br. J. Dermatol. 2006, 154 (Suppl. 1), 11–15. [Google Scholar] [CrossRef] [PubMed]
- Scherer, H.J. Structural Development in Gliomas. Am. J. Cancer 1938, 34, 333–351. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, C.; Mansouri, S.; Mora, C.; Nassiri, F.; Suppiah, S.; Martino, J.; Zadeh, G.; Fernandez-Luna, J.L. Molecular and Clinical Insights into the Invasive Capacity of Glioblastoma Cells. J. Oncol. 2019, 2019, 1740763. [Google Scholar] [CrossRef]
- Fabian, C.; Han, M.; Bjerkvig, R.; Niclou, S.P. Novel facets of glioma invasion. Int. Rev. Cell Mol. Biol. 2021, 360, 33–64. [Google Scholar] [CrossRef]
- Beauchesne, P. Extra-neural metastases of malignant gliomas: Myth or reality? Cancers 2011, 3, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Lun, M.; Lok, E.; Gautam, S.; Wu, E.; Wong, E.T. The natural history of extracranial metastasis from glioblastoma multiforme. J. Neuro-Oncol. 2011, 105, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.D.; Rapp, M.; Schneiderhan, T.; Sabel, M.; Hayman, A.; Scherer, A.; Kröpil, P.; Budach, W.; Gerber, P.; Kretschmar, U.; et al. Glioblastoma multiforme metastasis outside the CNS: Three case reports and possible mechanisms of escape. J. Clin. Oncol. 2014, 32, e80–e84. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Holdhoff, M.; Ye, X.; Supko, J.G.; Nabors, L.B.; Desai, A.S.; Walbert, T.; Lesser, G.J.; Read, W.L.; Lieberman, F.S.; Lodge, M.A.; et al. Timed sequential therapy of the selective T-type calcium channel blocker mibefradil and temozolomide in patients with recurrent high-grade gliomas. Neuro-Oncology 2017, 19, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Eisele, G.; Wick, A.; Eisele, A.C.; Clément, P.M.; Tonn, J.; Tabatabai, G.; Ochsenbein, A.; Schlegel, U.; Neyns, B.; Krex, D.; et al. Cilengitide treatment of newly diagnosed glioblastoma patients does not alter patterns of progression. J. Neuro-Oncol. 2014, 117, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Tonn, J.C.; Kerkau, S.; Hanke, A.; Bouterfa, H.; Mueller, J.G.; Wagner, S.; Vince, G.H.; Roosen, K. Effect of synthetic matrix-metalloproteinase inhibitors on invasive capacity and proliferation of human malignant gliomas in vitro. Int. J. Cancer 1999, 80, 764–772. [Google Scholar] [CrossRef]
- Koutroulis, I.; Zarros, A.; Theocharis, S. The role of matrix metalloproteinases in the pathophysiology and progression of human nervous system malignancies: A chance for the development of targeted therapeutic approaches? Expert. Opin. Ther. Targets 2008, 12, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.A.; Ye, X.; Chamberlain, M.; Mikkelsen, T.; Batchelor, T.; Desideri, S.; Piantadosi, S.; Fisher, J.; Fine, H.A. Talampanel with standard radiation and temozolomide in patients with newly diagnosed glioblastoma: A multicenter phase II trial. J. Clin. Oncol. 2009, 27, 4155–4161. [Google Scholar] [CrossRef]
- Iwamoto, F.M.; Kreisl, T.N.; Kim, L.; Duic, J.P.; Butman, J.A.; Albert, P.S.; Fine, H.A. Phase 2 trial of talampanel, a glutamate receptor inhibitor, for adults with recurrent malignant gliomas. Cancer 2010, 116, 1776–1782. [Google Scholar] [CrossRef]
- Drappatz, J.; Norden, A.D.; Wen, P.Y. Therapeutic strategies for inhibiting invasion in glioblastoma. Expert. Rev. Neurother. 2009, 9, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, Z.; Herz, J.; Kipnis, J. Meningeal Lymphatics: From Anatomy to Central Nervous System Immune Surveillance. J. Immunol. 2020, 204, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Kivisäkk, P.; Mahad, D.J.; Callahan, M.K.; Trebst, C.; Tucky, B.; Wei, T.; Wu, L.; Baekkevold, E.S.; Lassmann, H.; Staugaitis, S.M.; et al. Human cerebrospinal fluid central memory CD4+ T cells: Evidence for trafficking through choroid plexus and meninges via P-selectin. Proc. Natl. Acad. Sci. USA 2003, 100, 8389–8394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, L.; Zhang, H.; Zhang, Y.; Ju, H.; Wang, X.; Ren, H.; Zhu, X.; Dong, Y. The immunosuppressive microenvironment and immunotherapy in human glioblastoma. Front. Immunol. 2022, 13, 1003651. [Google Scholar] [CrossRef]
- Gustafson, M.P.; Lin, Y.; New, K.C.; Bulur, P.A.; O’Neill, B.P.; Gastineau, D.A.; Dietz, A.B. Systemic immune suppression in glioblastoma: The interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro-Oncology 2010, 12, 631–644. [Google Scholar] [CrossRef]
- Parney, I.F. Basic concepts in glioma immunology. Adv. Exp. Med. Biol. 2012, 746, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Andaloussi, A.E.; Han, Y.; Lesniak, M.S. Progression of intracranial glioma disrupts thymic homeostasis and induces T-cell apoptosis in vivo. Cancer Immunol. Immunother. 2008, 57, 1807–1816. [Google Scholar] [CrossRef]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- Ayasoufi, K.; Pfaller, C.K.; Evgin, L.; Khadka, R.H.; Tritz, Z.P.; Goddery, E.N.; Fain, C.E.; Yokanovich, L.T.; Himes, B.T.; Jin, F.; et al. Brain cancer induces systemic immunosuppression through release of non-steroid soluble mediators. Brain 2020, 143, 3629–3652. [Google Scholar] [CrossRef]
- Rodrigues, J.C.; Gonzalez, G.C.; Zhang, L.; Ibrahim, G.; Kelly, J.J.; Gustafson, M.P.; Lin, Y.; Dietz, A.B.; Forsyth, P.A.; Yong, V.W.; et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro-Oncology 2010, 12, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Noell, S.; Fallier-Becker, P.; Mack, A.F.; Wolburg-Buchholz, K. The disturbed blood-brain barrier in human glioblastoma. Mol. Asp. Med. 2012, 33, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.G.; Campanati, L.; Righy, C.; D’Andrea-Meira, I.; Spohr, T.C.; Porto-Carreiro, I.; Pereira, C.M.; Balca-Silva, J.; Kahn, S.A.; DosSantos, M.F.; et al. Gliomas and the vascular fragility of the blood brain barrier. Front. Cell Neurosci. 2014, 8, 418. [Google Scholar] [CrossRef]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef]
- Chen, Z.; Mou, L.; Pan, Y.; Feng, C.; Zhang, J.; Li, J. CXCL8 Promotes Glioma Progression By Activating The JAK/STAT1/HIF-1alpha/Snail Signaling Axis. Onco Targets Ther. 2019, 12, 8125–8138. [Google Scholar] [CrossRef] [PubMed]
- Groblewska, M.; Litman-Zawadzka, A.; Mroczko, B. The Role of Selected Chemokines and Their Receptors in the Development of Gliomas. Int. J. Mol. Sci. 2020, 21, 3704. [Google Scholar] [CrossRef]
- Oushy, S.; Hellwinkel, J.E.; Wang, M.; Nguyen, G.J.; Gunaydin, D.; Harland, T.A.; Anchordoquy, T.J.; Graner, M.W. Glioblastoma multiforme-derived extracellular vesicles drive normal astrocytes towards a tumour-enhancing phenotype. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160477. [Google Scholar] [CrossRef]
- Simon, T.; Jackson, E.; Giamas, G. Breaking through the glioblastoma micro-environment via extracellular vesicles. Oncogene 2020, 39, 4477–4490. [Google Scholar] [CrossRef] [PubMed]
- Constam, D.B.; Philipp, J.; Malipiero, U.V.; ten Dijke, P.; Schachner, M.; Fontana, A. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J. Immunol. 1992, 148, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Huettner, C.; Paulus, W.; Roggendorf, W. Messenger RNA expression of the immunosuppressive cytokine IL-10 in human gliomas. Am. J. Pathol. 1995, 146, 317–322. [Google Scholar] [PubMed]
- Takeshima, H.; Kuratsu, J.; Takeya, M.; Yoshimura, T.; Ushio, Y. Expression and localization of messenger RNA and protein for monocyte chemoattractant protein-1 in human malignant glioma. J. Neurosurg. 1994, 80, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sinha, S.; Jiang, X.; Murphy, L.; Fitch, S.; Wilson, C.; Grant, G.; Yang, F. Matrix Stiffness Modulates Patient-Derived Glioblastoma Cell Fates in Three-Dimensional Hydrogels. Tissue Eng. Part A 2021, 27, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Platten, M.; Weller, M. Glioma cell invasion: Regulation of metalloproteinase activity by TGF-beta. J. Neuro-Oncol. 2001, 53, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, H.K. Current Understanding of Hypoxia in Glioblastoma Multiforme and Its Response to Immunotherapy. Cancers 2022, 14, 1176. [Google Scholar] [CrossRef]
- Hu, M.; Li, Y.; Lu, Y.; Wang, M.; Li, Y.; Wang, C.; Li, Q.; Zhao, H. The regulation of immune checkpoints by the hypoxic tumor microenvironment. PeerJ 2021, 9, e11306. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strahle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-Oncology 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.A.; Pomella, N.; Rosser, G.; Guglielmi, L.; Vinel, C.; Millner, T.O.; Rees, J.; Aley, N.; Sheer, D.; Wei, J.; et al. Microglia promote glioblastoma via mTOR-mediated immunosuppression of the tumour microenvironment. Embo J 2020, 39, e103790. [Google Scholar] [CrossRef] [PubMed]
- Suzumura, A.; Sawada, M.; Yamamoto, H.; Marunouchi, T. Transforming growth factor-beta suppresses activation and proliferation of microglia in vitro. J. Immunol. 1993, 151, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro-Oncology 2006, 8, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Kilian, M.; Sheinin, R.; Tan, C.L.; Friedrich, M.; Krämer, C.; Kaminitz, A.; Sanghvi, K.; Lindner, K.; Chih, Y.-C.; Cichon, F.; et al. MHC class II-restricted antigen presentation is required to prevent dysfunction of cytotoxic T cells by blood-borne myeloids in brain tumors. Cancer Cell 2023, 41, 235–251. [Google Scholar] [CrossRef]
- Nagaraj, S.; Gabrilovich, D.I. Myeloid-derived suppressor cells. Adv. Exp. Med. Biol. 2007, 601, 213–223. [Google Scholar] [CrossRef]
- Dubinski, D.; Wölfer, J.; Hasselblatt, M.; Schneider-Hohendorf, T.; Bogdahn, U.; Stummer, W.; Wiendl, H.; Grauer, O.M. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro-Oncology 2016, 18, 807–818. [Google Scholar] [CrossRef]
- Crane, C.A.; Ahn, B.J.; Han, S.J.; Parsa, A.T. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: Implications for immunotherapy. Neuro-Oncology 2012, 14, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Abou-Ghazal, M.; Reina-Ortiz, C.; Yang, D.S.; Sun, W.; Qiao, W.; Hiraoka, N.; Fuller, G.N. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin. Cancer Res. 2008, 14, 5166–5172. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro-Oncology 2010, 12, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.J.; Giles, A.J.; Gilbert, M. T lymphocyte-targeted immune checkpoint modulation in glioma. J. Immunother. Cancer 2020, 8, e000379. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Yagita, H.; Ikawa, Y.; Oyaizu, N. Fas drives cell cycle progression in glioma cells via extracellular signal-regulated kinase activation. Cancer Res. 2000, 60, 1766–1772. [Google Scholar] [PubMed]
- Wischhusen, J.; Jung, G.; Radovanovic, I.; Beier, C.; Steinbach, J.P.; Rimner, A.; Huang, H.; Schulz, J.B.; Ohgaki, H.; Aguzzi, A.; et al. Identification of CD70-mediated apoptosis of immune effector cells as a novel immune escape pathway of human glioblastoma. Cancer Res. 2002, 62, 2592–2599. [Google Scholar] [PubMed]
- Chahlavi, A.; Rayman, P.; Richmond, A.L.; Biswas, K.; Zhang, R.; Vogelbaum, M.; Tannenbaum, C.; Barnett, G.; Finke, J.H. Glioblastomas induce T-lymphocyte death by two distinct pathways involving gangliosides and CD70. Cancer Res. 2005, 65, 5428–5438. [Google Scholar] [CrossRef]
- Pham, T.; Roth, S.; Kong, J.; Guerra, G.; Narasimhan, V.; Pereira, L.; Desai, J.; Heriot, A.; Ramsay, R. An Update on Immunotherapy for Solid Tumors: A Review. Ann. Surg. Oncol. 2018, 25, 3404–3412. [Google Scholar] [CrossRef]
- Philip, M.; Schietinger, A. CD8(+) T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 2022, 22, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthelemy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Zakharia, Y.; Johnson, T.S.; Colman, H.; Vahanian, N.N.; Link, C.J.; Kennedy, E.; Sadek, R.F.; Kong, F.M.; Vender, J.; Munn, D.; et al. A phase I/II study of the combination of indoximod and temozolomide for adult patients with temozolomide-refractory primary malignant brain tumors. J. Clin. Oncol. 2014, 32 (Suppl. 15), TPS2107. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; Lopez-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inoges, S.; de Andrea, C.; Lopez-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Sanborn, R.E.; Pishvaian, M.J.; Callahan, M.K.; Weise, A.; Sikic, B.I.; Rahma, O.; Cho, D.C.; Rizvi, N.A.; Sznol, M.; Lutzky, J.; et al. Safety, tolerability and efficacy of agonist anti-CD27 antibody (varlilumab) administered in combination with anti-PD-1 (nivolumab) in advanced solid tumors. J. Immunother. Cancer 2022, 10, e005147. [Google Scholar] [CrossRef]
- Jensen, C.; Maarup, S.B.; Poulsen, H.S.; Hasselbalch, B.; Karsdal, M.A.; Svane, I.M.; Lassen, U.N.; Willumsen, N. Indirect assessment of tumor-infiltrating lymphocyte activity in serum for predicting outcome in patients with glioblastoma treated with immunotherapy in the recurrent setting. J. Clin. Oncol. 2022, 40 (Suppl. 16), 2059. [Google Scholar] [CrossRef]
- Sim, H.-W.; Lwin, Z.; Barnes, E.; McDonald, K.; Yip, S.; Verhaak, R.; Heimberger, A.; Hall, M.; Wong, M.; Jennens, R.; et al. CTIM-24. Nutmeg: A randomized phase II study of nivolumab and temozolomide versus temozolomide alone in newly diagnosed elderly patients with glioblastoma. Neuro-Oncology 2022, 24 (Suppl. 7), vii65. [Google Scholar] [CrossRef]
