

Novel Avenues for the Detection of Cancer-Associated Viral Genome Integrations Using Long-Read Sequencing Technologies



Simple Summary
Abstract
1. Introduction
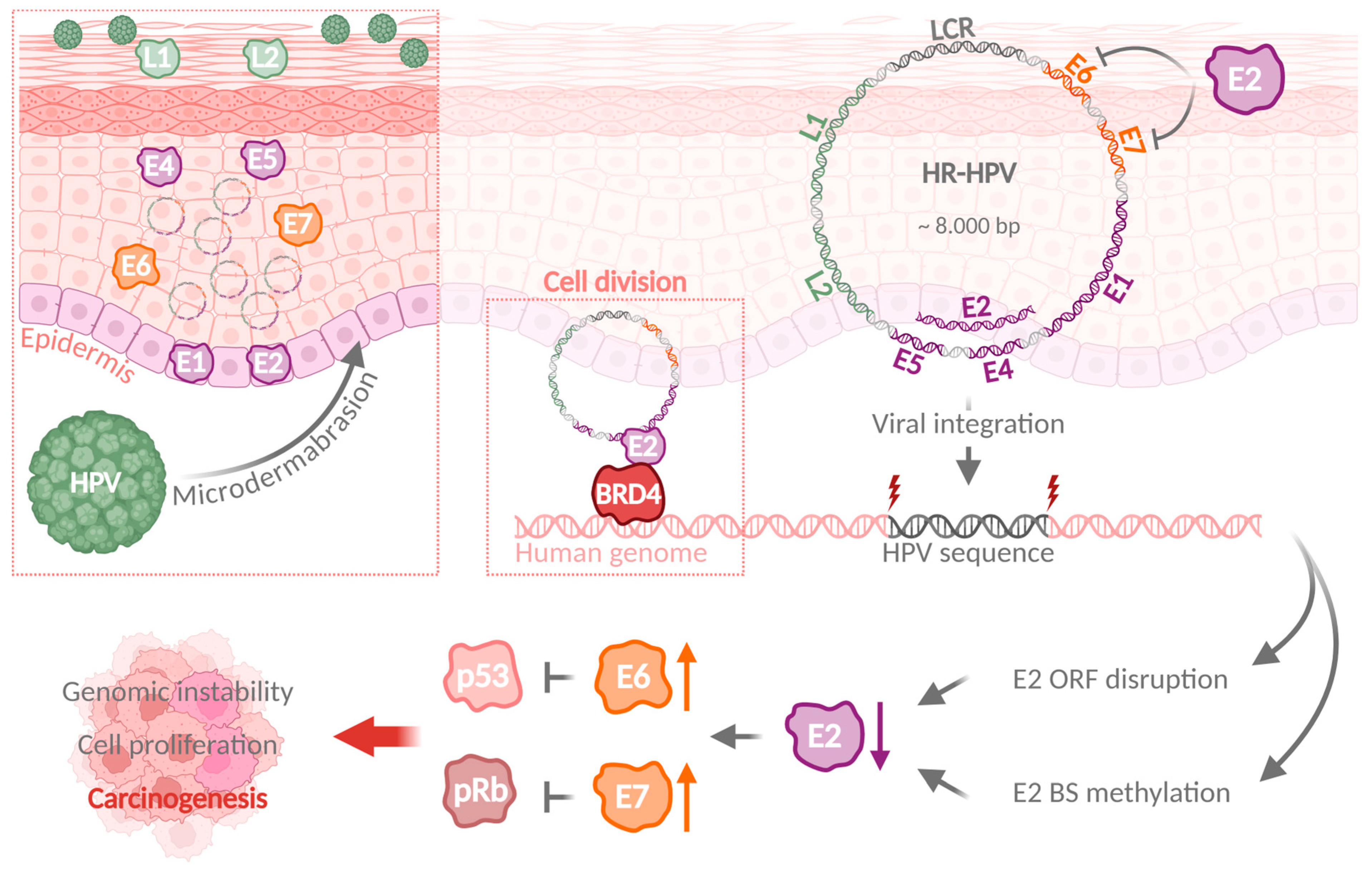
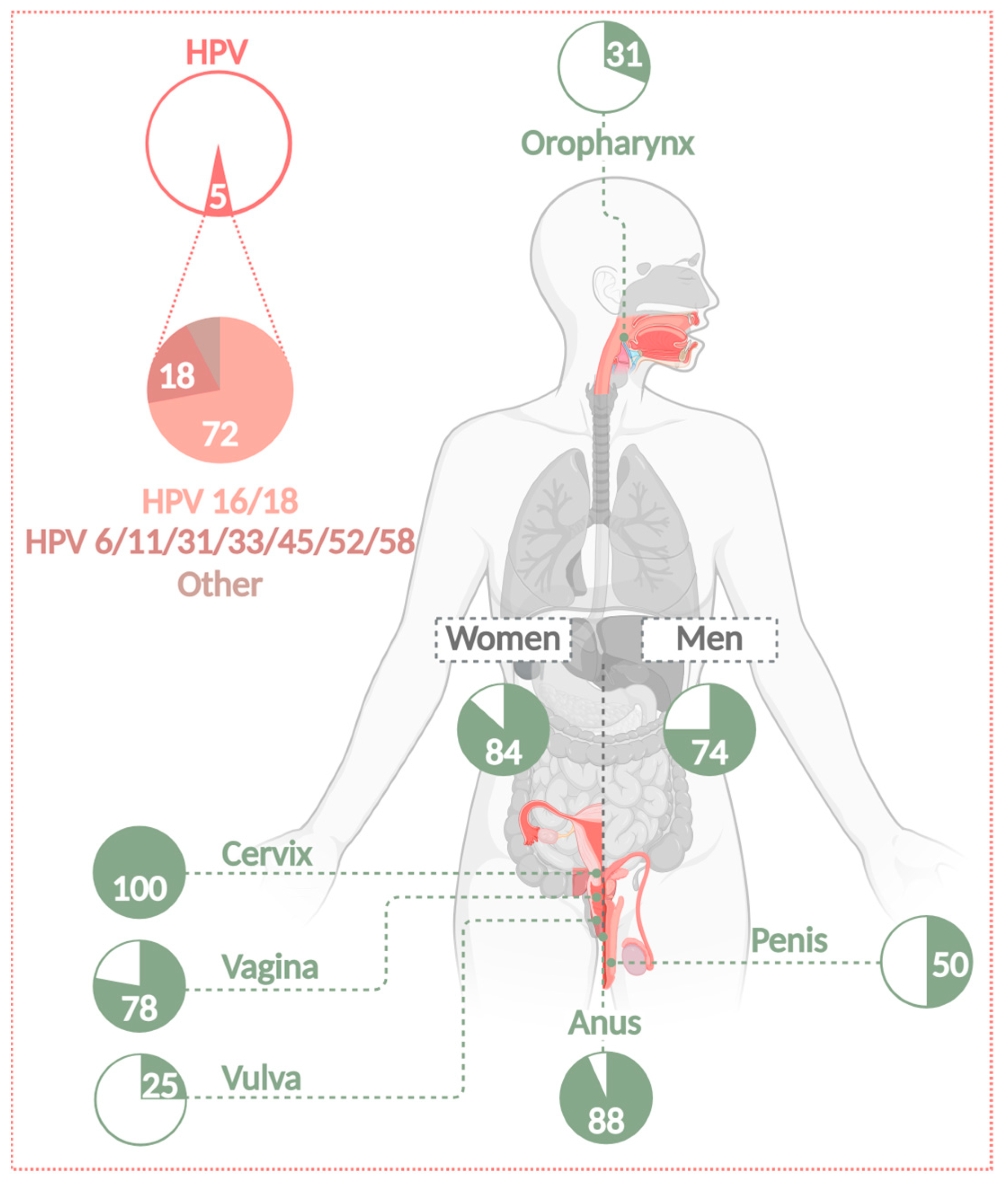
2. Classification and Occurrence of Human Papillomaviruses
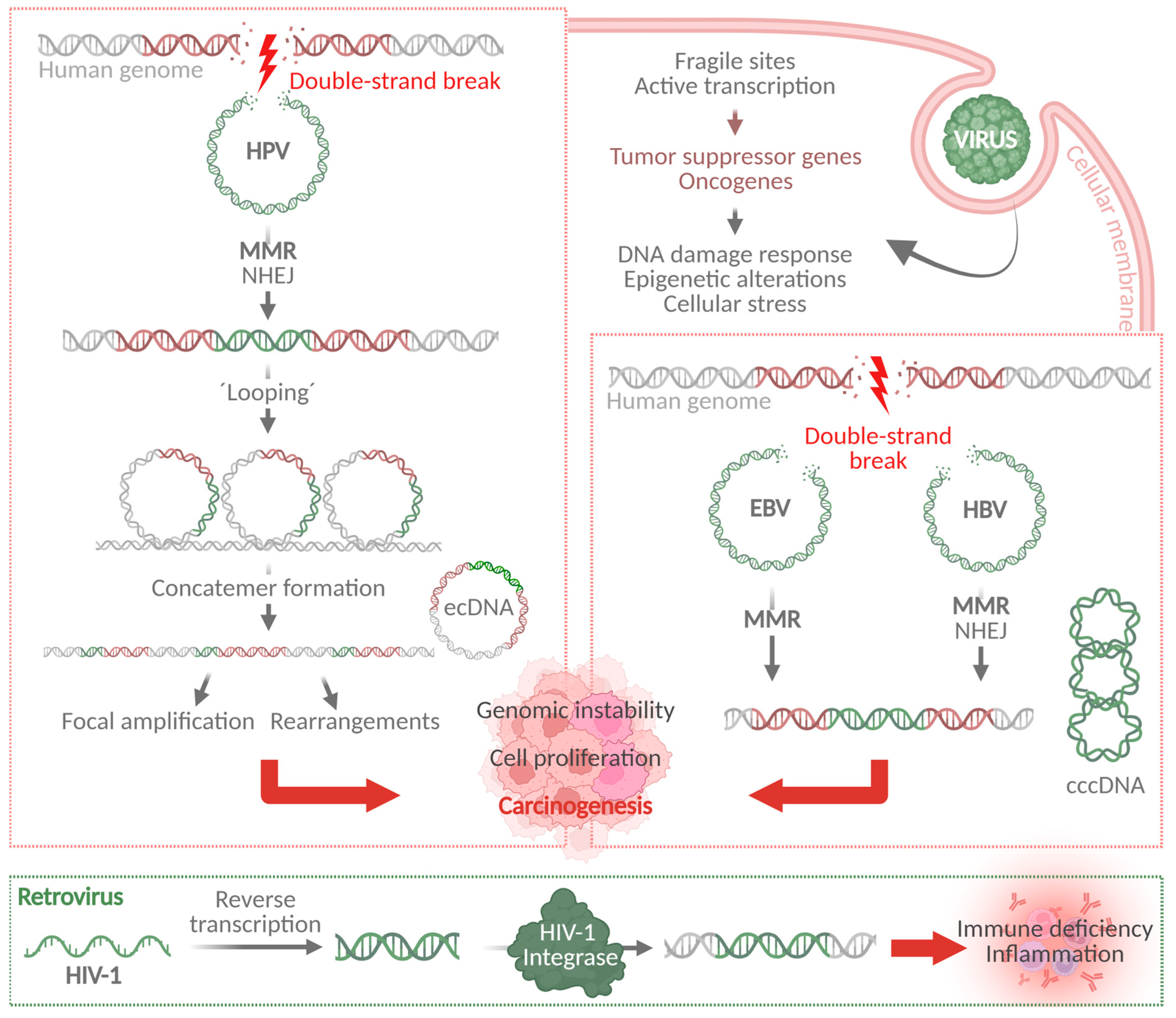
3. Mechanisms of Viral Genome Integration Are Shared Between Different Oncogenic Viruses
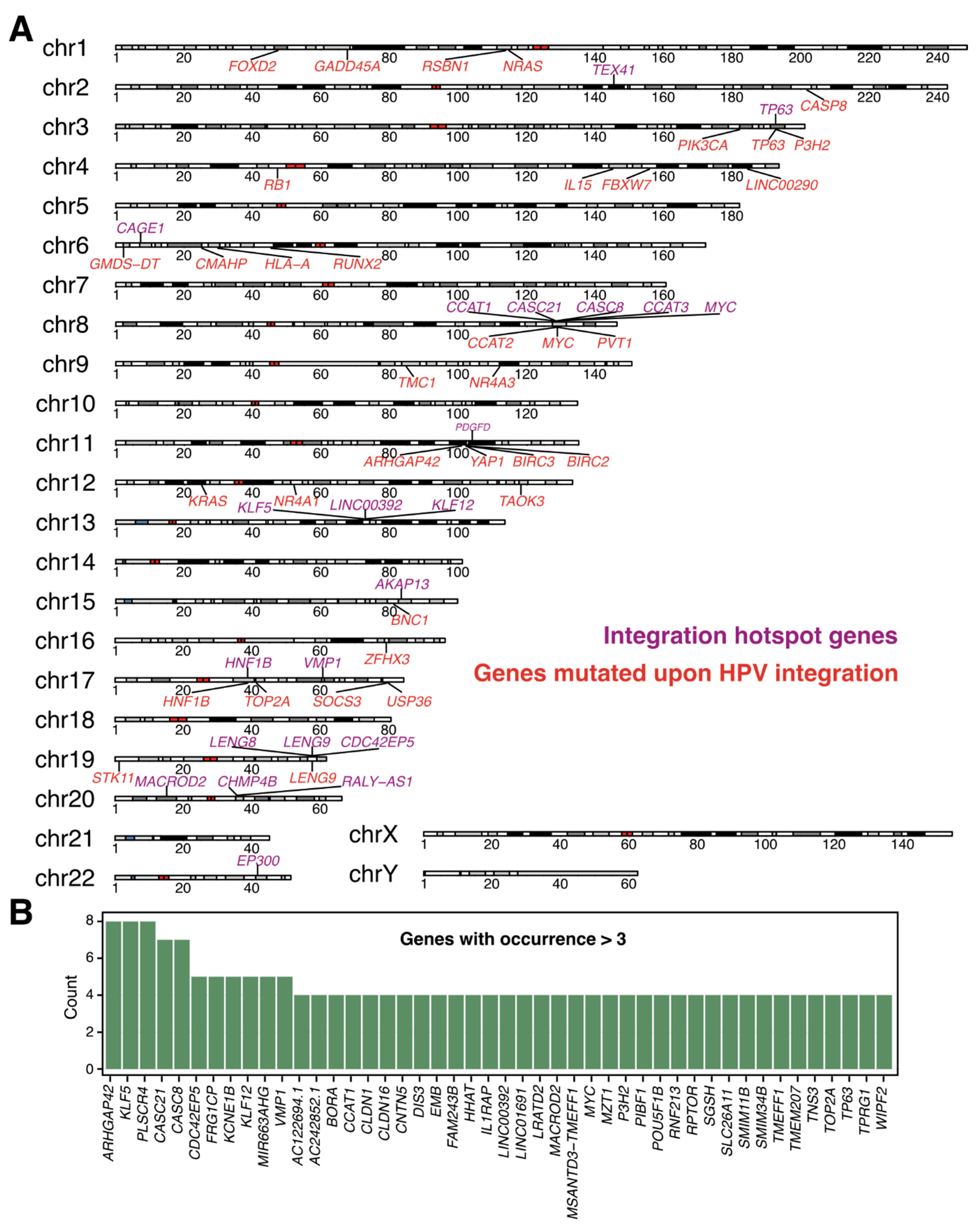
4. Recurrent HPV Integration Sites Are Associated with Tumor Progression
5. Technologies Can Identify Viral Integration Sites with Variable Resolution
6. LR-Seq Efficiently Resolves Structural and Functional Complexities of HPV Integration
7. HPV Frequently Integrates into Repetitive and Non-Coding Regions of the Host Genome
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- de Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Warburton, A.; Markowitz, T.E.; Katz, J.P.; Pipas, J.M.; McBride, A.A. Recurrent integration of human papillomavirus genomes at transcriptional regulatory hubs. NPJ Genom. Med. 2021, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Catalán-Castorena, O.; Garibay-Cerdenares, O.L.; Illades-Aguiar, B.; Rodríguez-Ruiz, H.A.; Zubillaga-Guerrero, M.I.; Leyva-Vázquez, M.A.; Encarnación-Guevara, S.; Alarcón-Romero, L.D.C. The role of HR-HPV integration in the progression of premalignant lesions into different cancer types. Heliyon 2024, 10, e34999. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K.; Symer, D.E.; Mahmoud, M.; Jiang, B.; Goodwin, S.; Wangsa, D.; Li, Z.; Xiao, W.; Dunn, J.D.; Ried, T.; et al. Intratumoral Heterogeneity and Clonal Evolution Induced by HPV Integration. Cancer Discov. 2023, 13, 910–927. [Google Scholar] [CrossRef]
- Aganezov, S.; Yan, S.M.; Soto, D.C.; Kirsche, M.; Zarate, S.; Avdeyev, P.; Taylor, D.J.; Shafin, K.; Shumate, A.; Xiao, C.; et al. A complete reference genome improves analysis of human genetic variation. Science 2022, 376, eabl3533. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32 (Suppl. 1), S7–S15. [Google Scholar] [CrossRef]
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360. [Google Scholar] [CrossRef]
- Della Fera, A.N.; Warburton, A.; Coursey, T.L.; Khurana, S.; McBride, A.A. Persistent Human Papillomavirus Infection. Viruses 2021, 13, 321. [Google Scholar] [CrossRef]
- Egawa, N.; Wang, Q.; Griffin, H.M.; Murakami, I.; Jackson, D.; Mahmood, R.; Doorbar, J. HPV16 and 18 genome amplification show different E4-dependence, with 16E4 enhancing E1 nuclear accumulation and replicative efficiency via its cell cycle arrest and kinase activation functions. PLoS Pathog. 2017, 13, e1006282. [Google Scholar] [CrossRef]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006, 110, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. 1), 2–23. [Google Scholar] [CrossRef] [PubMed]