- Ahluwalia, M.; Peereboom, D.; Schilero, C.; Forst, D.; Wong, E.; Wen, P.; Reardon, D. RBTT-01. randomized phase 2 open label study of nivolumab plus standard dose bevacizumab versus nivolumab plus low dose bevacizumab in recurrent glioblastoma. Neuro-Oncology 2018, 20 (Suppl. 6), vi234. [Google Scholar] [CrossRef][Green Version]
- Ahluwalia, M.S.; Rauf, Y.; Li, H.; Wen, P.Y.; Peereboom, D.M.; Reardon, D.A. Randomized phase 2 study of nivolumab (nivo) plus either standard or reduced dose bevacizumab (bev) in recurrent glioblastoma (rGBM). J. Clin. Oncol. 2021, 39 (Suppl. 15), 2015. [Google Scholar] [CrossRef]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; van den Bent, M.; Bähr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro-Oncology 2023, 25, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro-Oncology 2022, 24, 1935–1949. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Kim, T.M.; Frenel, J.S.; Simonelli, M.; Lopez, J.; Subramaniam, D.S.; Siu, L.L.; Wang, H.; Krishnan, S.; Stein, K.; et al. Treatment with pembrolizumab in programmed death ligand 1-positive recurrent glioblastoma: Results from the multicohort phase 1 KEYNOTE-028 trial. Cancer 2021, 127, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.K.; Cher, L.; Bowyer, S.; Gan, H.K.; Long, A.P.; Balasubramanian, A.; Lee, S.Y.; Lee, W.S.; Yoo, J.-S. Phase Ib study of olinvacimab (O) with pembrolizumab (P) in patients with recurrent glioblastoma (rGBM). J. Clin. Oncol. 2020, 38 (Suppl. 15), e14545. [Google Scholar] [CrossRef]
- Sahebjam, S.; Forsyth, P.; Tran, N.; Mokhtari, S.; Arrington, J.; Jaglal, M.; Etame, A.; Liu, J.; Wicklund, M.; Gatewood, T.; et al. ATIM-08. A phase I trial of pembrolizumab and vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma (NCT03426891). Neuro-Oncology 2018, 20 (Suppl. 6), vi2. [Google Scholar] [CrossRef][Green Version]
- Hwang, H.; Huang, J.; Khaddour, K.; Butt, O.H.; Ansstas, G.; Chen, J.; Katumba, R.G.; Kim, A.H.; Leuthardt, E.C.; Campian, J.L. Prolonged response of recurrent IDH-wild-type glioblastoma to laser interstitial thermal therapy with pembrolizumab. CNS Oncol. 2022, 11, Cns81. [Google Scholar] [CrossRef] [PubMed]
- Campian, J.; Butt, O.; Ghinaseddin, A.; Rahman, M.; Chheda, M.; Johanns, T.; Ansstas, G.; Huang, J.; Liu, J.; Talcott, G.; et al. Ctim-26. Phase I/II study of the combination of pembrolizumab (Mk-3475) and laser interstitial thermal therapy (litt) in recurrent glioblastoma. Neuro-Oncology 2021, 23 (Suppl. 6), vi56. [Google Scholar] [CrossRef]
- Sloan, A.E.; Rogers, L.R.; Machtay, M. Phase I/II study of laser interstitial thermotherapy (LITT) combined with checkpoint inhibitor for recurrent glioblastoma (rGBM). J. Clin. Oncol. 2018, 36 (Suppl. 15), TPS2074. [Google Scholar] [CrossRef]
- Giordano, F.A.; Layer, J.P.; Leonardelli, S.; Friker, L.L.; Seidel, C.; Schaub, C.; Turiello, R.; Sperk, E.; Grau, F.; Paech, D.; et al. Radiotherapy and olaptesed pegol (NOX-A12) in partially resected or biopsy-only MGMT-unmethylated glioblastoma: Interim data from the German multicenter phase 1/2 GLORIA trial. J. Clin. Oncol. 2022, 40 (Suppl. 16), 2050. [Google Scholar] [CrossRef]
- Giordano, F.A.; Layer, J.P.; Leonardelli, S.; Friker, L.L.; Turiello, R.; Corvino, D.; Zeyen, T.; Schaub, C.; Mueller, W.; Sperk, E.; et al. Potential predictive biomarker for response to radiotherapy and CXCL12 inhibition in glioblastoma in the phase I/II GLORIA trial. J. Clin. Oncol. 2023, 41 (Suppl. 16), 2048. [Google Scholar] [CrossRef]
- Baldini, C.; Cassier, P.A.; Delord, J.-P.; Simonelli, M.; Touat, M.; Yao, L.; Duic, J.P.; Gozman, A.; Marabelle, A. CTIM-03. pembrolizumab monotherapy for microsatellite instability-high (MSI-H) or mismatch repair deficient (DMMR) recurrent gliomas: Results from the multicohort phase 2 keynote-158 study. Neuro-Oncology 2022, 24 (Suppl. 7), vii59–vii60. [Google Scholar] [CrossRef]
- Reardon, D.A.; Nayak, L.; Peters, K.B.; Clarke, J.L.; Jordan, J.T.; Groot, J.F.D.; Nghiemphu, P.L.; Kaley, T.J.; Colman, H.; Gaffey, S.C.; et al. Phase II study of pembrolizumab or pembrolizumab plus bevacizumab for recurrent glioblastoma (rGBM) patients. J. Clin. Oncol. 2018, 36, 2006. [Google Scholar] [CrossRef]
- Nayak, L.; Molinaro, A.M.; Peters, K.; Clarke, J.L.; Jordan, J.T.; de Groot, J.; Nghiemphu, L.; Kaley, T.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057. [Google Scholar] [CrossRef]
- Iwamoto, F.; Tanguturi, S.; Desai, A.; Nayak, L.; Uhlmann, E.; Wang, T.; Lustig, R.; Hertan, L.; Bagley, S.; Hayden, J.; et al. CTIM-18. PHASE 2 STUDY OF PD-1 BLOCKADE WITH PEMBROLIZUMAB PLUS RE-IRRADIATION FOR RECURRENT GLIOBLASTOMA (RGBM) (NCT03661723). Neuro-Oncology 2022, 24 (Suppl. 7), vii63–vii64. [Google Scholar] [CrossRef]
- Tran, D.D.; Ghiaseddin, A.P.; Chen, D.D.; Le, S.B. Final analysis of 2-THE-TOP: A phase 2 study of TTFields (Optune) plus pembrolizumab plus maintenance temozolomide (TMZ) in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2023, 41 (Suppl. 16), 2024. [Google Scholar] [CrossRef]
- de Groot, J.; Penas-Prado, M.; Alfaro-Munoz, K.; Hunter, K.; Pei, B.L.; O’Brien, B.; Weathers, S.P.; Loghin, M.; Kamiya Matsouka, C.; Yung, W.K.A.; et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro-Oncology 2020, 22, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Webb, M.; Burns, T.C.; Twohy, E.; Sener, U.; Kizilbash, S.H.; Ruff, M.W.; Uhm, J.H.; Galanis, E.; D’Andre, S.D.; Riviere-Cazaux, C.; et al. Efficacy and safety study of neoadjuvant efineptakin alfa (NT-I7) and pembrolizumab in recurrent glioblastoma. J. Clin. Oncol. 2023, 41 (Suppl. 16), TPS2085. [Google Scholar] [CrossRef]
- Lwin, Z.; Gomez-Roca, C.; Saada-Bouzid, E.; Yanez, E.; Muñoz, F.L.; Im, S.A.; Castanon, E.; Senellart, H.; Graham, D.; Voss, M.; et al. LBA41 LEAP-005: Phase II study of lenvatinib (len) plus pembrolizumab (pembro) in patients (pts) with previously treated advanced solid tumours. Ann. Oncol. 2020, 31, S1170. [Google Scholar] [CrossRef]
- Jacques, F.H.; Nicholas, G.; Lorimer, I.A.J.; Sikati Foko, V.; Prevost, J.; Dumais, N.; Milne, K.; Nelson, B.H.; Woulfe, J.; Jansen, G.; et al. Avelumab in newly diagnosed glioblastoma. Neuro-Oncol. Adv. 2021, 3, vdab118. [Google Scholar] [CrossRef]
- Tiu, C.; Yau, W.H.; Welsh, L.C.; Jones, T.L.; Zachariou, A.; Prout, T.; Parmar, M.; Turner, A.J.; Daly, R.W.; Yap, C.; et al. Abstract CT093: Preliminary evidence of antitumor activity of Ipatasertib (Ipat) and Atezolizumab (A) in glioblastoma (GBM) patients (pts) with PTEN loss in the Phase 1 Ice-CAP trial (NCT03673787). Cancer Res. 2023, 83 (Suppl. 8), CT093. [Google Scholar] [CrossRef]
- Weathers, S.-P.S.; Kamiya-Matsuoka, C.; Harrison, R.A.; Liu, D.D.; Dervin, S.; Yun, C.; Loghin, M.E.; Penas-Prado, M.; Majd, N.; Yung, W.K.A.; et al. Phase I/II study to evaluate the safety and clinical efficacy of atezolizumab (atezo; aPDL1) in combination with temozolomide (TMZ) and radiation in patients with newly diagnosed glioblastoma (GBM). J. Clin. Oncol. 2020, 38 (Suppl. 15), 2511. [Google Scholar] [CrossRef]
- Reardon, D.A.; Kaley, T.J.; Dietrich, J.; Clarke, J.L.; Dunn, G.; Lim, M.; Cloughesy, T.F.; Gan, H.K.; Park, A.J.; Schwarzenberger, P.; et al. Phase II study to evaluate safety and efficacy of MEDI4736 (durvalumab) + radiotherapy in patients with newly diagnosed unmethylated MGMT glioblastoma (new unmeth GBM). J. Clin. Oncol. 2019, 37 (Suppl. 15), 2032. [Google Scholar] [CrossRef]
- Ranjan, S.; Quezado, M.; Garren, N.; Boris, L.; Siegel, C.; Lopes Abath Neto, O.; Theeler, B.J.; Park, D.M.; Nduom, E.; Zaghloul, K.A.; et al. Clinical decision making in the era of immunotherapy for high grade-glioma: Report of four cases. BMC Cancer 2018, 18, 239. [Google Scholar] [CrossRef]
- Duerinck, J.; Schwarze, J.K.; Awada, G.; Tijtgat, J.; Vaeyens, F.; Bertels, C.; Geens, W.; Klein, S.; Seynaeve, L.; Cras, L.; et al. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: A phase I clinical trial. J. Immunother. Cancer 2021, 9, e002296. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Omuro, A.; Reardon, D.A.; Sampson, J.H.; Baehring, J.; Sahebjam, S.; Cloughesy, T.F.; Chalamandaris, A.-G.; Potter, V.; Butowski, N.; Lim, M. Nivolumab plus radiotherapy with or without temozolomide in newly diagnosed glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro-Oncol. Adv. 2022, 4, vdac025. [Google Scholar] [CrossRef] [PubMed]
- Bagley, S.J.; Mathew, D.; Desai, A.S.; Chen, K.; Long, Q.; Shabason, J.E.; Lustig, R.A.; Kurtz, G.; Alonso-Basanta, M.; Maloney, E.; et al. PD1 inhibition and GITR agonism in combination with fractionated stereotactic radiotherapy for the treatment of recurrent glioblastoma: A phase 2, multi-arm study. J. Clin. Oncol. 2023, 41 (Suppl. 16), 2004. [Google Scholar] [CrossRef]
- Lukas, R.; Sachdev, S.; Kumthekar, P.; Dixit, K.; Grimm, S.; Gondi, V.; Sharp, L.; Lezon, R.; James, D.; Lesniak, M.; et al. Ctim-12. a phase 1 trial of immunoradiotherapy with the ido enzyme inhibitor (bms-986205) and nivolumab in patients with newly diagnosed mgmt promoter unmethylated idhwt glioblastoma. Neuro-Oncology 2021, 23 (Suppl. 6), vi51–vi52. [Google Scholar] [CrossRef]
- Daud, A.; Saleh, M.N.; Hu, J.; Bleeker, J.S.; Riese, M.J.; Meier, R.; Zhou, L.; Serbest, G.; Lewis, K.D. Epacadostat plus nivolumab for advanced melanoma: Updated phase 2 results of the ECHO-204 study. J. Clin. Oncol. 2018, 36, 9511. [Google Scholar] [CrossRef]
- Lynes, J.; Jackson, S.; Sanchez, V.; Dominah, G.; Wang, X.; Kuek, A.; Hayes, C.P.; Benzo, S.; Scott, G.C.; Chittiboina, P.; et al. Cytokine Microdialysis for Real-Time Immune Monitoring in Glioblastoma Patients Undergoing Checkpoint Blockade. Neurosurgery 2019, 84, 945–953. [Google Scholar] [CrossRef]
- Lim, M.; Ye, X.; Piotrowski, A.F.; Desai, A.S.; Ahluwalia, M.S.; Walbert, T.; Fisher, J.D.; Desideri, S.; Nabors, L.B.; Wen, P.Y.; et al. Updated safety phase I trial of anti-LAG-3 alone and in combination with anti-PD-1 in patients with recurrent GBM. J. Clin. Oncol. 2020, 38 (Suppl. 15), 2512. [Google Scholar] [CrossRef]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Orpilla, J.; Moughon, D.; Shin, N.; Sedighim, S.; Yong, W.H.; Li, G.; Cloughesy, T.F.; et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 2016, 1, e87059. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Lim, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.; Ashby, L.; Ansstas, G.; Baehring, J.; Taylor, J.; et al. CTIM-25. A randomized phase 3 study of nivolumab or placebo combined with radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma with methylated mgmt promoter: Checkmate 548. Neuro-Oncology 2021, 23 (Suppl. 6), vi55–vi56. [Google Scholar] [CrossRef]
- Lombardi, G.; Barresi, V.; Indraccolo, S.; Simbolo, M.; Fassan, M.; Mandruzzato, S.; Simonelli, M.; Caccese, M.; Pizzi, M.; Fassina, A.; et al. Pembrolizumab Activity in Recurrent High-Grade Gliomas with Partial or Complete Loss of Mismatch Repair Protein Expression: A Monocentric, Observational and Prospective Pilot Study. Cancers 2020, 12, 2283. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wohrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-Oncology 2015, 17, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Carter, T.; Shaw, H.; Cohn-Brown, D.; Chester, K.; Mulholland, P. Ipilimumab and Bevacizumab in Glioblastoma. Clin. Oncol. 2016, 28, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A. Immune-checkpoint inhibitors for glioblastoma: What have we learned? Arq. Neuro-Psiquiatr. 2022, 80 (Suppl. 1), 266–269. [Google Scholar] [CrossRef]
- Lim, M.; Ye, X.; Piotrowski, A.F.; Desai, A.S.; Ahluwalia, M.S.; Walbert, T.; Fisher, J.D.; Desideri, S.; Belcaid, Z.; Jackson, C.; et al. Updated phase I trial of anti-LAG-3 or anti-CD137 alone and in combination with anti-PD-1 in patients with recurrent GBM. J. Clin. Oncol. 2019, 37, 2017. [Google Scholar] [CrossRef]
- Arrieta, V.A.; Dmello, C.; McGrail, D.J.; Brat, D.J.; Lee-Chang, C.; Heimberger, A.B.; Chand, D.; Stupp, R.; Sonabend, A.M. Immune checkpoint blockade in glioblastoma: From tumor heterogeneity to personalized treatment. J. Clin. Investig. 2023, 133, e163447. [Google Scholar] [CrossRef] [PubMed]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro-Oncology 2017, 19, 1047–1057. [Google Scholar] [CrossRef]
- Leuthardt, E.C.; Duan, C.; Kim, M.J.; Campian, J.L.; Kim, A.H.; Miller-Thomas, M.M.; Shimony, J.S.; Tran, D.D. Hyperthermic Laser Ablation of Recurrent Glioblastoma Leads to Temporary Disruption of the Peritumoral Blood Brain Barrier. PLoS ONE 2016, 11, e0148613. [Google Scholar] [CrossRef]
- Salehi, A.; Paturu, M.R.; Patel, B.; Cain, M.D.; Mahlokozera, T.; Yang, A.B.; Lin, T.H.; Leuthardt, E.C.; Yano, H.; Song, S.K.; et al. Therapeutic enhancement of blood-brain and blood-tumor barriers permeability by laser interstitial thermal therapy. Neuro-Oncol. Adv. 2020, 2, vdaa071. [Google Scholar] [CrossRef]