- Porter, V.L.; Marra, M.A. The Drivers, Mechanisms, and Consequences of Genome Instability in HPV-Driven Cancers. Cancers 2022, 14, 4623. [Google Scholar] [CrossRef] [PubMed]
- de Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef]
- Muñoz, N.; Bosch, F.X.; de Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef]
- Saraiya, M.; Unger, E.R.; Thompson, T.D.; Lynch, C.F.; Hernandez, B.Y.; Lyu, C.W.; Steinau, M.; Watson, M.; Wilkinson, E.J.; Hopenhayn, C.; et al. US assessment of HPV types in cancers: Implications for current and 9-valent HPV vaccines. J. Natl. Cancer Inst. 2015, 107, djv086. [Google Scholar] [CrossRef]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Z.; Xu, R.; Ke, Y. Comprehensive mapping of the human papillomavirus (HPV) DNA integration sites in cervical carcinomas by HPV capture technology. Oncotarget 2016, 7, 5852–5864. [Google Scholar] [CrossRef]
- Garza-Rodríguez, M.L.; Oyervides-Muñoz, M.A.; Pérez-Maya, A.A.; Sánchez-Domínguez, C.N.; Berlanga-Garza, A.; Antonio-Macedo, M.; Valdés-Chapa, L.D.; Vidal-Torres, D.; Vidal-Gutiérrez, O.; Pérez-Ibave, D.C.; et al. Analysis of HPV Integrations in Mexican Pre-Tumoral Cervical Lesions Reveal Centromere-Enriched Breakpoints and Abundant Unspecific HPV Regions. Int. J. Mol. Sci. 2021, 22, 3242. [Google Scholar] [CrossRef]
- Koneva, L.A.; Zhang, Y.; Virani, S.; Hall, P.B.; McHugh, J.B.; Chepeha, D.B.; Wolf, G.T.; Carey, T.E.; Rozek, L.S.; Sartor, M.A. HPV Integration in HNSCC Correlates with Survival Outcomes, Immune Response Signatures, and Candidate Drivers. Mol. Cancer Res. 2018, 16, 90–102. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.; Lameiras, S.; Jeannot, E.; Marie, Y.; Castera, L.; Sastre-Garau, X.; Nicolas, A. Mechanistic signatures of HPV insertions in cervical carcinomas. NPJ Genom. Med. 2016, 1, 16004. [Google Scholar] [CrossRef] [PubMed]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Zhu, W.; Zhou, J.; Li, H.; Xu, X.; Zhang, B.; Gao, X. Repetitive DNA sequence detection and its role in the human genome. Commun. Biol. 2023, 6, 954. [Google Scholar] [CrossRef]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef]
- Porter, V.L.; Ng, M.; O’Neill, K.; MacLennan, S.; Corbett, R.D.; Culibrk, L.; Hamadeh, Z.; Iden, M.; Schmidt, R.; Tsaih, S.W.; et al. Rearrangements of viral and human genomes at human papillomavirus integration events and their allele-specific impacts on cancer genome regulation. Genome Res. 2025, 35, 653–670. [Google Scholar] [CrossRef]