- Frederico, S.C.; Hancock, J.C.; Brettschneider, E.E.S.; Ratnam, N.M.; Gilbert, M.R.; Terabe, M. Making a Cold Tumor Hot: The Role of Vaccines in the Treatment of Glioblastoma. Front. Oncol. 2021, 11, 672508. [Google Scholar] [CrossRef]
- Khong, H.; Overwijk, W.W. Adjuvants for peptide-based cancer vaccines. J. Immunother. Cancer 2016, 4, 56. [Google Scholar] [CrossRef]
- Rudnick, J.D.; Fink, K.L.; Landolfi, J.C.; Markert, J.; Piccioni, D.E.; Glantz, M.J.; Swanson, S.J.; Gringeri, A.; Yu, J. Immunological targeting of CD133 in recurrent glioblastoma: A multi-center phase I translational and clinical study of autologous CD133 dendritic cell immunotherapy. J. Clin. Oncol. 2017, 35, 2059. [Google Scholar] [CrossRef]
- Reap, E.A.; Suryadevara, C.M.; Batich, K.A.; Sanchez-Perez, L.; Archer, G.E.; Schmittling, R.J.; Norberg, P.K.; Herndon, J.E., 2nd; Healy, P.; Congdon, K.L.; et al. Dendritic Cells Enhance Polyfunctionality of Adoptively Transferred T Cells That Target Cytomegalovirus in Glioblastoma. Cancer Res. 2018, 78, 256–264. [Google Scholar] [CrossRef]
- Vlahovic, G.; Archer, G.E.; Reap, E.; Desjardins, A.; Peters, K.B.; Randazzo, D.; Healy, P.; Herndon, J.E.; Friedman, A.H.; Friedman, H.S.; et al. Phase I trial of combination of antitumor immunotherapy targeted against cytomegalovirus (CMV) plus regulatory T-cell inhibition in patients with newly-diagnosed glioblastoma multiforme (GBM). J. Clin. Oncol. 2016, 34 (Suppl. 15), e13518. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E., 2nd; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Ghiaseddin, A.; Deleyrolle, P.; Peters, K.B.; Archer, G.; Sampson, J.; Mitchell, D. CTIM-07 – Phase II randomized, blinded, placebo-controlled trial testing pp65 CMV mRNA dendritic cell vaccine and tetanus-diphtheria toxoid for newly diagnosed GBM (ATTAC II, NCT02465268). Neuro-Oncology 2022, 24 (Suppl. 7), vii60–vii61. [Google Scholar] [CrossRef]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E., 2nd; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303. [Google Scholar] [CrossRef]
- Sampson, J.H.; Batich, K.A.; Mitchell, D.A.; Herndon, J.E.; Broadwater, G.; Healy, P.; Sanchez-Perez, L.; Nair, S.; Congdon, K.; Norberg, P.; et al. Reproducibility of outcomes in sequential trials using CMV-targeted dendritic cell vaccination for glioblastoma. J. Clin. Oncol. 2022, 40 (Suppl. 16), 2005. [Google Scholar] [CrossRef]
- Sampson, J.H.; Schmittling, R.J.; Archer, G.E.; Congdon, K.L.; Nair, S.K.; Reap, E.A.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; Herndon, J.E., 2nd; et al. A pilot study of IL-2Rα blockade during lymphopenia depletes regulatory T-cells and correlates with enhanced immunity in patients with glioblastoma. PLoS ONE 2012, 7, e31046. [Google Scholar] [CrossRef]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; O’Rourke, D.M.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; et al. Rindopepimut with Bevacizumab for Patients with Relapsed EGFRvIII-Expressing Glioblastoma (ReACT): Results of a Double-Blind Randomized Phase II Trial. Clin. Cancer Res. 2020, 26, 1586–1594. [Google Scholar] [CrossRef]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro-Oncology 2015, 17, 854–861. [Google Scholar] [CrossRef]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E., 2nd; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef]
- Sampson, J.H.; Aldape, K.D.; Archer, G.E.; Coan, A.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-Oncology 2011, 13, 324–333. [Google Scholar] [CrossRef]
- Schmittling, R.J.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.; Pegram, C.; Herndon, J.E., 2nd; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Detection of humoral response in patients with glioblastoma receiving EGFRvIII-KLH vaccines. J. Immunol. Methods 2008, 339, 74–81. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Crane, C.A.; Han, S.J.; Ahn, B.; Oehlke, J.; Kivett, V.; Fedoroff, A.; Butowski, N.; Chang, S.M.; Clarke, J.; Berger, M.S.; et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin. Cancer Res. 2013, 19, 205–214. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; McDermott, M.W.; et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro-Oncology 2014, 16, 274–279. [Google Scholar] [CrossRef]
- Bloch, O.; Lim, M.; Sughrue, M.E.; Komotar, R.J.; Abrahams, J.M.; O’Rourke, D.M.; D’Ambrosio, A.; Bruce, J.N.; Parsa, A.T. Autologous Heat Shock Protein Peptide Vaccination for Newly Diagnosed Glioblastoma: Impact of Peripheral PD-L1 Expression on Response to Therapy. Clin. Cancer Res. 2017, 23, 3575–3584. [Google Scholar] [CrossRef]
- Bloch, O.; Shi, Q.; Anderson, S.K.; Knopp, M.; Raizer, J.; Clarke, J.; Waziri, A.; Colman, H.; Bruce, J.; Olson, J.J.; et al. ATIM-14. Alliance A071101: A phase II randomized trial comparing the efficacy of heat shock protein peptide complex-96 (HSPPC-96) vaccine given with bevacizumab versus bevacizumab alone in the treatment of surgically resectable recurrent glioblastoma. Neuro-Oncology 2017, 19 (Suppl. 6), vi29. [Google Scholar] [CrossRef]
- Carpentier, A.F.; Verlut, C.; Ghiringhelli, F.; Bronnimann, C.; Ursu, R.; Fumet, J.D.; Gherga, E.; Lefort, F.; Belin, C.; Vernerey, D.; et al. Anti-telomerase vaccine in patients with newly diagnosed, unmethylated MGMT glioblastoma: A phase II study. J. Clin. Oncol. 2023, 41 (Suppl. 16), 2005. [Google Scholar] [CrossRef]
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol. Immunother. 2016, 65, 1339–1352. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Ciesielski, M.; Abad, A.; Reardon, D.; Aiken, R.; Barbaro, M.; Sinicrope, K.; Peereboom, D.M.; Odia, Y.; Brenner, A.; et al. P07.09.B A randomized phase 2B study of survivin vaccine survaxm plus adjuvant temozolomide for newly-diagnosed glioblastoma (survive). Neuro-Oncology 2023, 25 (Suppl. 2), ii52–ii53. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Figel, S.A.; Mechtler, L.L.; Peereboom, D.M.; Hutson, A.D.; Withers, H.G.; et al. Phase IIa Study of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2023, 41, 1453–1465. [Google Scholar] [CrossRef]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.J.; Glantz, M.; Peereboom, D.M.; et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin. Cancer Res. 2019, 25, 5799–5807. [Google Scholar] [CrossRef]
- Rampling, R.; Peoples, S.; Mulholland, P.J.; James, A.; Al-Salihi, O.; Twelves, C.J.; McBain, C.; Jefferies, S.; Jackson, A.; Stewart, W.; et al. A Cancer Research UK First Time in Human Phase I Trial of IMA950 (Novel Multipeptide Therapeutic Vaccine) in Patients with Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2016, 22, 4776–4785. [Google Scholar] [CrossRef]
- Migliorini, D.; Dutoit, V.; Allard, M.; Grandjean Hallez, N.; Marinari, E.; Widmer, V.; Philippin, G.; Corlazzoli, F.; Gustave, R.; Kreutzfeldt, M.; et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro-Oncology 2019, 21, 923–933. [Google Scholar] [CrossRef]
- Boydell, E.; Marinari, E.; Migliorini, D.; Dietrich, P.Y.; Patrikidou, A.; Dutoit, V. Exploratory Study of the Effect of IMA950/Poly-ICLC Vaccination on Response to Bevacizumab in Relapsing High-Grade Glioma Patients. Cancers 2019, 11, 464. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brem, S.; Desai, A.S.; Bagley, S.J.; Kurz, S.C.; Fuente, M.I.D.L.; Nagpal, S.; Welch, M.R.; Hormigo, A.; Forsyth, P.A.J.; et al. Intramuscular (IM) INO-5401 + INO-9012 with electroporation (EP) in combination with cemiplimab (REGN2810) in newly diagnosed glioblastoma. J. Clin. Oncol. 2022, 40 (Suppl. 16), 2004. [Google Scholar] [CrossRef]
- Olin, M.R.; Low, W.; McKenna, D.H.; Haines, S.J.; Dahlheimer, T.; Nascene, D.; Gustafson, M.P.; Dietz, A.B.; Clark, H.B.; Chen, W.; et al. Vaccination with dendritic cells loaded with allogeneic brain tumor cells for recurrent malignant brain tumors induces a CD4+IL17+ response. J. ImmunoTherapy Cancer 2014, 2, 4. [Google Scholar] [CrossRef]
- Prins, R.M.; Wang, X.; Soto, H.; Young, E.; Lisiero, D.N.; Fong, B.; Everson, R.; Yong, W.H.; Lai, A.; Li, G.; et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J. Immunother. 2013, 36, 152–157. [Google Scholar] [CrossRef]
- Hu, J.L.; Omofoye, O.A.; Rudnick, J.D.; Kim, S.; Tighiouart, M.; Phuphanich, S.; Wang, H.; Mazer, M.; Ganaway, T.; Chu, R.M.; et al. A Phase I Study of Autologous Dendritic Cell Vaccine Pulsed with Allogeneic Stem-like Cell Line Lysate in Patients with Newly Diagnosed or Recurrent Glioblastoma. Clin. Cancer Res. 2022, 28, 689–696. [Google Scholar] [CrossRef]
- Erhart, F.; Buchroithner, J.; Reitermaier, R.; Fischhuber, K.; Klingenbrunner, S.; Sloma, I.; Hibsh, D.; Kozol, R.; Efroni, S.; Ricken, G.; et al. Immunological analysis of phase II glioblastoma dendritic cell vaccine (Audencel) trial: Immune system characteristics influence outcome and Audencel up-regulates Th1-related immunovariables. Acta Neuropathol. Commun. 2018, 6, 135. [Google Scholar] [CrossRef]
- Inogés, S.; Tejada, S.; de Cerio, A.L.; Gállego Pérez-Larraya, J.; Espinós, J.; Idoate, M.A.; Domínguez, P.D.; de Eulate, R.G.; Aristu, J.; Bendandi, M.; et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J. Transl. Med. 2017, 15, 104. [Google Scholar] [CrossRef]
- Fadul, C.E.; Fisher, J.L.; Hampton, T.H.; Lallana, E.C.; Li, Z.; Gui, J.; Szczepiorkowski, Z.M.; Tosteson, T.D.; Rhodes, C.H.; Wishart, H.A.; et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J. Immunother. 2011, 34, 382–389. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination with Extension of Survival Among Patients with Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef]
- Wick, W.; Dietrich, P.-Y.; Kuttruff, S.; Hilf, N.; Frenzel, K.; Admon, A.; Burg, S.H.v.d.; Deimling, A.v.; Gouttefangeas, C.; Kroep, J.R.; et al. GAPVAC-101: First-in-human trial of a highly personalized peptide vaccination approach for patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2018, 36 (Suppl. 15), 2000. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Kodysh, J.; Rubinsteyn, A.; Blazquez, A.; Mandeli, J.; Bhardwaj, N.; Hormigo, A. CTIM-17. phase I study of the safety and immunogenicity of personalized neoantigen vaccines and tumor treating fields in patients with newly diagnosed glioblastoma. Neuro-Oncology 2020, 22 (Suppl. 2), ii36. [Google Scholar]
- Narita, Y.; Arakawa, Y.; Yamasaki, F.; Nishikawa, R.; Aoki, T.; Kanamori, M.; Nagane, M.; Kumabe, T.; Hirose, Y.; Ichikawa, T.; et al. A randomized, double-blind, phase III trial of personalized peptide vaccination for recurrent glioblastoma. Neuro-Oncology 2019, 21, 348–359. [Google Scholar] [CrossRef]
- Dunn-Pirio, A.; Peters, K.; DesJardins, A.; Randazzo, D.; Friedman, H.; Healy, P.; II, J.H.; Reap, E.; Archer, G.; Li, Q.-J.; et al. Tumor stem cell RNA-loaded dendritic cell vaccine for recurrent glioblastoma: A phase 1 trial (S41.004). Neurology 2017, 88, S41.004. [Google Scholar] [CrossRef]
- Vik-Mo, E.O.; Nyakas, M.; Mikkelsen, B.V.; Moe, M.C.; Due-Tønnesen, P.; Suso, E.M.; Sæbøe-Larssen, S.; Sandberg, C.; Brinchmann, J.E.; Helseth, E.; et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 2013, 62, 1499–1509. [Google Scholar] [CrossRef]
- Jouanneau, E.; Black, K.L.; Veiga, L.; Cordner, R.; Goverdhana, S.; Zhai, Y.; Zhang, X.X.; Panwar, A.; Mardiros, A.; Wang, H.; et al. Intrinsically de-sialylated CD103(+) CD8 T cells mediate beneficial anti-glioma immune responses. Cancer Immunol. Immunother. 2014, 63, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Vandermeulen, G.; Préat, V. Cancer DNA vaccines: Current preclinical and clinical developments and future perspectives. J. Exp. Clin. Cancer Res. 2019, 38, 146. [Google Scholar] [CrossRef]
- Melnick, K.; Dastmalchi, F.; Mitchell, D.; Rahman, M.; Sayour, E.J. Contemporary RNA Therapeutics for Glioblastoma. Neuromolecular Med. 2022, 24, 8–12. [Google Scholar] [CrossRef]
- Herrada, A.A.; Rojas-Colonelli, N.; González-Figueroa, P.; Roco, J.; Oyarce, C.; Ligtenberg, M.A.; Lladser, A. Harnessing DNA-induced immune responses for improving cancer vaccines. Hum. Vaccin. Immunother. 2012, 8, 1682–1693. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brem, S.; Desai, A.S.; Bagley, S.J.; Kurz, S.C.; Fuente, M.I.D.L.; Nagpal, S.; Welch, M.R.; Hormigo, A.; Carroll, N.; et al. INO-5401 and INO-9012 delivered intramuscularly (IM) with electroporation (EP) in combination with cemiplimab (REGN2810) in newly diagnosed glioblastoma (GBM): Interim results. J. Clin. Oncol. 2020, 38 (Suppl. 15), 2514. [Google Scholar] [CrossRef]
- Huang, B.; Li, X.; Li, Y.; Zhang, J.; Zong, Z.; Zhang, H. Current Immunotherapies for Glioblastoma Multiforme. Front. Immunol. 2020, 11, 603911. [Google Scholar] [CrossRef]
- Uematsu, M.; Ohsawa, I.; Aokage, T.; Nishimaki, K.; Matsumoto, K.; Takahashi, H.; Asoh, S.; Teramoto, A.; Ohta, S. Prognostic significance of the immunohistochemical index of survivin in glioma: A comparative study with the MIB-1 index. J. Neuro-Oncol. 2005, 72, 231–238. [Google Scholar] [CrossRef]
- Caudill, M.M.; Li, Z. HSPPC-96: A personalised cancer vaccine. Expert. Opin. Biol. Ther. 2001, 1, 539–547. [Google Scholar] [CrossRef]