- Zhou, L.; Qiu, Q.; Zhou, Q.; Li, J.; Yu, M.; Li, K.; Xu, L.; Ke, X.; Xu, H.; Lu, B.; et al. Long-read sequencing unveils high-resolution HPV integration and its oncogenic progression in cervical cancer. Nat. Commun. 2022, 13, 2563. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- Groves, I.J.; Coleman, N. Human papillomavirus genome integration in squamous carcinogenesis: What have next-generation sequencing studies taught us? J. Pathol. 2018, 245, 9–18. [Google Scholar] [CrossRef]
- MacLennan, S.A.; Marra, M.A. Oncogenic Viruses and the Epigenome: How Viruses Hijack Epigenetic Mechanisms to Drive Cancer. Int. J. Mol. Sci. 2023, 24, 9543. [Google Scholar] [CrossRef]
- Arora, P.; Kim, E.O.; Jung, J.K.; Jang, K.L. Hepatitis C virus core protein downregulates E-cadherin expression via activation of DNA methyltransferase 1 and 3b. Cancer Lett. 2008, 261, 244–252. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, Z.J.; Jolly, C.; Androphy, E.J.; Mercer, A.; Matthews, C.M.; Hibma, M.H. Transcriptional repression of E-cadherin by human papillomavirus type 16 E6. PLoS ONE 2012, 7, e48954. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.O.; Kwun, H.J.; Jung, J.K.; Choi, K.H.; Min, D.S.; Jang, K.L. Hepatitis B virus X protein represses E-cadherin expression via activation of DNA methyltransferase 1. Oncogene 2005, 24, 6617–6625. [Google Scholar] [CrossRef]
- Tsai, C.L.; Li, H.P.; Lu, Y.J.; Hsueh, C.; Liang, Y.; Chen, C.L.; Tsao, S.W.; Tse, K.P.; Yu, J.S.; Chang, Y.S. Activation of DNA methyltransferase 1 by EBV LMP1 Involves c-Jun NH(2)-terminal kinase signaling. Cancer Res. 2006, 66, 11668–11676. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Wang, Y.; Li, W.; Cui, Z.; Pan, T.; Jin, Z.; Huang, Z.; Li, L.; Lang, B.; Wu, J.; et al. Genome-wide virus-integration analysis reveals a common insertional mechanism of HPV, HBV and EBV. Clin. Transl. Med. 2022, 12, e971. [Google Scholar] [CrossRef]
- Jiang, C.; Lian, X.; Gao, C.; Sun, X.; Einkauf, K.B.; Chevalier, J.M.; Chen, S.M.Y.; Hua, S.; Rhee, B.; Chang, K.; et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 2020, 585, 261–267. [Google Scholar] [CrossRef]
- Artesi, M.; Hahaut, V.; Cole, B.; Lambrechts, L.; Ashrafi, F.; Marçais, A.; Hermine, O.; Griebel, P.; Arsic, N.; van der Meer, F.; et al. PCIP-seq: Simultaneous sequencing of integrated viral genomes and their insertion sites with long reads. Genome Biol. 2021, 22, 97. [Google Scholar] [CrossRef]
- Molina, M.A.; Steenbergen, R.D.M.; Pumpe, A.; Kenyon, A.N.; Melchers, W.J.G. HPV integration and cervical cancer: A failed evolutionary viral trait. Trends Mol. Med. 2024, 30, 890–902. [Google Scholar] [CrossRef]
- McBride, A.A. Human malignancies associated with persistent HPV infection. Oncol. 2024, 29, 457–464. [Google Scholar] [CrossRef]
- Fan, J.; Fu, Y.; Peng, W.; Li, X.; Shen, Y.; Guo, E.; Lu, F.; Zhou, S.; Liu, S.; Yang, B.; et al. Multi-omics characterization of silent and productive HPV integration in cervical cancer. Cell Genom. 2023, 3, 100211. [Google Scholar] [CrossRef]