- Amato, R.J. Heat shock protein-peptide complex-96 (Vitespen) for the treatment of cancer. Oncol. Rev. 2008, 2, 29–35. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Filley, A.C.; Dey, M. Dendritic cell based vaccination strategy: An evolving paradigm. J. Neuro-Oncol. 2017, 133, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Lynes, J.; Sanchez, V.; Dominah, G.; Nwankwo, A.; Nduom, E. Current Options and Future Directions in Immune Therapy for Glioblastoma. Front. Oncol. 2018, 8, 578. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, G.; Märten, A.; Kiske, S.M.; Feil, F.; Bieber, T.; Schmidt-Wolf, I.G. Generation of dendritic cell-based vaccines for cancer therapy. Br. J. Cancer 2002, 86, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- De Vries, I.J.; Krooshoop, D.J.; Scharenborg, N.M.; Lesterhuis, W.J.; Diepstra, J.H.; Van Muijen, G.N.; Strijk, S.P.; Ruers, T.J.; Boerman, O.C.; Oyen, W.J.; et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003, 63, 12–17. [Google Scholar] [PubMed]
- Mitchell, D.A.; Xie, W.; Schmittling, R.; Learn, C.; Friedman, A.; McLendon, R.E.; Sampson, J.H. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro-Oncology 2008, 10, 10–18. [Google Scholar] [CrossRef]
- Nair, S.K.; De Leon, G.; Boczkowski, D.; Schmittling, R.; Xie, W.; Staats, J.; Liu, R.; Johnson, L.A.; Weinhold, K.; Archer, G.E.; et al. Recognition and killing of autologous, primary glioblastoma tumor cells by human cytomegalovirus pp65-specific cytotoxic T cells. Clin. Cancer Res. 2014, 20, 2684–2694. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef]
- Saikali, S.; Avril, T.; Collet, B.; Hamlat, A.; Bansard, J.Y.; Drenou, B.; Guegan, Y.; Quillien, V. Expression of nine tumour antigens in a series of human glioblastoma multiforme: Interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for immunotherapy. J. Neuro-Oncol. 2007, 81, 139–148. [Google Scholar] [CrossRef]
- Pasqualetti, F.; Zanotti, S. Nonrandomised controlled trial in recurrent glioblastoma patients: The promise of autologous tumour lysate-loaded dendritic cell vaccination. Br. J. Cancer 2023, 129, 895–896. [Google Scholar] [CrossRef]
- Preusser, M.; van den Bent, M.J. Autologous tumor lysate-loaded dendritic cell vaccination (DCVax-L) in glioblastoma: Breakthrough or fata morgana? Neuro-Oncology 2023, 25, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Gatto, L.; Di Nunno, V.; Tosoni, A.; Bartolini, S.; Ranieri, L.; Franceschi, E. DCVax-L Vaccination in Patients with Glioblastoma: Real Promise or Negative Trial? The Debate Is Open. Cancers 2023, 15, 3251. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Ventz, S.; Trippa, L. External Control Arms and Data Analysis Methods in Nonrandomized Trial of Patients with Glioblastoma. JAMA Oncol. 2023, 9, 1006–1007. [Google Scholar] [CrossRef] [PubMed]
- Mandel, J.J.; de Groot, J.F. External Control Arms and Data Analysis Methods in Nonrandomized Trial of Patients with Glioblastoma. JAMA Oncol. 2023, 9, 1006. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, S.W.; Makalowski, J.; Kampers, L.F.C.; Van de Vliet, P.; Sprenger, T.; Schirrmacher, V.; Stucker, W. Dendritic cell vaccination for glioblastoma multiforme patients: Has a new milestone been reached? Transl. Cancer Res. 2023, 12, 2224–2228. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, F.; Yao, Y.; Wang, L.L.; Zhu, Y.; Hu, J. Adoptive Cell Therapy: A Novel and Potential Immunotherapy for Glioblastoma. Front. Oncol. 2020, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed]
- Durgin, J.S.; Henderson, F., Jr.; Nasrallah, M.P.; Mohan, S.; Wang, S.; Lacey, S.F.; Melenhorst, J.J.; Desai, A.S.; Lee, J.Y.K.; Maus, M.V.; et al. Case Report: Prolonged Survival Following EGFRvIII CAR T Cell Treatment for Recurrent Glioblastoma. Front. Oncol. 2021, 11, 669071. [Google Scholar] [CrossRef]
- Tang, O.Y.; Tian, L.; Yoder, T.; Xu, R.; Kulikovskaya, I.; Gupta, M.; Melenhorst, J.J.; Lacey, S.F.; O’Rourke, D.M.; Binder, Z.A. PD1 Expression in EGFRvIII-Directed CAR T Cell Infusion Product for Glioblastoma Is Associated with Clinical Response. Front. Immunol. 2022, 13, 872756. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Desai, R.; Abel, M.L.; Riccione, K.A.; Batich, K.A.; Shen, S.H.; Chongsathidkiet, P.; Gedeon, P.C.; Elsamadicy, A.A.; Snyder, D.J.; et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. Oncoimmunology 2018, 7, e1434464. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients with Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, J.; Yang, X.; Liu, Y.; Zou, C.; Lv, W.; Chen, C.; Cheng, K.K.-y.; Chen, T.; Chang, L.-J.; et al. Safety and antitumor activity of GD2-Specific 4SCAR-T cells in patients with glioblastoma. Mol. Cancer 2023, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Badhiwala, J.; Decker, W.K.; Berens, M.E.; Bhardwaj, R.D. Clinical trials in cellular immunotherapy for brain/CNS tumors. Expert. Rev. Neurother. 2013, 13, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed]
- Litten, J.B.; Ramakrishnan, A.; Astrow, S.H.; Harrison, C.; Aliki, A.; Badie, B. Phase 1b multicenter study to evaluate CHM 1101 in patients with recurrent or progressive glioblastoma. J. Clin. Oncol. 2023, 41 (Suppl. 16), TPS2086. [Google Scholar] [CrossRef]
- Lin, Q.; Ba, T.; Ho, J.; Chen, D.; Cheng, Y.; Wang, L.; Xu, G.; Xu, L.; Zhou, Y.; Wei, Y.; et al. First-in-Human Trial of EphA2-Redirected CAR T-Cells in Patients with Recurrent Glioblastoma: A Preliminary Report of Three Cases at the Starting Dose. Front. Oncol. 2021, 11, 694941. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Chen, D.; Tang, C.; Ji, C.; Li, Z.; Qian, Q. Safety, efficacy, and biomarker analysis of response to engineered tumor-infiltrating lymphocytes secreting anti-PD-1 antibody in recurrent glioblastoma: An open-label, two-arms, phase 1 study. J. Clin. Oncol. 2023, 41 (Suppl. 16), 2042. [Google Scholar] [CrossRef]
- Quattrocchi, K.B.; Miller, C.H.; Cush, S.; Bernard, S.A.; Dull, S.T.; Smith, M.; Gudeman, S.; Varia, M.A. Pilot study of local autologous tumor infiltrating lymphocytes for the treatment of recurrent malignant gliomas. J. Neuro-Oncol. 1999, 45, 141–157. [Google Scholar] [CrossRef]
- Sims, J.S.; Grinshpun, B.; Feng, Y.; Ung, T.H.; Neira, J.A.; Samanamud, J.L.; Canoll, P.; Shen, Y.; Sims, P.A.; Bruce, J.N. Diversity and divergence of the glioma-infiltrating T-cell receptor repertoire. Proc. Natl. Acad. Sci. USA 2016, 113, E3529–E3537. [Google Scholar] [CrossRef]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef] [PubMed]
- Karachi, A.; Dastmalchi, F.; Nazarian, S.; Huang, J.; Sayour, E.J.; Jin, L.; Yang, C.; Mitchell, D.A.; Rahman, M. Optimizing T Cell-Based Therapy for Glioblastoma. Front. Immunol. 2021, 12, 705580. [Google Scholar] [CrossRef]
- Luksik, A.S.; Yazigi, E.; Shah, P.; Jackson, C.M. CAR T Cell Therapy in Glioblastoma: Overcoming Challenges Related to Antigen Expression. Cancers 2023, 15, 1414. [Google Scholar] [CrossRef]
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front. Neurosci. 2021, 15, 662064. [Google Scholar] [CrossRef]
- Brown, C.E.; Warden, C.D.; Starr, R.; Deng, X.; Badie, B.; Yuan, Y.C.; Forman, S.J.; Barish, M.E. Glioma IL13Ralpha2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS ONE 2013, 8, e77769. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-shelf, steroid-resistant, IL13Ralpha2-specific CAR T cells for treatment of glioblastoma. Neuro-Oncology 2022, 24, 1318–1330. [Google Scholar] [CrossRef]
- Mineo, J.F.; Bordron, A.; Baroncini, M.; Maurage, C.A.; Ramirez, C.; Siminski, R.M.; Berthou, C.; Dam Hieu, P. Low HER2-expressing glioblastomas are more often secondary to anaplastic transformation of low-grade glioma. J. Neuro-Oncol. 2007, 85, 281–287. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology 2018, 20, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.K.; Ryan, A.J.; Seymour, L.W. Beyond cancer cells: Targeting the tumor microenvironment with gene therapy and armed oncolytic virus. Mol. Ther. 2021, 29, 1668–1682. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Ahmed, A.U.; Ulasov, I.V.; Sonabend, A.M.; Miska, J.; Lee-Chang, C.; Balyasnikova, I.V.; Chandler, J.P.; Portnow, J.; Tate, M.C.; et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: A first-in-human, phase 1, dose-escalation trial. Lancet Oncol. 2021, 22, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.R.; Chen, M.M.; Ene, C.; Lang, F.F.; Kan, P. Perfusion-guided endovascular super-selective intra-arterial infusion for treatment of malignant brain tumors. J. Neurointerv Surg. 2022, 14, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Rivera-Molina, Y.; Fueyo, J.; Jiang, H.; Nguyen, T.; Ho Shin, D.; Youssef, G.; Fan, X.; Gumin, J.; Alonso, M.M.; Phadnis, S.; et al. EXTH-27. Activating the immunity within the tumor using viroimmunotherapy: Delta-24-RGD oncolytic adenovirus armed with the immunopositive regulator gitrl. Neuro-Oncology 2019, 21 (Suppl. 6), vi87. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Abbed, K.M.; Tatter, S.; Louis, D.N.; Hochberg, F.H.; Barker, F.; Kracher, J.; Grossman, S.A.; Fisher, J.D.; Carson, K.; et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol. Ther. 2004, 10, 958–966. [Google Scholar] [CrossRef]
- Umemura, Y.; Orringer, D.; Junck, L.; Varela, M.L.; West, M.E.J.; Faisal, S.M.; Comba, A.; Heth, J.; Sagher, O.; Leung, D.; et al. Combined cytotoxic and immune-stimulatory gene therapy for primary adult high-grade glioma: A phase 1, first-in-human trial. Lancet Oncol. 2023, 24, 1042–1052. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Aguilar, L.K.; Bell, S.D.; Kaur, B.; Hardcastle, J.; Cavaliere, R.; McGregor, J.; Lo, S.; Ray-Chaudhuri, A.; Chakravarti, A.; et al. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J. Clin. Oncol. 2011, 29, 3611–3619. [Google Scholar] [CrossRef]
- Ji, N.; Weng, D.; Liu, C.; Gu, Z.; Chen, S.; Guo, Y.; Fan, Z.; Wang, X.; Chen, J.; Zhao, Y.; et al. Adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of recurrent high-grade glioma. Oncotarget 2016, 7, 4369–4378. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, L.A.; Manzanera, A.G.; Bell, S.D.; Cavaliere, R.; McGregor, J.M.; Grecula, J.C.; Newton, H.B.; Lo, S.S.; Badie, B.; Portnow, J.; et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro-Oncology 2016, 18, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Yu, J.S.; Lukas, R.V.; Solomon, I.H.; Ligon, K.L.; Nakashima, H.; Triggs, D.A.; Reardon, D.A.; Wen, P.; Stopa, B.M.; et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci. Transl. Med. 2019, 11, eaaw5680. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.J.; Peters, K.B.; Vredenburgh, J.; Bokstein, F.; Blumenthal, D.T.; Yust-Katz, S.; Peretz, I.; Oberman, B.; Freedman, L.S.; Ellingson, B.M.; et al. Safety and efficacy of VB-111, an anticancer gene therapy, in patients with recurrent glioblastoma: Results of a phase I/II study. Neuro-Oncology 2020, 22, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Brenner, A.; de Groot, J.F.; Butowski, N.A.; Zach, L.; Campian, J.L.; Ellingson, B.M.; Freedman, L.S.; Cohen, Y.C.; Lowenton-Spier, N.; et al. A randomized controlled phase III study of VB-111 combined with bevacizumab vs bevacizumab monotherapy in patients with recurrent glioblastoma (GLOBE). Neuro-Oncology 2020, 22, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Rampling, R.; Cruickshank, G.; Papanastassiou, V.; Nicoll, J.; Hadley, D.; Brennan, D.; Petty, R.; MacLean, A.; Harland, J.; McKie, E.; et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000, 7, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Papanastassiou, V.; Rampling, R.; Fraser, M.; Petty, R.; Hadley, D.; Nicoll, J.; Harland, J.; Mabbs, R.; Brown, M. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: A proof of principle study. Gene Ther. 2002, 9, 398–406. [Google Scholar] [CrossRef]
- Harrow, S.; Papanastassiou, V.; Harland, J.; Mabbs, R.; Petty, R.; Fraser, M.; Hadley, D.; Patterson, J.; Brown, S.M.; Rampling, R. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: Safety data and long-term survival. Gene Ther. 2004, 11, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef]
- Markert, J.M.; Liechty, P.G.; Wang, W.; Gaston, S.; Braz, E.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Lakeman, A.D.; Palmer, C.A.; et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol. Ther. 2009, 17, 199–207. [Google Scholar] [CrossRef]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: Results of a phase I trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Nakashima, H.; Kasai, K.; Fernandez, S.A.; Oglesbee, M. Preclinical Toxicology of rQNestin34.5v.2: An Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol. Ther. Methods Clin. Dev. 2020, 17, 871–893. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Solomon, I.; Nakashima, H.; Lawler, S.E.; Triggs, D.; Zhang, A.; Grant, J.; Reardon, D.A.; Wen, P.Y.; Lee, E.Q.; et al. First-in-human CAN-3110 (ICP-34.5 expressing HSV-1 oncolytic virus) in patients with recurrent high-grade glioma. J. Clin. Oncol. 2021, 39 (Suppl. 15), 2009. [Google Scholar] [CrossRef]