- Chaiwongkot, A.; Vinokurova, S.; Pientong, C.; Ekalaksananan, T.; Kongyingyoes, B.; Kleebkaow, P.; Chumworathayi, B.; Patarapadungkit, N.; Reuschenbach, M.; von Knebel Doeberitz, M. Differential methylation of E2 binding sites in episomal and integrated HPV 16 genomes in preinvasive and invasive cervical lesions. Int. J. Cancer 2013, 132, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Shen, K.; McBride, A.A. Papillomavirus genomes associate with BRD4 to replicate at fragile sites in the host genome. PLoS Pathog. 2014, 10, e1004117. [Google Scholar] [CrossRef] [PubMed]
- Bodelon, C.; Untereiner, M.E.; Machiela, M.J.; Vinokurova, S.; Wentzensen, N. Genomic characterization of viral integration sites in HPV-related cancers. Int. J. Cancer 2016, 139, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, I.K.; Sandve, G.K.; Schmitz, M.; Dürst, M.; Hovig, E. Transcriptionally active regions are the preferred targets for chromosomal HPV integration in cervical carcinogenesis. PLoS ONE 2015, 10, e0119566. [Google Scholar] [CrossRef]
- Gao, G.; Johnson, S.H.; Vasmatzis, G.; Pauley, C.E.; Tombers, N.M.; Kasperbauer, J.L.; Smith, D.I. Common fragile sites (CFS) and extremely large CFS genes are targets for human papillomavirus integrations and chromosome rearrangements in oropharyngeal squamous cell carcinoma. Genes Chromosomes Cancer 2017, 56, 59–74. [Google Scholar] [CrossRef]
- Thorland, E.C.; Myers, S.L.; Gostout, B.S.; Smith, D.I. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene 2003, 22, 1225–1237. [Google Scholar] [CrossRef]
- Balaji, H.; Demers, I.; Wuerdemann, N.; Schrijnder, J.; Kremer, B.; Klussmann, J.P.; Huebbers, C.U.; Speel, E.M. Causes and Consequences of HPV Integration in Head and Neck Squamous Cell Carcinomas: State of the Art. Cancers 2021, 13, 4089. [Google Scholar] [CrossRef] [PubMed]
- Demers, I.; Balaji, H.; Feitsma, H.; Stelloo, E.; Swennenhuis, J.; Sergeeva, I.; Wuerdemann, N.; van den Hout, M.; Wagner, S.; Kremer, B.; et al. Proximity ligation-based sequencing for the identification of human papillomavirus genomic integration sites in formalin-fixed paraffin embedded oropharyngeal squamous cell carcinomas. J. Med. Virol. 2024, 96, e29837. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, C.; Liu, W.; Lv, X.; Hu, T.; Yang, F.; Yang, W.; He, L.; Huang, X. Long-read sequencing reveals the structural complexity of genomic integration of HPV DNA in cervical cancer cell lines. BMC Genom. 2024, 25, 198. [Google Scholar] [CrossRef]
- Schmitt, M.; Bravo, I.G.; Snijders, P.J.; Gissmann, L.; Pawlita, M.; Waterboer, T. Bead-based multiplex genotyping of human papillomaviruses. J. Clin. Microbiol. 2006, 44, 504–512. [Google Scholar] [CrossRef]
- Luft, F.; Klaes, R.; Nees, M.; Dürst, M.; Heilmann, V.; Melsheimer, P.; von Knebel Doeberitz, M. Detection of integrated papillomavirus sequences by ligation-mediated PCR (DIPS-PCR) and molecular characterization in cervical cancer cells. Int. J. Cancer 2001, 92, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Klaes, R.; Woerner, S.M.; Ridder, R.; Wentzensen, N.; Duerst, M.; Schneider, A.; Lotz, B.; Melsheimer, P.; von Knebel Doeberitz, M. Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res. 1999, 59, 6132–6136. [Google Scholar] [PubMed]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef]
- Sigurpalsdottir, B.D.; Stefansson, O.A.; Holley, G.; Beyter, D.; Zink, F.; Hardarson, M.; Sverrisson, S.; Kristinsdottir, N.; Magnusdottir, D.N.; Magnusson, O.; et al. A comparison of methods for detecting DNA methylation from long-read sequencing of human genomes. Genome Biol. 2024, 25, 69. [Google Scholar] [CrossRef]
- Ho, D.W.-H.; Lyu, X.; Ng, I.O.-L. Viral integration detection strategies and a technical update on Virus-Clip. Biocell 2021, 45, 1495–1500. [Google Scholar] [CrossRef]
- Cameron, D.L.; Jacobs, N.; Roepman, P.; Priestley, P.; Cuppen, E.; Papenfuss, A.T. VIRUSBreakend: Viral Integration Recognition Using Single Breakends. Bioinformatics 2021, 37, 3115–3119. [Google Scholar] [CrossRef]
- Kojima, R.; Nakamoto, S.; Kogure, T.; Ma, Y.; Ogawa, K.; Iwanaga, T.; Qiang, N.; Ao, J.; Nakagawa, R.; Muroyama, R.; et al. Re-analysis of hepatitis B virus integration sites reveals potential new loci associated with oncogenesis in hepatocellular carcinoma. World J. Virol. 2023, 12, 209–220. [Google Scholar] [CrossRef]
- Church, D.M.; Schneider, V.A.; Graves, T.; Auger, K.; Cunningham, F.; Bouk, N.; Chen, H.C.; Agarwala, R.; McLaren, W.M.; Ritchie, G.R.; et al. Modernizing reference genome assemblies. PLoS Biol. 2011, 9, e1001091. [Google Scholar] [CrossRef]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef]
- Zhao, Q.; Yang, S.; Hao, S.; Chen, Z.; Tang, L.; Wu, Z.; Wu, J.; Xu, M.; Ma, Z.; Zhou, L.; et al. Identification of transcriptionally-active human papillomavirus integrants through nanopore sequencing reveals viable targets for gene therapy against cervical cancer. J. Med. Virol. 2024, 96, e29769. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Li, Y.; Zhang, C.; Qi, Y.; Sun, Y.; Li, W. Multiple HPV integration mode in the cell lines based on long-reads sequencing. Front. Microbiol. 2023, 14, 1294146. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.A.; Kadam, A.; Leveque, G.; Golabi, N.; Zeitouni, A.; Richardson, K.; Mascarella, M.; Sadeghi, N.; Loganathan, S.K. Long-read sequencing of oropharyngeal squamous cell carcinoma tumors reveal diverse patterns of high-risk Human Papillomavirus integration. Front. Oncol. 2023, 13, 1264646. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, I.; Rossi, N.M.; Keskus, A.; Xie, Y.; Ahmad, T.; Bryant, A.; Lou, H.; Paredes, J.G.; Milano, R.; Rao, N.; et al. Insights into the Mechanisms and Structure of Breakage-Fusion-Bridge Cycles in Cervical Cancer using Long-Read Sequencing. Medrxiv Prepr. Serv. Health Sci. 2023, 111, 544–561. [Google Scholar] [CrossRef]