- Rainov, N.G. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 2000, 11, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- Kalkanis, S.; Jolly, D.J.; Pertschuk, D.; Ostertag, D.; Robbins, J.M.; Huang, T.T.; Gruber, H.; Mikkelsen, T. AT-29: INTRAVENOUS ADMINISTRATION OF TOCA 511 IN PATIENTS WITH RECURRENT GLIOBLASTOMA. Neuro-Oncology 2014, 16 (Suppl. 5), v15. [Google Scholar] [CrossRef][Green Version]
- Merchan, J.R.; Ahnert, J.R.; Falchook, G.; Ostertag, D.; Tejera, D.; Gruber, H.E.; Jolly, D.J.; Shorr, J. Toca 6: A phase 1b study of Toca 511 and Toca FC in patients with advanced solid tumors or lymphoma. J. Clin. Oncol. 2018, 36 (Suppl. 15), TPS2613. [Google Scholar] [CrossRef]
- Ostertag, D.; Amundson, K.K.; Lopez Espinoza, F.; Martin, B.; Buckley, T.; Galvão da Silva, A.P.; Lin, A.H.; Valenta, D.T.; Perez, O.D.; Ibañez, C.E.; et al. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro-Oncology 2012, 14, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Landolfi, J.; Vogelbaum, M.A.; Ostertag, D.; Elder, J.B.; Bloomfield, S.; Carter, B.; Chen, C.C.; Kalkanis, S.N.; Kesari, S.; et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro-Oncology 2018, 20, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Aghi, M.; Vogelbaum, M.A.; Kesari, S.; Chen, C.C.; Liau, L.M.; Piccioni, D.; Portnow, J.; Chang, S.; Robbins, J.M.; Boyce, T.; et al. AT-02 Intratumoral delivery of the retroviral replicating vector (RRV) TOCA 511 in subjects with recurrent high grade glioma: Interim report of phase 1 study (NCT 01156584). Neuro-Oncology 2014, 16 (Suppl. 5), v8. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Petrecca, K.; Walbert, T.; Butowski, N.; Salacz, M.; Perry, J.; Damek, D.; Bota, D.; Bettegowda, C.; Zhu, J.-J.; et al. Effect of Vocimagene Amiretrorepvec in Combination with Flucytosine vs Standard of Care on Survival Following Tumor Resection in Patients with Recurrent High-Grade Glioma: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1939–1946. [Google Scholar] [CrossRef]
- Ahluwalia, M.; Pugh, S.; Ellingson, B.; Kotecha, R.; Cloughesy, T.; Vogelbaum, M.; Aldape, K.; Cui, Y.; Armstrong, T.; Mehta, M. RBTT-11. NRG Oncology NRG-BN006: A Phase II/III randomized, open-label study of Toca 511 and Toca FC with standard of care compared to standard of care in patients with newly diagnosed glioblastoma. Neuro-Oncology 2019, 21 (Suppl. 6), vi220–vi221. [Google Scholar] [CrossRef]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Csatary, L.K.; Gosztonyi, G.; Szeberenyi, J.; Fabian, Z.; Liszka, V.; Bodey, B.; Csatary, C.M. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J. Neuro-Oncol. 2004, 67, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.; Rommelaere, J. Immune System Stimulation by Oncolytic Rodent Protoparvoviruses. Viruses 2019, 11, 415. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, A.; Gromeier, M.; Herndon, J.E., 2nd; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, P.; Roldán, G.; George, D.; Wallace, C.; Palmer, C.A.; Morris, D.; Cairncross, G.; Matthews, M.V.; Markert, J.; Gillespie, Y.; et al. A Phase I Trial of Intratumoral Administration of Reovirus in Patients with Histologically Confirmed Recurrent Malignant Gliomas. Mol. Ther. 2008, 16, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Kicielinski, K.P.; Chiocca, E.A.; Yu, J.S.; Gill, G.M.; Coffey, M.; Markert, J.M. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol. Ther. 2014, 22, 1056–1062. [Google Scholar] [CrossRef]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef] [PubMed]
- Trask, T.W.; Trask, R.P.; Aguilar-Cordova, E.; Shine, H.D.; Wyde, P.R.; Goodman, J.C.; Hamilton, W.J.; Rojas-Martinez, A.; Chen, S.H.; Woo, S.L.; et al. Phase I study of adenoviral delivery of the HSV-tk gene and ganciclovir administration in patients with current malignant brain tumors. Mol. Ther. 2000, 1, 195–203. [Google Scholar] [CrossRef]
- Smitt, P.S.; Driesse, M.; Wolbers, J.; Kros, M.; Avezaat, C. Treatment of relapsed malignant glioma with an adenoviral vector containing the herpes simplex thymidine kinase gene followed by ganciclovir. Mol. Ther. 2003, 7, 851–858. [Google Scholar] [CrossRef]
- Sandmair, A.M.; Loimas, S.; Puranen, P.; Immonen, A.; Kossila, M.; Puranen, M.; Hurskainen, H.; Tyynelä, K.; Turunen, M.; Vanninen, R.; et al. Thymidine kinase gene therapy for human malignant glioma, using replication-deficient retroviruses or adenoviruses. Hum. Gene Ther. 2000, 11, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Germano, I.M.; Fable, J.; Gultekin, S.H.; Silvers, A. Adenovirus/herpes simplex-thymidine kinase/ganciclovir complex: Preliminary results of a phase I trial in patients with recurrent malignant gliomas. J. Neuro-Oncol. 2003, 65, 279–289. [Google Scholar] [CrossRef]
- Immonen, A.; Vapalahti, M.; Tyynelä, K.; Hurskainen, H.; Sandmair, A.; Vanninen, R.; Langford, G.; Murray, N.; Ylä-Herttuala, S. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: A randomised, controlled study. Mol. Ther. 2004, 10, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Bögler, O.; Su Huang, H.-J.; Kleihues, P.; Cavenee, W.K. The p53 gene and its role in human brain tumors. Glia 1995, 15, 308–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.W.; Alemany, R.; Wang, J.; Koch, P.E.; Ordonez, N.G.; Roth, J.A. Safety evaluation of Ad5CMV-p53 in vitro and in vivo. Hum. Gene Ther. 1995, 6, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.F.; Bruner, J.M.; Fuller, G.N.; Aldape, K.; Prados, M.D.; Chang, S.; Berger, M.S.; McDermott, M.W.; Kunwar, S.M.; Junck, L.R.; et al. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: Biological and clinical results. J. Clin. Oncol. 2003, 21, 2508–2518. [Google Scholar] [CrossRef]
- Barrett, J.A.; Cai, H.; Miao, J.; Khare, P.D.; Gonzalez, P.; Dalsing-Hernandez, J.; Sharma, G.; Chan, T.; Cooper, L.J.N.; Lebel, F. Regulated intratumoral expression of IL-12 using a RheoSwitch Therapeutic System((R)) (RTS((R))) gene switch as gene therapy for the treatment of glioma. Cancer Gene Ther. 2018, 25, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Lesniak, M.S. Adenoviral virotherapy for malignant brain tumors. Expert. Opin. Biol. Ther. 2009, 9, 737–747. [Google Scholar] [CrossRef]
- Cheng, P.-H.; Wechman, S.L.; McMasters, K.M.; Zhou, H.S. Oncolytic Replication of E1b-Deleted Adenoviruses. Viruses 2015, 7, 5767–5779. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Heise, C.C.; Williams, A.M.; Xue, S.; Propst, M.; Kirn, D.H. Intravenous administration of ONYX-015, a selectively replicating adenovirus, induces antitumoral efficacy. Cancer Res. 1999, 59, 2623–2628. [Google Scholar] [PubMed]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.X.; Levin, V.A.; Yung, W.K.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef]
- Suzuki, K.; Fueyo, J.; Krasnykh, V.; Reynolds, P.N.; Curiel, D.T.; Alemany, R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin. Cancer Res. 2001, 7, 120–126. [Google Scholar] [PubMed]
- Stepanenko, A.A.; Sosnovtseva, A.O.; Valikhov, M.P.; Chernysheva, A.A.; Cherepanov, S.A.; Yusubalieva, G.M.; Ruzsics, Z.; Lipatova, A.V.; Chekhonin, V.P. Superior infectivity of the fiber chimeric oncolytic adenoviruses Ad5/35 and Ad5/3 over Ad5-delta-24-RGD in primary glioma cultures. Mol. Ther. Oncolytics 2022, 24, 230–248. [Google Scholar] [CrossRef]
- Alonso, M.M.; García-Moure, M.; Gonzalez-Huarriz, M.; Marigil, M.; Hernandez-Alcoceba, R.; Buñales, M.; Hervás, S.; Gallego, J.; Gomez-Manzano, C.; Fueyo, J.; et al. Abstract CT027: Oncolytic virus DNX-2401 with a short course of temozolomide for glioblastoma at first recurrence: Clinical data and prognostic biomarkers. Cancer Res. 2017, 77 (Suppl. 13), CT027. [Google Scholar] [CrossRef]
- Lang, F.F.; Tran, N.D.; Puduvalli, V.K.; Elder, J.B.; Fink, K.L.; Conrad, C.A.; Yung, W.K.A.; Penas-Prado, M.; Gomez-Manzano, C.; Peterkin, J.; et al. Phase 1b open-label randomized study of the oncolytic adenovirus DNX-2401 administered with or without interferon gamma for recurrent glioblastoma. J. Clin. Oncol. 2017, 35 (Suppl. 15), 2002. [Google Scholar] [CrossRef]
- van Putten, E.H.P.; Kleijn, A.; van Beusechem, V.W.; Noske, D.; Lamers, C.H.J.; de Goede, A.L.; Idema, S.; Hoefnagel, D.; Kloezeman, J.J.; Fueyo, J.; et al. Convection Enhanced Delivery of the Oncolytic Adenovirus Delta24-RGD in Patients with Recurrent GBM: A Phase I Clinical Trial Including Correlative Studies. Clin. Cancer Res. 2022, 28, 1572–1585. [Google Scholar] [CrossRef]
- Nassiri, F.; Patil, V.; Yefet, L.S.; Singh, O.; Liu, J.; Dang, R.M.A.; Yamaguchi, T.N.; Daras, M.; Cloughesy, T.F.; Colman, H.; et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: A phase 1/2 trial. Nat. Med. 2023, 29, 1370–1378. [Google Scholar] [CrossRef]
- Jiang, H.; Rivera-Molina, Y.; Gomez-Manzano, C.; Clise-Dwyer, K.; Bover, L.; Vence, L.M.; Yuan, Y.; Lang, F.F.; Toniatti, C.; Hossain, M.B.; et al. Oncolytic Adenovirus and Tumor-Targeting Immune Modulatory Therapy Improve Autologous Cancer Vaccination. Cancer Res. 2017, 77, 3894–3907. [Google Scholar] [CrossRef]
- Ulasov, I.V.; Rivera, A.A.; Sonabend, A.M.; Rivera, L.B.; Wang, M.; Zhu, Z.B.; Lesniak, M.S. Comparative evaluation of survivin, midkine and CXCR4 promoters for transcriptional targeting of glioma gene therapy. Cancer Biol. Ther. 2007, 6, 679–685. [Google Scholar] [CrossRef][Green Version]
- Ulasov, I.V.; Zhu, Z.B.; Tyler, M.A.; Han, Y.; Rivera, A.A.; Khramtsov, A.; Curiel, D.T.; Lesniak, M.S. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum. Gene Ther. 2007, 18, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Auffinger, B.; Spencer, D.A.; Miska, J.; Chang, A.L.; Kane, J.R.; Young, J.S.; Kanojia, D.; Qiao, J.; Mann, J.F.; et al. Single dose GLP toxicity and biodistribution study of a conditionally replicative adenovirus vector, CRAd-S-pk7, administered by intracerebral injection to Syrian hamsters. J. Transl. Med. 2016, 14, 134. [Google Scholar] [CrossRef]
- Perez, O.D.; Logg, C.R.; Hiraoka, K.; Diago, O.; Burnett, R.; Inagaki, A.; Jolson, D.; Amundson, K.; Buckley, T.; Lohse, D.; et al. Design and selection of Toca 511 for clinical use: Modified retroviral replicating vector with improved stability and gene expression. Mol. Ther. 2012, 20, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.A.; Lopez Espinoza, F.; Mendoza, D.; Kato, Y.; Inagaki, A.; Hiraoka, K.; Kasahara, N.; Gruber, H.E.; Jolly, D.J.; Robbins, J.M. Toca 511 gene transfer and treatment with the prodrug, 5-fluorocytosine, promotes durable antitumor immunity in a mouse glioma model. Neuro-Oncology 2017, 19, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, S.; Masui, K.; Sakamoto, T.; Nakatani, T.; Kikukawa, M.; Tsujinoue, H.; Mitoro, A.; Yamazaki, M.; Yoshiji, H.; Fukui, H.; et al. Bystander effect caused by cytosine deaminase gene and 5-fluorocytosine in vitro is substantially mediated by generated 5-fluorouracil. Anticancer. Res. 1998, 18, 3399–3406. [Google Scholar] [PubMed]
- McKie, E.A.; MacLean, A.R.; Lewis, A.D.; Cruickshank, G.; Rampling, R.; Barnett, S.C.; Kennedy, P.G.; Brown, S.M. Selective in vitro replication of herpes simplex virus type 1 (HSV-1) ICP34.5 null mutants in primary human CNS tumours--evaluation of a potentially effective clinical therapy. Br. J. Cancer 1996, 74, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef]
- Todo, T. Oncolytic virus therapy using genetically engineered herpes simplex viruses. Front. Biosci. 2008, 13, 2060–2064. [Google Scholar] [CrossRef]
- Coffin, R. Interview with Robert Coffin, inventor of T-VEC: The first oncolytic immunotherapy approved for the treatment of cancer. Immunotherapy 2016, 8, 103–106. [Google Scholar] [CrossRef]
- Gromeier, M.; Nair, S.K. Recombinant Poliovirus for Cancer Immunotherapy. Annu. Rev. Med. 2018, 69, 289–299. [Google Scholar] [CrossRef]
- Merrill, M.K.; Bernhardt, G.; Sampson, J.H.; Wikstrand, C.J.; Bigner, D.D.; Gromeier, M. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro-Oncology 2004, 6, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Russell, S.J. History of Oncolytic Viruses: Genesis to Genetic Engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.E.; Ungerechts, G. Measles Virus as an Oncolytic Immunotherapy. Cancers 2021, 13, 544. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Chen, Y.; Hong, X.; Liu, X.; Su, X.; Li, S.; Dong, X.; Zhao, G.; Li, Y. Newcastle disease virus enhances the growth-inhibiting and proapoptotic effects of temozolomide on glioblastoma cells in vitro and in vivo. Sci. Rep. 2018, 8, 11470. [Google Scholar] [CrossRef] [PubMed]
- Reichard, K.W.; Lorence, R.M.; Cascino, C.J.; Peeples, M.E.; Walter, R.J.; Fernando, M.B.; Reyes, H.M.; Greager, J.A. Newcastle disease virus selectively kills human tumor cells. J. Surg. Res. 1992, 52, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Daeffler, L.; Pozdeev, V.I.; Angelova, A.; Rommelaere, J. Immune Conversion of Tumor Microenvironment by Oncolytic Viruses: The Protoparvovirus H-1PV Case Study. Front. Immunol. 2019, 10, 1848. [Google Scholar] [CrossRef] [PubMed]
- Hartley, A.; Kavishwar, G.; Salvato, I.; Marchini, A. A Roadmap for the Success of Oncolytic Parvovirus-Based Anticancer Therapies. Annu. Rev. Virol. 2020, 7, 537–557. [Google Scholar] [CrossRef]