- Liu, M.; Han, Z.; Zhi, Y.; Ruan, Y.; Cao, G.; Wang, G.; Xu, X.; Mu, J.; Kang, J.; Dai, F.; et al. Long-read sequencing reveals oncogenic mechanism of HPV-human fusion transcripts in cervical cancer. Transl. Res. J. Lab. Clin. Med. 2023, 253, 80–94. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, Q.; Tang, L.; Chen, Z.; Wu, Z.; Li, K.; Lin, R.; Chen, Y.; Ou, D.; Zhou, L.; et al. Whole Genome Assembly of Human Papillomavirus by Nanopore Long-Read Sequencing. Front. Genet. 2021, 12, 798608. [Google Scholar] [CrossRef]
- Iden, M.; Tsaih, S.W.; Huang, Y.W.; Liu, P.; Xiao, M.; Flister, M.J.; Rader, J.S. Multi-omics mapping of human papillomavirus integration sites illuminates novel cervical cancer target genes. Br. J. Cancer 2021, 125, 1408–1419. [Google Scholar] [CrossRef]
- Yang, W.; Liu, Y.; Dong, R.; Liu, J.; Lang, J.; Yang, J.; Wang, W.; Li, J.; Meng, B.; Tian, G. Accurate Detection of HPV Integration Sites in Cervical Cancer Samples Using the Nanopore MinION Sequencer Without Error Correction. Front. Genet. 2020, 11, 660. [Google Scholar] [CrossRef]
- Pacholewska, A.; Lienhard, M.; Brüggemann, M.; Hänel, H.; Bilalli, L.; Königs, A.; Heß, F.; Becker, K.; Köhrer, K.; Kaiser, J.; et al. Long-read transcriptome sequencing of CLL and MDS patients uncovers molecular effects of SF3B1 mutations. Genome Res. 2024, 34, 1832–1848. [Google Scholar] [CrossRef]
- Karimzadeh, M.; Arlidge, C.; Rostami, A.; Lupien, M.; Bratman, S.V.; Hoffman, M.M. Human papillomavirus integration transforms chromatin to drive oncogenesis. Genome Biol. 2023, 24, 142. [Google Scholar] [CrossRef]
- Hannan, A.J. Tandem Repeats and Repeatomes: Delving Deeper into the ‘Dark Matter’ of Genomes. EBioMedicine 2018, 31, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, S.J.; Storer, J.M.; Hartley, G.A.; Grady, P.G.S.; Gershman, A.; de Lima, L.G.; Limouse, C.; Halabian, R.; Wojenski, L.; Rodriguez, M.; et al. From telomere to telomere: The transcriptional and epigenetic state of human repeat elements. Science 2022, 376, eabk3112. [Google Scholar] [CrossRef] [PubMed]
- Ting, D.T.; Lipson, D.; Paul, S.; Brannigan, B.W.; Akhavanfard, S.; Coffman, E.J.; Contino, G.; Deshpande, V.; Iafrate, A.J.; Letovsky, S.; et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 2011, 331, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Enukashvily, N.I.; Ponomartsev, N.V.; Ketkar, A.; Suezov, R.; Chubar, A.V.; Prjibelski, A.D.; Shafranskaya, D.D.; Elmshäuser, S.; Keber, C.U.; Stefanova, V.N.; et al. Pericentromeric satellite lncRNAs are induced in cancer-associated fibroblasts and regulate their functions in lung tumorigenesis. Cell Death Dis. 2023, 14, 19. [Google Scholar] [CrossRef]
- Ugarković, Đ.; Sermek, A.; Ljubić, S.; Feliciello, I. Satellite DNAs in Health and Disease. Genes 2022, 13, 1154. [Google Scholar] [CrossRef]
- Sharma, S.; Munger, K. The Role of Long Noncoding RNAs in Human Papillomavirus-associated Pathogenesis. Pathogens 2020, 9, 289. [Google Scholar] [CrossRef]
- Lajer, C.B.; Garnæs, E.; Friis-Hansen, L.; Norrild, B.; Therkildsen, M.H.; Glud, M.; Rossing, M.; Lajer, H.; Svane, D.; Skotte, L.; et al. The role of miRNAs in human papilloma virus (HPV)-associated cancers: Bridging between HPV-related head and neck cancer and cervical cancer. Br. J. Cancer 2012, 106, 1526–1534. [Google Scholar] [CrossRef]
- Salgado-Hernández, S.V.; Martínez-Retamoza, L.; Ocadiz-Delgado, R.; Pérez-Mora, S.; Cedeño-Arboleda, G.E.; Gómez-García, M.D.C.; Gariglio, P.; Pérez-Ishiwara, D.G. miRNAs Dysregulated in Human Papillomavirus-Associated Benign Prostatic Lesions and Prostate Cancer. Cancers 2024, 17, 26. [Google Scholar] [CrossRef]
- Gunasekharan, V.; Laimins, L.A. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J. Virol. 2013, 87, 6037–6043. [Google Scholar] [CrossRef]
- Li, B.; Guo, X.; Li, N.; Chen, Q.; Shen, J.; Huang, X.; Huang, G.; Wang, F. WNT1, a target of miR-34a, promotes cervical squamous cell carcinoma proliferation and invasion by induction of an E-P cadherin switch via the WNT/β-catenin pathway. Cell. Oncol. 2020, 43, 489–503. [Google Scholar] [CrossRef]
- Yaginuma, Y.; Yoshimoto, M.; Eguchi, A.; Tokuda, A.; Takahashi, S. The human papillomavirus18 E7 protein inhibits CENP-C binding to α-satellite DNA. Virus Res. 2015, 205, 27–32. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Zhang, R.; Cai, Y.; Yang, X.; Wang, Z.; Li, Y.; Cheng, X.; Ye, X.; Xiang, Y.; et al. Preferential sites for the integration and disruption of human papillomavirus 16 in cervical lesions. J. Clin. Virol. 2013, 56, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Altemose, N. A classical revival: Human satellite DNAs enter the genomics era. Semin. Cell Dev. Biol. 2022, 128, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Dyer, N.; Young, L.; Ott, S. Artifacts in the data of Hu et al. Nat. Genet. 2016, 48, 2–4. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Sequencing Method | Reference Genome | Integration Sites | Reference | |
|---|---|---|---|---|---|
| Hotspot Genes | Genomic Regions | ||||
| Cervical carcinoma | Oxford Nanopore, Oxford, UK | GRCh38 | I: MYC, KLF5, KLF12, TP63 M: NR4A1, NR4A3 | Introns, repeats, exons, CpG islands | Porter et al., 2025 [26] |