- Geletneky, K.; Angelova, A.; Leuchs, B.; Bartsch, A.; Capper, D.; Hajda, J.; Rommelaere, J. Atnt-07favorable Response of Patients with Glioblastoma at Second or Third Recurrence to Repeated Injection of Oncolytic Parvovirus H-1 in Combination with Bevacicumab. Neuro-Oncology 2015, 17 (Suppl. 5), v11. [Google Scholar] [CrossRef]
- Li, J.; Bonifati, S.; Hristov, G.; Marttila, T.; Valmary-Degano, S.; Stanzel, S.; Schnölzer, M.; Mougin, C.; Aprahamian, M.; Grekova, S.P.; et al. Synergistic combination of valproic acid and oncolytic parvovirus H-1PV as a potential therapy against cervical and pancreatic carcinomas. EMBO Mol. Med. 2013, 5, 1537–1555. [Google Scholar] [CrossRef]
- Geletneky, K.; Bartsch, A.; Weiss, C.; Bernhard, H.; Marchini, A.; Rommelaere, J. ATIM-40. High rate of objective anti-tumor response in 9 patients with glioblastoma after viro-immunotherapy with oncolytic parvovirus H-1 in combination with bevacicumab and PD-1 checkpoint blockade. Neuro-Oncology 2018, 20 (Suppl. 6), vi10. [Google Scholar] [CrossRef][Green Version]
- Geletneky, K.; Weiss, C.; Bernhard, H.; Capper, D.; Leuchs, B.; Marchini, A.; Rommelaere, J. ATIM-29. First clinical observation of improved anti-tumor effects of viro-immunotherapy with oncolytic parvovirus H-1 in combination with PD-1 checkpoint blockade and bevacicumab in patients with recurrent glioblastoma. Neuro-Oncology 2016, 18 (Suppl. 6), vi24. [Google Scholar] [CrossRef]
- Hengstschläger, M.; Knöfler, M.; Müllner, E.W.; Ogris, E.; Wintersberger, E.; Wawra, E. Different regulation of thymidine kinase during the cell cycle of normal versus DNA tumor virus-transformed cells. J. Biol. Chem. 1994, 269, 13836–13842. [Google Scholar] [CrossRef] [PubMed]
- Foloppe, J.; Kintz, J.; Futin, N.; Findeli, A.; Cordier, P.; Schlesinger, Y.; Hoffmann, C.; Tosch, C.; Balloul, J.M.; Erbs, P. Targeted delivery of a suicide gene to human colorectal tumors by a conditionally replicating vaccinia virus. Gene Ther. 2008, 15, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Dutoit, V.; Marinari, E.; Dietrich, P.-Y.; Migliorini, D. Combination of the Ima950/Poly-Iclc Multipeptide Vaccine with Pembrolizumab in Relapsing Glioblastoma Patients. Neuro-Oncology 2020, 22 (Suppl. 2), ii34. [Google Scholar] [CrossRef]
- Miller, A.; Kosaloglu-Yalcin, Z.; Westernberg, L.; Montero, L.; Bahmanof, M.; Frentzen, A.; Premlal, A.L.R.; Greenbaum, J.; Seumois, G.; Habbaba, R.; et al. A phase 1b study of personalized neoantigen vaccine plus pembrolizumab in adults with advanced cancer. J. Clin. Oncol. 2021, 39 (Suppl. 15), 2615. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Peereboom, D.M.; Ciolfi, M.; Schilero, C.; Hobbs, B.; Ciesielski, M.J.; Fenstermaker, R.A. Phase II study of pembrolizumab plus SurVaxM for glioblastoma at first recurrence. J. Clin. Oncol. 2020, 38 (Suppl. 15), TPS2581. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Gelb, A.B.; Chen, C.C.; Rao, G.; Reardon, D.A.; Wen, P.Y.; Bi, W.L.; Peruzzi, P.; Amidei, C.; Triggs, D.; et al. Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: An open-label, multi-institutional phase I trial. Neuro-Oncology 2022, 24, 951–963. [Google Scholar] [CrossRef]
- Sloan, A.E.; Buerki, R.A.; Murphy, C.; Kelly, A.T.; Ambady, P.; Brown, M.; Butowski, N.A.; Cavaliere, R.; Curry, W.T.; Desjardins, A.; et al. LUMINOS-101: Phase 2 study of PVSRIPO with pembrolizumab in recurrent glioblastoma. J. Clin. Oncol. 2021, 39 (Suppl. 15), TPS2065. [Google Scholar] [CrossRef]
- Awada, H.; Paris, F.; Pecqueur, C. Exploiting radiation immunostimulatory effects to improve glioblastoma outcome. Neuro-Oncology 2023, 25, 433–446. [Google Scholar] [CrossRef]
- De Martino, M.; Padilla, O.; Daviaud, C.; Wu, C.C.; Gartrell, R.D.; Vanpouille-Box, C. Exploiting Radiation Therapy to Restore Immune Reactivity of Glioblastoma. Front. Oncol. 2021, 11, 671044. [Google Scholar] [CrossRef] [PubMed]
- Slika, H.; Karimov, Z.; Alimonti, P.; Abou-Mrad, T.; De Fazio, E.; Alomari, S.; Tyler, B. Preclinical Models and Technologies in Glioblastoma Research: Evolution, Current State, and Future Avenues. Int. J. Mol. Sci. 2023, 24, 16316. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, A.A.; Sosnovtseva, A.O.; Valikhov, M.P.; Chernysheva, A.A.; Abramova, O.V.; Naumenko, V.A.; Chekhonin, V.P. The need for paradigm shift: Prognostic significance and implications of standard therapy-related systemic immunosuppression in glioblastoma for immunotherapy and oncolytic virotherapy. Front. Immunol. 2024, 15, 1326757. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]




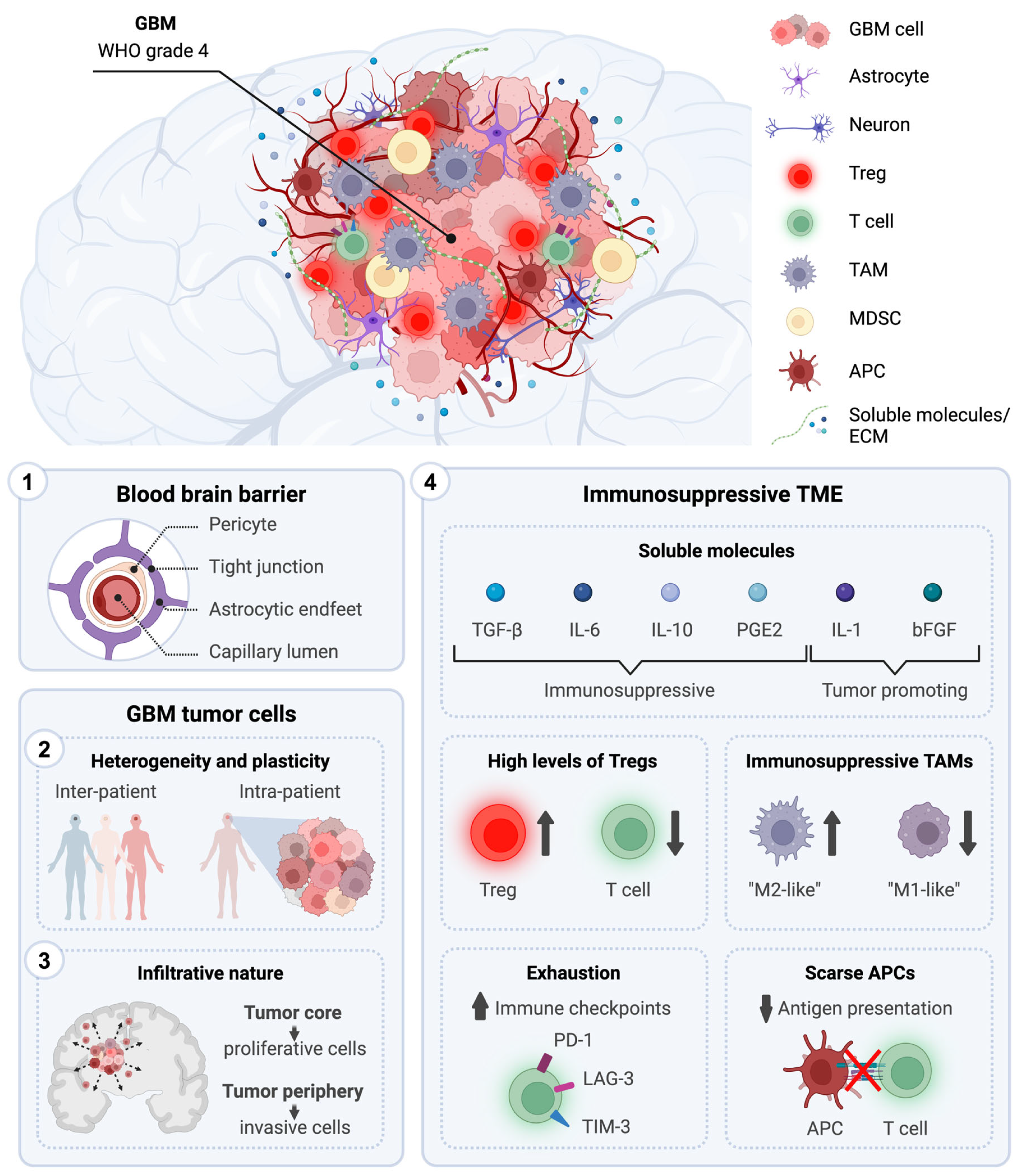{kind=link}
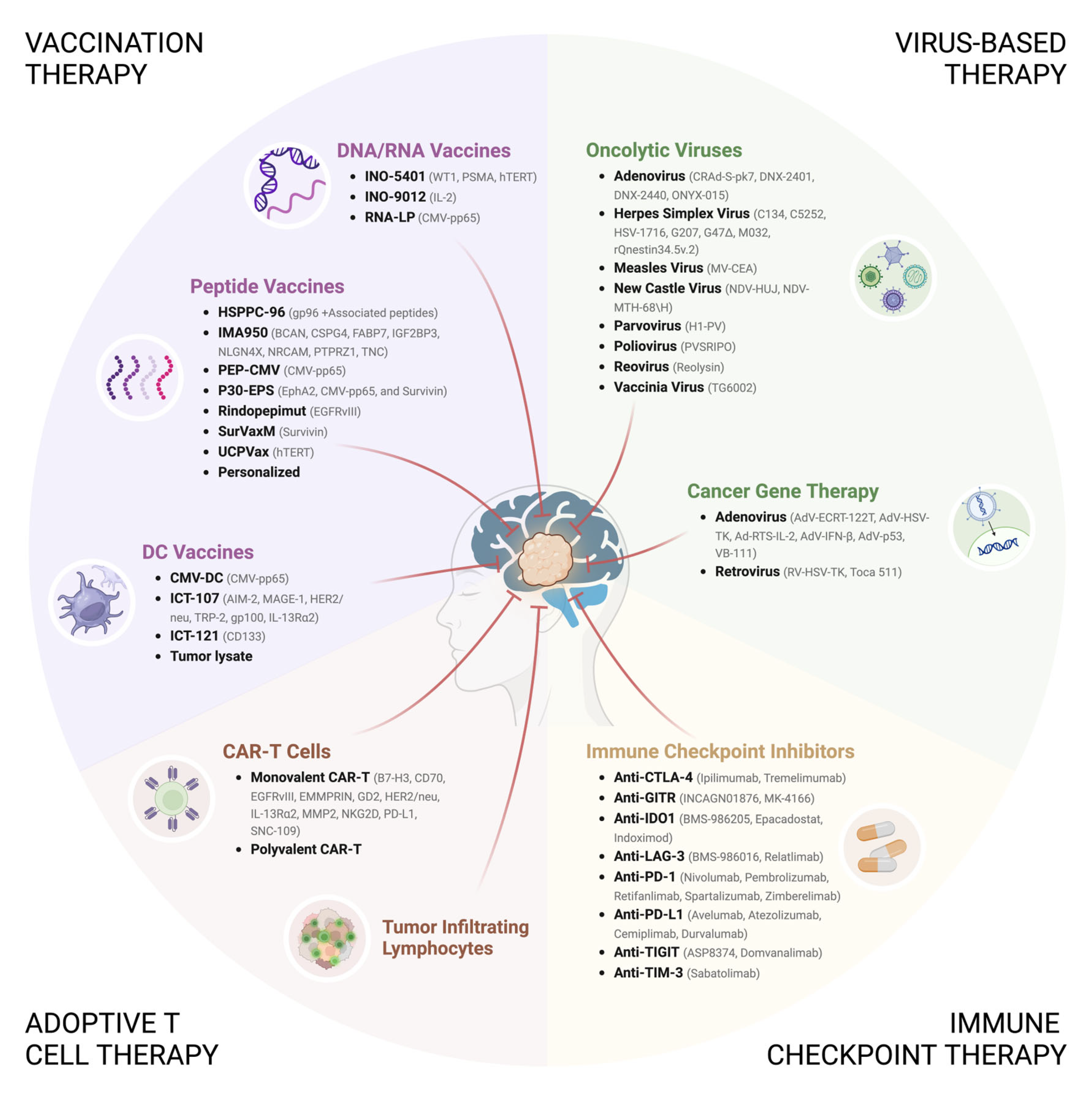{kind=link}
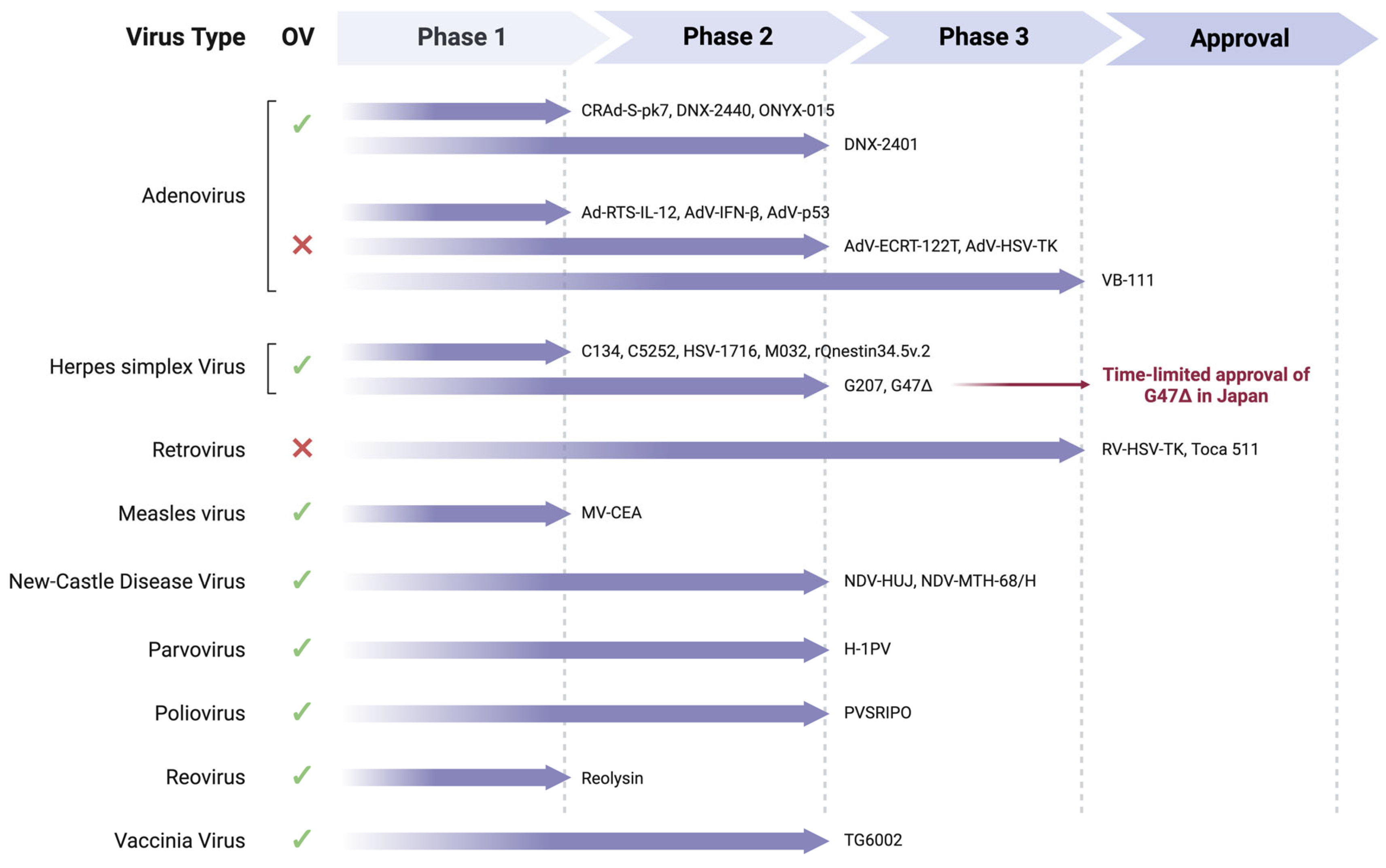{kind=link}
| Inhibitor | NCT Number | Phase | Study Status | Tumor Target | Intervention | Outcome |
|---|---|---|---|---|---|---|
| Anti-CTLA-4 (Ipilimumab) | NCT05074992 | 2 | Terminated | ndGBM | Ipi | |
| Anti-IDO1 (Indoximod) | NCT02052648 [130] | 1/2 | Completed | Malignant Brain Tumors | IND + TMZ | |
| IND + TMZ + Bev | ||||||
| IND + TMZ + Stereotactic RT | ||||||
| Anti-PD-1 (Nivolumab) | NCT02648633 | 1 | Terminated | rGBM | Valproate + Stereotactic RT + Nivo | |
| NCT02550249 [131] | 2 | Completed | GBM | Neoadjuvant Nivo | mOS: 7.3 months (95% CI, 5.4–7.9), mPFS: 4.1 months (95% CI, 2.8–5.5) | |
| NCT02335918 [132] | 2 | Completed | Advanced Solid Tumors | Nivo + Varlilumab | OS-12: 40.9% | |
| NCT03890952 [133] | 2 | Active Not Recruiting | rGBM | Nivo + Bev + Surgery | ||
| Nivo + Bev | ||||||
| NCT04195139 [134] | 2 | Active Not Recruiting | ndGBM | RT + TMZ + Nivo | mOS: 11.8 months, PFS-6: 64% | |
| RT + TMZ | mOS: 12.0 months, PFS-6: 49% | |||||
| NCT03743662 | 2 | Active Not Recruiting | rGBM (MGMT-M) | RT + Bev + Nivo | ||
| RT + Bev + Nivo + Surgery | ||||||
| NCT03452579 [135,136] | 2 | Active Not Recruiting | rGBM | Nivo + Bev (10 mg/Kg) | OS-12: 41.1%, OS-12 (age > 60 year): 46.2%, OS-12 (age ≤ 60 years): 35.6%. | |
| Nivo + Bev (3 mg/Kg) | OS-12: 37.7%, OS-12 (age > 60 year): 23.8%, OS-12 (age ≤ 60 years): 56.4%. | |||||
| NCT04704154 | 2 | Active Not Recruiting | Recurrent or Metastatic Tumors | Nivo + Regorafenib | ||
| NCT05909618 | 2 | Not Yet Recruiting | GBM and Brain Metastases (MGMT-UN) | Crizanlizumab | ||
| Crizanlizumab + Nivo | ||||||
| NCT02617589 [137] | 3 | Completed | ndGBM (MGMT-UN) | Nivo + RT | mPFS: 6.0 months (95% CI, 5.7–6.2), mOS: 13.4 months (95% CI, 12.6–14.3) | |
| TMZ + RT | mPFS: 6.2 months (95% CI, 5.9–6.7), mOS: 14.9 months (95% CI, 13.3–16.1) | |||||
| NCT02667587 [138] | 3 | Active Not Recruiting | ndGBM (MGMT-M) | RT + TMZ + Nivo | mPFS: 10.64 months (95% CI, 8.90–11.79), mOS: 28.91 months (95% CI, 24.38–31.57), | |
| RT + TMZ + Placebo | mPFS: 10.32 months (95% CI, 9.69–12.45), mOS: 32.07 months (95% CI, 29.37–33.77), | |||||
| Anti-PD-1 (Pembrolizumab) | NCT02852655 | 1 | Completed | rGBM | Pembro | |
| NCT02054806 [139] | 1 | Completed | Advanced Solid Tumors | Pembro | rGBM = mPFS: 2.8 months (95% CI, 1.9–8.1), mOS: 13.1 months (95% CI, 8.0–26.6) | |
| NCT05700955 | 1 | Recruiting | rGBM | Pembro + TMZ | ||
| NCT02530502 | 1 | Terminated | ndGBM | Pembro + TMZ + RT | ||
| NCT03722342 [140] | 1 | Active Not Recruiting | rGBM | Pembro + Olinvacimab | ||
| NCT03426891 [141] | 1 | Completed | ndGBM | Pembro + Vorinostat + TMZ + RT | ||
| NCT02311582 [142,143] | 1/2 | Active Not Recruiting | Recurrent Malignant Gliomas | Pembro + LITT | mPFS: 10.5 months, mOS: 11.4 months | |
| Pembro | mPFS: 2.1 months, mOS: 5.2 months | |||||
| NCT03277638 [144] | 1/2 | Recruiting | rGBM | Pembro (7 days before LITT) | ||
| Pembro (14 days after LITT) | ||||||
| Pembro (35 days after LITT) | ||||||
| NCT04977375 | 1/2 | Recruiting | rGBM | Pembro + Stereotactic RT + Surgery | ||
| NCT02430363 | 1/2 | Unknown | GBM or Gliosarcoma | Pembro | ||
| Pictilisib | ||||||
| NCT05053880 | 1/2 | Unknown | rGBM | Pembro | ||
| Pembro + ACT001 | ||||||
| NCT04121455 [145,146] | 1/2 | Active Not Recruiting | ndGBM (MGMT-UN) | NOX-A12 (200 mg) + RT | ||
| NOX-A12 (400 mg) + RT | ||||||
| NOX-A12 (600 mg) + RT | ||||||
| NOX-A12 (600 mg) + RT + Bev | ||||||
| NOX-A12 (600 mg) + RT | ||||||
| NOX-A12 (600 mg) + RT + Pembro | ||||||
| NCT05973903 | 1/2 | Not Yet Recruiting | rGBM | Lenvatinib + Pembro + TTF | ||
| NCT02628067 [147] | 2 | Recruiting | Advanced Solid Tumors | Pembro | Glioma = mPFS: 1.4 (95% CI, 1.0–2.1), mOS: 5.6 months (95% CI, 2.6–16.2) | |
| NCT02337491 [148,149] | 2 | Completed | rGBM | Pembro + Bev | PFS-6: 26% (95% CI, 16.3–41.5), mOS: 8.8 months (95% CI, 7.7–14.2) | |
| Pembro | PFS-6: 6.7% (95% CI, 1.7–25.4), mOS: 10.3 months (95% CI, 8.5–12.5) | |||||
| NCT03661723 [150] | 2 | Active Not Recruiting | rGBM | Pembro + RT (lead-in) | ORR: 3.3%, OS-6: 83.3 (95% CI, 6.5–92.7), OS-12: 40.0 (95% CI, 22.8–56.6) | |
| Pembro + Bev + RT (lead-in) | ORR: 10.0%, OS-6: 56.7 (95% CI, 37.3–72.1), OS-12: 16.6 (95% CI, 6.0–31.7) | |||||
| Pembro + RT | ||||||
| Pembro + Bev + RT | ||||||
| NCT05463848 | 2 | Recruiting | rGBM | Pembro + Olaparib + TMZ (Safety Lead In) | ||
| Pembro + Olaparib + TMZ (Surgical Cohort) | ||||||
| Pembro (Surgical Cohort) | ||||||
| NCT03347617 | 2 | Active Not Recruiting | ndGBM | Ferumoxytol MRI + Pembro | ||
| NCT03197506 | 2 | Suspended | ndGBM | Pembro + Surgery + TMZ + RT | ||
| Pembro + TMZ + RT | ||||||
| NCT05879120 | 2 | Not Yet Recruiting | rGBM | MRgFUS + Neoadjuvant Pembro + Adjuvant Pembro | ||
| Neoadjuvant Pembro + Adjuvant Pembro | ||||||
| NCT03405792 [151] | 2 | Active Not Recruiting | ndGBM | TTF + TMZ + Pembro | mPFS: 12.0 months, PFS-12: 50.0%, mOS: 24.8 months, OS-24: 52.4% | |
| TTF + TMZ | mPFS: 5.8 months, PFS-12: 28.2%, mOS: 14.7 months, OS-24: 12% | |||||
| NCT02337686 [152] | 2 | Active Not Recruiting | rGBM | Pembro + Surgery | mPFS: 4.5 months (95% CI, 2.27–6.83), PFS-6: 40%, mOS: 20 months, estimated OS-12: 63% | |
| NCT05465954 [153] | 2 | Recruiting | rGBM | Pembro + Efineptakin alfa | ||
| NCT03797326 [154] | 2 | Active Not Recruiting | Solid Tumors | Pembro + Lenvatinib | ||
| Lenvatinib | ||||||
| NCT05235737 | 4 | Recruiting | ndGBM | Neoadjuvant Pembro + Adjuvant Pembro + SOC | ||
| Neoadjuvant Pembro + SOC | ||||||
| SOC | ||||||
| Anti-PD-L1 (Avelumab) | NCT03047473 [155] | 2 | Completed | ndGBM | Avelumab | ORR: 23.3%, mPFS: 9.7 months (95% CI, 8.2–15.5), mOS: 15.3 months (95% CI, 10.7–21.5) |
| Anti-PD-L1 (Atezolizumab) | NCT05423210 | 1 | Active Not Recruiting | ndGBM | Atezo + Fractionated Stereotactic RT | |
| NCT04160494 | 1 | Active Not Recruiting | Recurrent Gliomas | D2C7-IT (6.92 μg/mL) + Atezo | ||
| D2C7-IT (4.61 μg/mL) + Atezo | ||||||
| NCT03158389 | 1/2 | Completed | ndGBM (MGMT-UN) | APG101 + RT | ||
| Alectinib + RT | ||||||
| Idasanutlin + RT | ||||||
| Atezo + RT | ||||||
| Vismodegib + RT | ||||||
| Temsirolimus + RT | ||||||
| Palbociclib + RT | ||||||
| NCT03673787 [156] | 1/2 | Recruiting | Advanced Solid Tumors | Atezo + Ipatasertib | ||
| NCT03174197 [157] | 1/2 | Active Not Recruiting | ndGBM | Atezo + TMZ + RT | mOS: 17.1 months (95% CI, 13.9-N/A), mPFS: 9.7 months (95% CI, 7.6–15), mPFS (MGMT-M): 16.7 months (95% CI, 7.85-N/A), mPFS (MGMT-UN): 7.9 months (95% CI, 6.70–12.4) | |
| NCT05039281 | 1/2 | Recruiting | rGBM | Atezo + Cabozantinib | ||
| NCT06069726 | 2 | Not Yet Recruiting | rGBM | Pre-Surgery Atezo | ||
| NCT04729959 | 2 | Suspended | rGBM | Atezo + Tocilizumab + Stereotactic RT | ||
| Atezo + Tocilizumab + Stereotactic RT + Surgery | ||||||
| Anti-PD-L1 (Durvalumab) | NCT02336165 [158] | 2 | Completed | GBM | ndGBM = Durva + RT | OS-12: 60% (90% CI, 46.1–71.4) |
| Bev-Naïve rGBM = Durva | PFS-6: 19.4% (90% CI, 9.3–32.1) | |||||
| Bev-Naïve rGBM = Durva + Bev (10 mg/Kg) | PFS-6: 15.2% (90% CI, 6.7–26.8) | |||||
| Bev-Naïve rGBM = Durva + Bev (3 mg/Kg) | PFS-6: 17.2% (90% CI, 7.7–29.7) | |||||
| Bev-Resistant rGBM = Durva + Bev | OS-6: 36.4% (80% CI, 23.5–49.3) | |||||
| Anti-PD-1 + Anti-CTLA-4 | NCT02311920 [159] | 1 | Completed | ndGBM or Gliosarcoma | TMZ + Ipi | |
| TMZ + Nivo | ||||||
| TMZ + Ipi + Nivo | ||||||
| NCT04606316 | 1 | Recruiting | rGBM | Nivo + Ipi | ||
| Nivo + Placebo | ||||||
| Placebo | ||||||
| NCT03233152 [160] | 1 | Active Not Recruiting | rGBM | Nivo + Ipi | mPFS: 11.7 weeks (2–152), mOS: 38 weeks (95% CI, 27–49), | |
| NCT06097975 | 1 | Not Yet Recruiting | rGBM | Nivo + Ipi | ||
| NCT03367715 | 2 | Completed | ndGBM (MGMT-UN) | Nivo + Ipi + Short-Course RT | OS-12: 80%, mOS: 16.85 months (4.5–32.9), mPFS: 5.92 months (1.5–13.9) | |
| NCT03430791 | 2 | Terminated | rGBM | TTF + Nivo | ||
| TTF + Nivo + Ipi | ||||||
| NCT04817254 | 2 | Recruiting | ndGBM | Nivo + Ipi (1 mg/Kg) + TMZ | ||
| Nivo + Ipi (3 mg/Kg) + TMZ | ||||||
| NCT04145115 | 2 | Recruiting | rGBM | Nivo + Ipi | ||
| NCT04396860 | 2/3 | Active, not recruiting | ndGBM (MGMT-UN) | RT + TMZ | ||
| RT + Nivo + Ipi | ||||||
| NCT02017717 [161,162] | 3 | Active, not recruiting | rGBM | Nivo | OS-12: 41.8% (95% CI, 34.7–48.8), mOS: 9.8 months (95% CI, 8.2–11.8), mPFS: 1.51 months (95% CI, 1.48–1.61) | |
| Nivo + Ipi | ||||||