| Cervical carcinoma, CaSki, HeLa | Oxford Nanopore | GRCh38 | I: RUNX2, CLIC5 | Introns, intergenic, exons, promotors, UTRs | Zhao et al., 2024 [61] |
| HeLa, SiHa | PacBio, Menlo Park, CA, USA Illumina WGS, San Diego, CA, USA | GRCh38 | I: KLF5, LINC00392, CCAT1, CASC21 | Intergenic, introns of ncRNAs | Wang et al., 2024 [49] |
| CaSki, HeLa, SiHa | Oxford Nanopore | GRCh37 | None reported | None reported | Cui et al., 2023 [62] |
| OPSCC | Oxford Nanopore | GRCh38 | None reported | Intergenic, introns, exons | Gauthier et al., 2023 [63] |
| Cervical carcinoma, HNSCC | Oxford Nanopore | GRCh38 | M: PIK3CA, STK11, CASP8, ZFHX3, RB1, HLA-A, FBXW7, KRAS, MYC, YAP1, RUNX2, BIRC2, BIRC3 | Intragenic, introns | Rodriguez et al., 2023 [64] |
| OPSCC, 93-VU-147T, GUMC-395, HeLa, HTEC | Oxford Nanopore PacBio HiFi | GRCh37 | I: EP300, MYC | None reported | Akagi et al., 2023 [4] |
| Cervical carcinoma | PacBio Iso-Seq | GRCh38 | M: TP63, P3H2, GMDS-DT, CMAHP | Intergenic, introns, exons, UTRs | Liu et al., 2023 [65] |
| Cervical carcinoma | Oxford Nanopore | GRCh37 | I: KLF5, LINC00392, CASC8, CASC21, MACROD2, TEX41, VMP1 M: LINC00290, LINC02500, LENG9, IL20RB, SOX14, LENG8, LENG9, CDC42EP5, CASC21, CCAT2, CASC8, AKAP13 | Introns, intergenic, introns of ncRNAs, exons, UTRs | Zhou et al., 2022 [27] |
| Cervical carcinoma, CaSki | Oxford Nanopore | GRCh38 | I: CSMD3, ZFHX3 | Introns, intergenic | Yang et al., 2021 [66] |
| Cervical carcinoma | PacBio | GRCh37 | M: BNC1, RSBN1, USP36, TAOK3, NRAS, PVT1, TOP2A, SOCS3, GADD45A | Introns, exons | Iden et al., 2021 [67] |
| Cervical carcinoma | Oxford Nanopore | GRCh37 | I: CHMP4B, RALY-AS1 | Intergenic, intros, introns of ncRNAs, exons | Yang et al., 2020 [68] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergmann, L.-A.; Pacholewska, A.; Schweiger, M.R. Novel Avenues for the Detection of Cancer-Associated Viral Genome Integrations Using Long-Read Sequencing Technologies. Cancers 2025, 17, 1740. https://doi.org/10.3390/cancers17111740
Bergmann L-A, Pacholewska A, Schweiger MR. Novel Avenues for the Detection of Cancer-Associated Viral Genome Integrations Using Long-Read Sequencing Technologies. Cancers. 2025; 17(11):1740. https://doi.org/10.3390/cancers17111740
Chicago/Turabian StyleBergmann, Larissa-Anna, Alicja Pacholewska, and Michal R. Schweiger. 2025. "Novel Avenues for the Detection of Cancer-Associated Viral Genome Integrations Using Long-Read Sequencing Technologies" Cancers 17, no. 11: 1740. https://doi.org/10.3390/cancers17111740
APA StyleBergmann, L.-A., Pacholewska, A., & Schweiger, M. R. (2025). Novel Avenues for the Detection of Cancer-Associated Viral Genome Integrations Using Long-Read Sequencing Technologies. Cancers, 17(11), 1740. https://doi.org/10.3390/cancers17111740