| Bev | OS-12: 42.4% (95% CI, 34.9–49.6), mOS: 10.05 months (95% CI, 9–11.99), mPFS: 3.61 months (95% CI, 2.99–4.6) | |||||
| Anti-PD-1 + Anti-GITR | NCT04225039 [163] | 2 | Active, not recruiting | rGBM | Retifanlimab + INCAGN01876 + Stereotactic RT | mPFS: 3.9 months (95% CI, 2.1–6.2), mOS: 9.4 months (95% CI, 8.2–10.6) |
| Retifanlimab + INCAGN01876 + Stereotactic RT prior to Surgery | mPFS: 11.7 months, mOS: 20.1 months | |||||
| Retifanlimab + INCAGN01876 prior to Surgery | mPFS: 2.0 months, mOS: 9.4 months | |||||
| Anti-PD-1 + Anti-IDO1 | NCT04047706 [164] | 1 | Active, not recruiting | ndGBM | RT + TMZ + Nivo + BMS-986205 | |
| RT + Nivo + BMS-986205 | ||||||
| NCT02327078 [165] | 1/2 | Completed | Advanced Tumors | Nivo + Epacadostat | ||
| Anti-PD-1 + Anti-LAG-3 | NCT03493932 [166] | 1 | Completed | GBM | Nivo + Relatlimab | |
| NCT02658981 [167] | 1 | Completed | rGBM | BMS-986016 | ||
| BMS-986016 + Nivo | ||||||
| Anti-PD-1 + Anti-TIGIT | NCT04656535 | 0/1 | Recruiting | GBM | Domvanalimab + Placebo | |
| Zimberelimab + Placebo | ||||||
| Domvanalimab + Zimberelimab | ||||||
| Placebo | ||||||
| NCT04826393 | 1 | Active Not Recruiting | Recurrent Gliomas | Cemiplimab + ASP8374 | ||
| Anti-PD-1 + Anti-TIM-3 | NCT03961971 | 1 | Active Not Recruiting | rGBM | Spartalizumab + Sabatolimab + Stereotactic RT | |
| Anti-PD-1 + Anti-GITR or Anti-IDO1 or Anti-CTLA-4 | NCT03707457 | 1 | Terminated | rGBM | Nivo + MK-4166 | |
| Nivo + Epacadostat | ||||||
| Nivo + Ipi | ||||||
| Anti-PD-L1 + Anti-CTLA-4 | NCT02794883 | 2 | Completed | Recurrent Malignant Gliomas | Surgery + Durva | mOS: 11.71 (95% CI, 8.332–32.71), mPFS: 4.356 (95% CI, 2.941–32.74) |
| Surgery + Tremelimumab | mOS: 7.246 (95% CI, 2.746–16.32), mPFS: 2.746 (95% CI, 2.68–8.727) | |||||
| Surgery + Durva + Tremelimumab | mOS: 7.703 (95% CI, 7.41–40.14), mPFS: 4.913 (95% CI, 2.905–120.4) | |||||
| Various | NCT06047379 | 1/2 | Not Yet Recruiting | Malignant Gliomas or Brain Metastases | NEO212 + Ipi | |
| NEO212 + Pembro | ||||||
| NEO212 + Nivo | ||||||
| NEO212 + Regorafenib | ||||||
| NEO212 + CarbolaUn + Paclitaxel | ||||||
| NEO212 + FOLFIRI + Bev | ||||||
| NEO212 | ||||||
| NEO212 + SOC |
| Antigen | Vaccine/ Delivery | NCT Number | Phase | Study Status | Tumor Target | Intervention | Outcome |
|---|---|---|---|---|---|---|---|
| CD133 | DC vaccine | NCT02049489 [186] | 1 | Completed | rGBM | ICT-121 | |
| CMV-pp65 | Peptide Vaccine | NCT01854099 | 1 | Withdrawn | ndGBM | TMZ (5 Day) + PEP-CMV (Day 6–8) | |
| TMZ (5 Day) + PEP-CMV (day 22–24) | |||||||
| TMZ (21 Day) + PEP-CMV (day 22–24) | |||||||
| Peptide Vaccine | NCT02864368 | 1 | Terminated | ndGBM | Td + TMZ (5 Day) + PEP-CMV (Component A + Component B) + Td | ||
| Td + TMZ (21 Day) + PEP-CMV (Component A + Component B) + Td | |||||||
| Td + TMZ (5 Day) + PEP-CMV (Safety Cohort) + Td | |||||||
| Td + TMZ (5 Day) + PEP-CMV (Component A) + Td | |||||||
| Td + TMZ (21 Day) + PEP-CMV (Component A) + Td | |||||||
| DC vaccine | NCT04963413 | 1 | Active, not recruiting | ndGBM | CMV-DC + GM-CSF | ||
| DC vaccine | NCT00693095 [187] | 1 | Completed | ndGBM | CMV-ALT + CMV-DC | ||
| CMV-ALT | |||||||
| DC vaccine | NCT00626483 [188] | 1 | Completed | ndGBM | CMV-DC + GM-CSF + Basiliximab | mOS: 5.6 months (95% CI, 3.6–9.9), mPFS: 7.7 months (95% CI, 3.4–13.8) | |
| DC vaccine | NCT04741984 | 1 | Withdrawn | ndGBM (MGMT-UN) | Monocyte loaded with mRNA encoding for CMV-pp65 (MT-201) | ||
| DC vaccine | NCT00639639 [189,190] | 1 | Completed | ndGBM | CMV-ALT + CMV-DC + Unpulsed DCs (or Td) | ||
| CMV-DC + Unpulsed DCs (or Td) | |||||||
| CMV-DC + GM-CSF + Unpulsed DCs (or Td) | |||||||
| DC vaccine | NCT02465268 [191] | 2 | Active, not recruiting | ndGBM | Td + TMZ + Short-Length CMV-DC + GM-CSF | ||
| Td + TMZ + Full-Length CMV-DC + GM-CSF | |||||||
| Unpulsed PBMCs | |||||||
| DC vaccine | NCT02366728 [192,193] | 2 | Completed | ndGBM | CMV-DC | mOS: 16 months (95% CI, 12.8–25.5), mPFS: 6.5 months (95% CI, 4.4–12.1) | |
| CMV-DC + Td | mOS: 20 months (95% CI, 16.7–25.6), mPFS: 6.7 months (95% CI, 4.6–15.2) | ||||||
| CMV-DC + Td + Basiliximab | mOS: 19 months (95% CI, 10.2-N/A), mPFS: 7.1 months (95% CI, 6-N/A) | ||||||
| Liposome | NCT04573140 | 1 | Recruiting | ndGBM (MGMT-UN) | Liposome loaded with mRNA encoding for CMV-pp65 (RNA-LP) | ||
| EGFRvIII | Peptide Vaccine | NCT00626015 [194] | 1 | Completed | ndGBM (EGFRvIII+) | Rindopepimut + TMZ + Daclizumab | |
| Rindopepimut + TMZ + Placebo | |||||||
| Rindopepimut + Basiliximab | |||||||
| Peptide Vaccine | NCT01498328 [195] | 2 | Completed | rGBM (EGFRvIII+) | Bev-Naïve = Bev + Rindopepimut + GM-CSF | PFS-6: 28%, ORR: 30%, mDOR: 7.8 months (95% CI, 3.5–22.2) | |
| Bev-Naïve = Bev + KLH | PFS-6: 16%, ORR: 18%, mDOR: 5.6 months (95% CI, 3.7–7.4) | ||||||
| Bev-Resistant = Bev + Rindopepimut + GM-CSF | |||||||
| Peptide Vaccine | NCT00458601 [196] | 2 | Completed | ndGBM (EGFRvIII+) | Rindopepimut + GM-CSF + TMZ | mOS: 21.8 months, OS-36: 26% | |
| Peptide Vaccine | NCT00643097 [197,198,199] | 2 | Completed | ndGBM (EGFRvIII+) | Rindopepimut + GM-CSF | mPFS: 14.2 (95% CI, 9.9–17.6) | |
| Rindopepimut + GM-CSF + TMZ (5 Day, 200 mg/m2) | mPFS: 12.1 (95% CI, 10.5–23.7) | ||||||
| Rindopepimut + GM-CSF + TMZ (21 Day, 100 mg/m2) | mPFS: 11.6 (95% CI, 8.1–12.7) | ||||||
| Peptide Vaccine | NCT01480479 [200] | 3 | Completed | ndGBM (EGFRvIII+) | Rindopepimut + GM-CSF + TMZ | mOS: 20.1 months (95% CI, 18.5–22.1) | |
| KLH + TMZ | mOS: 20.0 months (95% CI, 18.1–21.9) | ||||||
| HSPPC-96 | Peptide Vaccine | NCT00293423 [201,202] | 1/2 | Completed | Recurrent Gliomas | HSPPC-96 Vaccine | OS-12: 29.3% (95% CI, 16.6–45.7), mOS: 42.6 weeks (95% CI, 34.7–50.5) |
| Peptide Vaccine | NCT00905060 [203] | 2 | Completed | ndGBM | HSPPC-96 Vaccine + TMZ | mOS: 23.8 months (95% CI, 9.8–30.2), mPFS: 18 (95% CI, 12.4–21.8) | |
| Peptide Vaccine | NCT01814813 [204] | 2 | Terminated | rGBM | HSPPC-96 Vaccine + Concomitant Bev | mOS: 6.6 months (95% CI, 5.4–10.4), mPFS: 3.7 months (95% CI, 2.9–5.4) | |
| HSPPC-96 Vaccine + Bev At Progression | mOS: 9.2 months (95% CI, 5.7–11.6), mPFS: 2.5 months (95% CI, 2.0–3.5) | ||||||
| Bev | mOS: 10.7 months (95% CI, 8.8–17.2), mPFS: 5.3 months (95% CI, 3.7–8.0) | ||||||
| hTERT | Peptide Vaccine | NCT00069940 | 1 | Completed | Sarcoma and Brain Tumors (HLA-A2+) | 540–548 hTERT Vaccine + GM-CSF | |
| Peptide Vaccine | NCT04280848 [205] | 2 | Active, not recruiting | ndGBM (MGMT-UN) | MGMT-UN = UCPVax | mPFS: 8.9 months (95% CI, 7.6–10.6), mOS: 17.9 months (95% CI, 16–23), OS-24: 26% | |
| MGMT-UN or MGMT m = UCPVax + TMZ | |||||||
| Survivin | Peptide Vaccine | NCT01250470 [206] | 1 | Completed | Recurrent Malignant Gliomas | SurVaxM/Montanide ISA-51 + GM-CSF | mPFS: 17.6 weeks, mOS: 86.6 weeks |
| Peptide Vaccine | NCT05163080 [207] | 2 | Recruiting | ndGBM | SurVaxM/Montanide ISA-51 + GM-CSF + TMZ | ||
| Placebo/Montanide ISA-51 + GM-CSF + TMZ | |||||||
| Peptide Vaccine | NCT02455557 [208] | 2 | Active, not recruiting | ndGBM | SurVaxM/Montanide ISA-51 + GM-CSF + TMZ | PFS-6: 95% (95% CI, 86–98), mPFS: 11.4 months, mOS: 25.8 months (95% CI, 19.5–43.5) | |
| AIM-2, MAGE-1, HER2/neu, TRP-2, gp100, and IL-13Rα2 | DC vaccine | NCT01280552 [209] | 2 | Completed | ndGBM | ICT-107 | mOS: 18.3 months (95% CI, 14.9–21.2), mPFS: 11.2 months (95% CI, 8.2–13.0) |
| Unpulsed DCs | mOS: 16.7 months (95% CI, 12.3–23.0), mPFS: 9.0 months (95% CI, 5.5–10.3) | ||||||
| NCT02546102 | 3 | Suspended | ndGBM | ICT-107 + TMZ | |||
| Placebo + TMZ | |||||||
| EGFRvIII, IL-13Rα2, EphA2, HER2/neu, YKL-40 | Peptide Vaccine | NCT02754362 | 2 | Withdrawn | rGBM | Bev + Multipeptide Vaccine + Poly-ICLC | |
| EphA2, CMV-pp65, and Survivin | Peptide Vaccine | NCT05283109 | 1 | Recruiting | ndGBM (MGMT-UN) | P30-EPS + Poly-ICLC | |
| BCAN, CSPG4, FABP7, IGF2BP3, NLGN4X, NRCAM, PTPRZ1 (2 peptides), and TNC | Peptide Vaccine | NCT01403285 | 1 | Terminated | GBM (HLA-A2+) | IMA950 + GM-CSF + Imiquimod + Cyclophosphamide | |
| Peptide Vaccine | NCT01222221 [210] | 1 | Completed | ndGBM (HLA-A2+) | IMA950 + GM-CSF + Chemoradiotherapy (Vaccine before TMZ) | mOS: 14.4 months | |
| IMA950 + GM-CSF + Chemoradiotherapy (Vaccine after TMZ) | mOS: 15.7 months | ||||||
| Peptide Vaccine | NCT01920191 [211,212] | 1/2 | Completed | ndGBM (HLA-A2+) | IMA950 + Poly-ICLC | mOS: 19 months (95% CI: 17.25–27.87), PFS-6: 81%, mPFS: 9.5 months | |
| WT-1, PSMA, hTERT, IL-2 | Electroporation | NCT03491683 [213] | 1/2 | Active, not recruiting | ndGBM | MGMT-UN = INO-5401 + INO-9012 + Cemiplimab + RT + TMZ | mOS: 17.9 months (95% CI, 14.5–19.8) |
| MGMT m = INO-5401 + INO-9012 + Cemiplimab + RT + TMZ | mOS: 32.5 months (95% CI, 18.4-N/A) | ||||||
| Tumor Lysate | DC vaccine | NCT01171469 [214] | 1 | Completed | Recurrent or Progressive Malignant Gliomas | DCs pulsed with Tumor Lysate (from BTSCs) + Imiquimod | |
| DC vaccine | NCT00068510 [215] | 1 | Completed | Malignant Gliomas | DCs pulsed with Tumor Lysate | ||
| DC vaccine | NCT01808820 | 1 | Completed | Malignant Gliomas | DCs pulsed with Tumor Lysate + Imiquimod | ||
| DC vaccine | NCT02010606 [216] | 1 | Completed | GBM | ndGBM = DCs pulsed with Tumor Lysate (from Allogeneic Stem-like Cells) + RT + TMZ | mPFS: 8.75 months, mOS: 20.36 months | |
| rGBM = DCs pulsed with Tumor Lysate (from Allogeneic Stem-like Cells) + Bev (optional) | mPFS: 3.23 months, PFS-6: 24%, mOS: 11.97 months | ||||||
| DC vaccine | NCT01213407 [217] | 2 | Completed | Malignant Gliomas | SOC + DCs pulsed with Tumor Lysate (Trivax) | ||
| SOC | |||||||
| DC vaccine | NCT01006044 [218] | 2 | Completed | GBM | DCs pulsed with Tumor Lysate | mPFS: 12.7 months (95% CI, 7–16), mOS: 23.4 months (95% CI, 16–33.1) | |
| DC vaccine | NCT00323115 [219] | 2 | Completed | ndGBM | DCs pulsed with Tumor Lysate + RT + TMZ | PFS-6: 90%, mPFS: 9.5 months, mOS: 28 months | |
| DC vaccine | NCT00045968 [220,221] | 3 | Active, not recruiting | GBM | DCs pulsed with Tumor Lysate (DCVax-L) | ndGBM = mOS: 19.3 months (95% CI, 17.5–21.3) rGBM = mOS: 13.2 months (95% CI, 9.7–16.8) | |
| Unpulsed PBMCs | ndGBM = mOS: 16.5 months (95% CI, 16.0–17.5) rGBM = mOS: 7.8 months (95% CI, 7.2–8.2) | ||||||
| Personalized | Peptide Vaccine | NCT02149225 [222,223] | 1 | Completed | ndGBM | APVAC1/APVAC2 + Poly-ICLC + GM-CSF + TMZ | mPFS: 14.2 months, mOS: 29 months |
| Peptide Vaccine | NCT02510950 | 1 | Terminated | ndGBM | Personalized Peptide Vaccine + Poly-ICLC + TMZ | ||
| Peptide Vaccine | NCT03223103 [224] | 1 | Active, not recruiting | ndGBM | Mutation-derived Tumor Antigen Vaccine + Poly-ICLC + TTF | Estimated PFS-12: 62.5%, estimated OS-12: 83.3% | |
| Peptide Vaccine | NCT05557240 | 1 | Recruiting | ndGBM | NPVAC1 + Poly-ICLC + TMZ | ||
| NPVAC2 + Poly-ICLC + TMZ | |||||||
| Electroporation | NCT04015700 | 1 | Active, not recruiting | ndGBM (MGMT-UN) | Personalized DNA Vaccine (GNOS-PV01) + INO-9012 | ||
| Peptide Vaccine | [225] | 3 | Concluded | rGBM (HLA-A24+) | Personalized Peptide Vaccine | mOS: 8.4 months (95% CI, 6.6–10.6) | |
| Placebo | mOS: 8.0 months (95% CI, 4.8–12.9) | ||||||
| N/A | Peptide Vaccine | NCT04842513 | 1 | Recruiting | ndGBM (HLA-A2+, MGMT-M) | Multipeptide Vaccine + XS15 + Montanide ISA-51 | |
| DC vaccine | NCT04968366 | 1 | Recruiting | ndGBM | DCs pulsed with Multiple Neopeptides + TMZ | ||
| DC vaccine | NCT00612001 [215] | 1 | Completed | Malignant Gliomas | DCs pulsed with Multiple Glioma-associated Peptides | ||
| DC vaccine | NCT00890032 [226] | 1 | Completed | rGBM | DCs pulsed with mRNA from BTSCs | mPFS: 3.2 months (95.0% CI, 1.8–7.2), mOS: 11 months (95.0% CI, 8.2–14.8) | |
| DC vaccine | NCT02820584 | 1 | Completed | rGBM | DCs pulsed with mRNA from Glioma Stem Cells | ||
| DC vaccine | NCT00846456 [227] | 1/2 | Completed | GBM | DCs pulsed with mRNA from Glioma Stem Cells | mOS (treated group): 759 days, mOS (control group): 585 days | |
| DC vaccine | NCT00576641 [228] | 1 | Completed | Brain Stem Glioma and GBM | DCs pulsed with Tumor Peptides |
| Antigen | NCT Number | Phase | Study Status | Tumor Target | Intervention | Outcome | |
|---|---|---|---|---|---|---|---|
| Monovalent CAR-T | B7-H3 | NCT05241392 | 1 | Recruiting | rGBM | B7-H3 CAR-T | |
| NCT05366179 | 1 | Recruiting | rGBM | B7-H3 CAR-T | |||
| NCT05474378 | 1 | Recruiting | rGBM | B7-H3 CAR-T | |||
| NCT04385173 | 1 | Recruiting | rGBM or Refractory GBM | B7-H3 CAR-T + TMZ | |||
| NCT04077866 | 1/2 | Recruiting | rGBM or Refractory GBM | TMZ | |||
| TMZ + B7-H3 CAR-T | |||||||
| CD70 | NCT05353530 | 1 | Recruiting | ndGBM (MGMT-UN, CD70+) | CD70 CAR-T | ||
| EGFRvIII | NCT05802693 | 1 | Not yet recruiting | rGBM (EGFRvIII+) | EGFRvIII CAR-T | ||
| NCT02209376 [253,254,255] | 1 | Terminated | rGBM | EGFRvIII CAR-T | mOS: 251 days | ||
| NCT02664363 [256] | 1 | Terminated | ndGBM (EGFRvIII+) | EGFRvIII CAR-T | |||
| NCT02844062 | 1 | Unknown | rGBM (EGFRvIII+) | EGFRvIII CAR-T | |||
| NCT03283631 | 1 | Terminated | rGBM | EGFRvIII CAR-T | |||
| NCT05063682 | 1 | Unknown | Leptomeningeal GBM (EGFRvIII+) | EGFRvIII CAR-T | |||
| NCT05660369 | 1 | Recruiting | GBM | EGFRvIII BiTE-secreting CAR-T | |||
| NCT05024175 | Observational | Not yet recruiting | GBM | / | |||
| NCT01454596 [257] | 1/2 | Completed | Malignant Gliomas (EGFRvIII+) | EGFRvIII CAR-T | mOS: 6.9 months (2.8–10) | ||
| NCT03941626 | 1/2 | Unknown | Solid Tumors (EGFRvIII+) | EGFRvIII CAR-T | |||
| NCT03638206 | 1/2 | Unknown | Solid Tumors (EGFRvIII+) | EGFRvIII CAR-T | |||
| EMMPRIN | NCT04045847 | 1 | Unknown | Recurrent Malignant Gliomas (CD147+) | EMMPRIN CAR-T | ||
| GD2 | NCT03170141 [258] | 1 | Enrolling by invitation | rGBM (GD2+) | GD2 CAR-T | mOS = 10 months (3–24) | |
| HER2/neu | NCT01109095 [259] | 1 | Completed | GBM | HER2 CAR-T | ||
| NCT03389230 | 1 | Active, not recruiting | Recurrent or Refractory Malignant Gliomas | HER2 CAR-T | |||
| IL-13Rα2 | NCT02208362 [260] | 1 | Active, not Recruiting | Recurrent Malignant Gliomas | IL-13Rα2 CAR-T (intratumoral) | ||
| IL-13Rα2 CAR-T (intracavitary) | |||||||
| IL-13Rα2 CAR-T (intraventricular) | |||||||
| IL-13Rα2 CAR-T (intratumoral/intraventricular) | |||||||
| NCT04661384 | 1 | Recruiting | Leptomeningeal GBM, Ependymoma, or Medulloblastoma | IL-13Rα2 CAR-T | |||
| NCT05540873 | 1 | Recruiting | Recurrent or Refractory Malignant Gliomas | IL-13Rα2 CAR-T | |||
| NCT00730613 [261] | 1 | Completed | Recurrent or Refractory Malignant Gliomas | IL-13Rα2 CTLs | |||
| MMP2 (Chlorotoxin) | NCT04214392 | 1 | Recruiting | rGBM (MMP2+) | MMP2 CAR-T (intratumoral) | ||
| MMP2 CAR-T (intratumoral/intraventricular) | |||||||
| NCT05627323 [262] | 1 | Recruiting | rGBM (MMP2+) | MMP2 CAR-T | |||
| NKG2D | NCT04270461 | 1 | Withdrawn | Recurrent Solid Tumors (NKG2DL+) | NKG2D CAR-T | ||
| NCT05131763 | 1 | Recruiting | Recurrent Solid Tumors (NKG2DL+) | NKG2D CAR-T | |||
| NCT04717999 | N/A | Not yet recruiting | rGBM | NKG2D CAR-T | |||
| NCT04550663 | 1 | Unknown | Relapsed or Refractory Solid Tumors (NKG2DL+) | NKG2D CAR-T | |||
| PD-L1 | NCT02937844 | 1 | Unknown | rGBM | PD-L1 CAR-T | ||
| SNC-109 | NCT05868083 | 1 | Recruiting | rGBM | SNC-109 CAR-T | ||
| Polyvalent CAR-T | IL-7Ra, CD44 and CD133 | NCT05577091 | 1 | Not yet recruiting | rGBM | Tris-CAR-T | |
| EGFRvIII, IL-13Rα2, HER2/neu, EphA2, CD133, GD2 | NCT03423992 [263] | 1 | Unknown | Recurrent Malignant Gliomas | Personalized CAR-T | mOS (EphA2-specific CAR-T) = 86–181 days | |
| TILs | NCT05333588 | 1 | Recruiting | GBM | TILs | ||
| NCT03347097 [264] | 1 | Unknown | rGBM | TILs | mOS: 16.1 months | ||
| PD-1-TILs | mOS: 11.2 months | ||||||
| NCT04943913 | 1 | Recruiting | Gliomas | TILs | |||
| Virus Name | Virus Type |
NCT Number | Phase |
Study Status | Tumor Target | Intervention | Outcome | |
|---|---|---|---|---|---|---|---|---|
| Adenovirus | OV | CRAd-S-pk7 | NCT05139056 | 1 | Recruiting | Recurrent Malignant Gliomas | NSC-expressing CRAd-S-pk7 | |
| NCT03072134 [281] | 1 | Completed | Newly Diagnosed Malignant Gliomas | NSC-expressing CRAd-S-pk7 | mPFS: 9.1 months (95% CI, 8.5–36), mOS: 18.4 months (95% CI, 6.5–36) | |||
| DNX-2401 | NCT03896568 [282] | 1 | Recruiting | Recurrent Malignant Gliomas | BM-hMSCs loaded with DNX-2401 | |||
| NCT02197169 [283] | 1 | Completed | rGBM or Gliosarcoma | DNX-2401 | ||||
| DNX-2401 + IFN-γ | ||||||||
| NCT01956734 [284] | 1 | Completed | rGBM | DNX-2401 + TMZ | ||||
| NCT01582516 | 1/2 | Completed | rGBM | DNX-2401 | ||||
| NCT00805376 [283] | 1 | Completed | Recurrent Malignant Gliomas | DNX-2401 | mOS: 9.5 months | |||
| DNX-2401 + Surgery | mOS: 13 months | |||||||
| DNX-2440 | NCT03714334 | 1 | Terminated | rGBM | DNX-2440 | |||
| ONYX-015 | [285] | 1 | Completed | Recurrent Malignant Gliomas | ONYX-015 | mOS (all patients): 6.2 months (1.3–28.0), mOS (GBM patients): 4.9 months | ||
| Non-Lytic | AdV-ECRT-122T | NCT06102525 | 1/2 | Not yet recruiting | GBM (hTERT+) | AdV-ECRT-122T + Valganciclovir | ||
| AdV-HSV-TK | NCT00002824 | 1 | Completed | Primary Brain Tumors | AdV-HSV-TK + Ganciclovir | |||
| NCT01811992 [286] | 1 | Completed | Malignant Gliomas | AdV-HSV-TK + AdV-Flt3L + Valacyclovir | mOS: 21.3 months (95% CI, 11.1–26.1) | |||
| NCT00751270 [287] | 1 | Completed | Malignant Gliomas | Resectable Gliomas = AdV-HSV-TK + Valacyclovir + RT | ||||
| Unresectable Gliomas = AdV-HSV-TK + Valacyclovir + RT | ||||||||
| NCT03596086 | 1/2 | Recruiting | rGBM | AdV-HSV-TK + Valacyclovir + Radiochemotherapy | ||||
| NCT03603405 | 1/2 | Recruiting | ndGBM | AdV-HSV-TK + Valacyclovir + Radiochemotherapy | ||||
| NCT00870181 [288] | 2 | Completed | Recurrent Malignant Gliomas | AdV-HSV-TK + Ganciclovir | PFS-6: 71.4%, mPFS: 34.9 weeks (9.0–238.4), mOS: 45.7 weeks (9.0–238.4) | |||
| SOC | PFS-6: 5.6%, mPFS: 7.4 weeks (1.1–35.3), mOS: 8.6 weeks (1.1–45.0) | |||||||
| NCT00589875 [289] | 2 | Completed | Malignant Gliomas | AdV-HSV-TK + Valacyclovir + RT | mOS: 17.1 months | |||
| SOC | mOS: 13.5 months | |||||||
| Ad-RTS-IL-12 | NCT02026271 [290] | 1 | Completed | Malignant Gliomas | Ad-RTS-IL-12 + Veledimex | |||
| AdV-IFN-β | NCT05914935 | 1 | Recruiting | rGBM | AdV-IFN-β | |||
| NCT00031083 | 1 | Completed | Malignant Gliomas | AdV-IFN-β | ||||
| AdV-p53 | NCT00004041 | 1 | Completed | Recurrent Malignant Gliomas | AdV-p53 | |||
| NCT00004080 | 1 | Completed | Recurrent or Progressive Brain Tumors | AdV-p53 | ||||
| VB-111 | NCT01260506 [291] | 1/2 | Completed | rGBM | VB-111 until progression | mOS: 223 days, OS-12: 18% | ||
| VB-111 upon progression + Bev (primed combination) | mOS: 414 days, OS-12: 57% | |||||||
| VB-111 + Bev (unprimed combination) | mOS: 141.5 days, OS-12: 10% | |||||||
| NCT02511405 [292] | 3 | Completed | rGBM | VB-111 + Bev | mOS: 6.8 months, ORR: 27.3% | |||
| Bev | mOS: 7.9 months, ORR: 21.9% | |||||||
| Herpes Simplex Virus | OV | C134 | NCT03657576 | 1 | Recruiting | rGBM | C134 | |
| C5252 | NCT05095441 | 1 | Not yet recruiting | rGBM or Progressive GBM | C5252 | |||
| HSV-1716 | NCT02031965 | 1 | Terminated | Recurrent Malignant Gliomas | HSV-1716 | |||
| [293] | 1 | Completed | Recurrent Malignant Gliomas | HSV-1716 | ||||
| [294] | 1 | Completed | Malignant Gliomas | HSV-1716 | ||||
| [295] | 1 | Completed | Malignant Gliomas | HSV-1716 | ||||
| G207 | NCT00157703 [296] | 1 | Completed | Malignant Gliomas | G207 + RT | mOS: 7.5 months (95% CI, 3.0–12.7) | ||
| NCT00028158 [297] | 1/2 | Completed | Recurrent Brain Tumors | G207 | ||||
| NCT00036699 [298] | 1/2 | Completed | Recurrent Brain Tumors | G207 | ||||
| G47Δ | UMIN000002661 [299] | 1/2 | Completed | rGBM or Progressive GBM | G47Δ | mOS: 30.5 (95% CI, 19.2–52.7) | ||
| M032 | NCT02062827 | 1 | Active, not recruiting | Recurrent Malignant Gliomas | M032 (NSC 733972) | |||
| rQnestin34.5v.2 | NCT03152318 [300,301] | 1 | Recruiting | Recurrent Malignant Gliomas | rQNestin34.5v.2 | |||
| rQNestin34.5v.2 + Cyclophosphamide | ||||||||
| rQNestin34.5v.2 (Multiple doses) | ||||||||
| Retrovirus | Non-Lytic | RV-HSV-TK | [302] | 3 | Completed | ndGBM | SOC | mOS: 354 days (95% CI, 315–372), OS-12: 55% |
| SOC + RV-HSV-TK + Ganciclovir | mOS: 365 days (95% CI, 334–416), OS-12: 50% | |||||||
| Toca 511 | NCT01985256 [303] | 1 | Completed | Recurrent Brain Tumors | Toca 511 + 5-FC | |||
| NCT02576665 [304] | 1 | Terminated | Solid Tumors or Lymphomas | Toca 511 + 5-FC | ||||
| NCT01470794 [305,306] | 1 | Completed | Recurrent Malignant Brain Tumors | Toca 511 + 5-FC | ||||
| NCT01156584 [307] | 1 | Completed | Recurrent Malignant Gliomas | Toca 511 + 5-FC | ||||
| NCT04327011 | 1 | Terminated | / | Toca 511 + 5-FC (Long term safety follow-up) | ||||
| NCT02414165 [308] | 2/3 | Terminated | Recurrent Malignant Gliomas | Toca 511 + 5-FC | mOS: 11.10 months | |||
| Lomustine, TMZ or Bev | mOS: 12.22 months | |||||||
| NCT04105374 [309] | 2/3 | Withdrawn | ndGBM | SOC | ||||
| SOC + Toca 511 + 5-FC | ||||||||
| Measles Virus | OV | MV-CEA | NCT00390299 | 1 | Completed | rGBM | MV-CEA (Intracavitary) | PFS-6: 22.2% (95% CI, 6.6–75.4), mOS: 11.8 months (95% CI, 4.4-N/A) |
| MV CEA (Intratumoral/Intracavitary) | PFS-6: 23.1% (95% CI, 8.6–62.3), mOS: 11.4 months (95% CI, 4.3-N/A) | |||||||
| Newcastle Disease Virus | OV | NDV-HUJ | NCT01174537 [310] | 1/2 | Withdrawn | rGBM, Sarcoma or Neuroblastoma | NDV (HUJ strain) | |
| NDV-MTH-68/H | [311] | / | / | Malignant Gliomas | NDV (MTH-68/H strain) | |||
| Parvovirus | OV | H-1PV | NCT01301430 [312,313] | 1/2 | Completed | rGBM or Progressive GBM | H-1PV | |
| Poliovirus | OV | PVSRIPO | NCT01491893 [314] | 1 | Completed | rGBM | PVSRIPO | mOS (PVSRIPO): 12.5 months (95% CI, 9.9–15.2), mOS (historical controls): 11.3 months (95% CI, 9.8–12.5) |
| NCT02986178 | 2 | Active, not recruiting | Recurrent Malignant Gliomas | PVSRIPO | ||||
| Reovirus | OV | Reolysin | NCT00528684 | 1 | Completed | Malignant Gliomas | Reolysin | |
| [315] | 1 | Completed | Recurrent Malignant Gliomas | Reolysin | mOS: 21 weeks (6 to 234) | |||
| [316] | 1 | Completed | Recurrent Malignant Gliomas | Reolysin | mOS: 140 days (97 to 989) | |||
| [317] | 1 | Completed | Malignant Gliomas and Brain Metastases | Reolysin | mOS: 469 days (118 to 1079) | |||
| Vaccinia Virus | OV | TG6002 | NCT03294486 | 1/2 | Completed | rGBM | TG6002 + 5-FC | |
| Combination | NCT Number | Phase | Study Status | Tumor Target | Intervention | Outcome | |
|---|---|---|---|---|---|---|---|
| ICT + ACT | Anti-PD-1 + CAR-T | NCT03726515 | 1 | Completed | ndGBM (MGMT-UN) | EGFRvIII CAR-T + Pembro | |
| NCT04003649 | 1 | Recruiting | rGBM or Refractory GBM | Nivo + IL-13Rα2 CAR-T + Ipi | |||
| Nivo + IL-13Rα2 CAR-T | |||||||
| IL-13Rα2 CAR-T | |||||||
| ICT + Vaccine | Anti-PD-1 + CMV-DC | NCT02529072 | 1 | Completed | Recurrent Brain Tumors | Nivo + Surgery + Nivo&CMV-DC | |
| Nivo&CMV-DC + Surgery + Nivo&CMV-DC | |||||||
| Anti-PD-1 + HSPPC-96 | NCT03018288 | 2 | Completed | ndGBM (MGMT-UN) | RT + TMZ | ||
| RT + TMZ + Pembro | |||||||
| RT + TMZ + Pembro + HSPPC-96 Vaccine | |||||||
| RT + TMZ + Pembro + Placebo | |||||||
| Anti-PD-1 + IMA950 | NCT03665545 [364] | 1/2 | Active, not recruiting | rGBM | IMA950 + Poly-ICLC | ||
| IMA950 + Poly-ICLC + Pembro | |||||||
| Anti-PD-1 or Anti-CTLA-4 + NeoVax | NCT03422094 | 1 | Terminated | ndGBM (MGMT-UN) | NeoVax + Nivo (start at time of progression) | ||
| NeoVax + Nivo (start with Cycle 1) | |||||||
| NeoVax + Nivo (start with Cycle 2) | |||||||
| NeoVax + Ipi + Nivo (start with Cycle 3) | |||||||
| NeoVax + Ipi + Nivo (day 1&15 each cycle) | |||||||
| NCT02287428 [237,365] | 1 | Recruiting | ndGBM | RT + NeoVax | mPFS: 7.6 months (90% CI, 6.2–9.5), mOS: 16.8 months (90% CI, 9.6–21.3) | ||
| RT + Pembro followed by NeoVax + Pembro | |||||||
| RT followed by NeoVax + Pembro | |||||||
| RT + 1 dose Pembro followed by NeoVax + Pembro | |||||||
| MGMT m = RT + TMZ Followed by TMZ + NeoVax + Pembro | |||||||
| Anti-PD-1 + SurVaxM | NCT04013672 [366] | Phase 2 | Active, not recruiting | rGBM | Pembro + SurVaxM/Montanide ISA-51 + GM-CSF (no prior immunotherapy) | ||
| Pembro + SurVaxM/Montanide ISA-51 + GM-CSF (prior failed immunotherapy) | |||||||
| Anti-PD-1 + DC-Tumor Lysate | NCT03014804 | 2 | Withdrawn | rGBM | DCVax-L | ||
| DCVax-L + Nivo | |||||||
| NCT04201873 | 1 | Recruiting | rGBM | Pembro + ATL-DC + Poly-ICLC | |||
| Placebo + ATL-DC + Poly-ICLC | |||||||
| ICT + Virus | Anti-PD-1 + AdV | NCT03576612 | 1 | Active, not recruiting | Newly Diagnosed Malignant Gliomas | MGMT-UN = AdV-HSV-TK/Valacyclovir + RT + TMZ + Nivo | |
| MGMT m and undetermined = AdV-HSV-TK/Valacyclovir + RT + TMZ + Nivo | |||||||
| NCT03636477 [367] | 1 | Completed | rGBM or Progressive GBM | Ad-RTS-IL-12 + Veledimex + Nivo | mOS: 16.9 months | ||
| Nivo | mOS: 9.8 months | ||||||
| Anti-PD-1 + HSV | NCT05084430 | 1/2 | Recruiting | Recurrent Malignant Gliomas | rGBM = Pembro + M032 | ||
| ndGBM = Pembro + M032 | |||||||
| NCT02798406 [337] | 2 | Completed | rGBM or Gliosarcoma | DNX-2401 + Pembro | ORR: 10.4% (90% CI, 4.2–20.7), OS-12: 52.7% (95% CI, 40.1–69.2), mOS: 12.5 months (10.7–13.5) | ||
| Anti-PD-1 + Poliovirus | NCT04479241 [368] | 2 | Active, not recruiting | rGBM | PVSRIPO + Pembro | ||
| Anti-PD-L1 + Poliovirus | NCT03973879 | 1/2 | Withdrawn | Recurrent Malignant Gliomas | PVSRIPO + Atezo | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvato, I.; Marchini, A. Immunotherapeutic Strategies for the Treatment of Glioblastoma: Current Challenges and Future Perspectives. Cancers 2024, 16, 1276. https://doi.org/10.3390/cancers16071276
Salvato I, Marchini A. Immunotherapeutic Strategies for the Treatment of Glioblastoma: Current Challenges and Future Perspectives. Cancers. 2024; 16(7):1276. https://doi.org/10.3390/cancers16071276
Chicago/Turabian StyleSalvato, Ilaria, and Antonio Marchini. 2024. "Immunotherapeutic Strategies for the Treatment of Glioblastoma: Current Challenges and Future Perspectives" Cancers 16, no. 7: 1276. https://doi.org/10.3390/cancers16071276
APA StyleSalvato, I., & Marchini, A. (2024). Immunotherapeutic Strategies for the Treatment of Glioblastoma: Current Challenges and Future Perspectives. Cancers, 16(7), 1276. https://doi.org/10.3390/cancers16071276