
Molecular Genetic Profile of Myelofibrosis: Implications in the Diagnosis, Prognosis, and Treatment Advancements


Simple Summary
Abstract
1. Introduction
2. Mutation Profile and Clonal Evolution of MF
2.1. The Driver Mutations
2.2. Additional Mutations

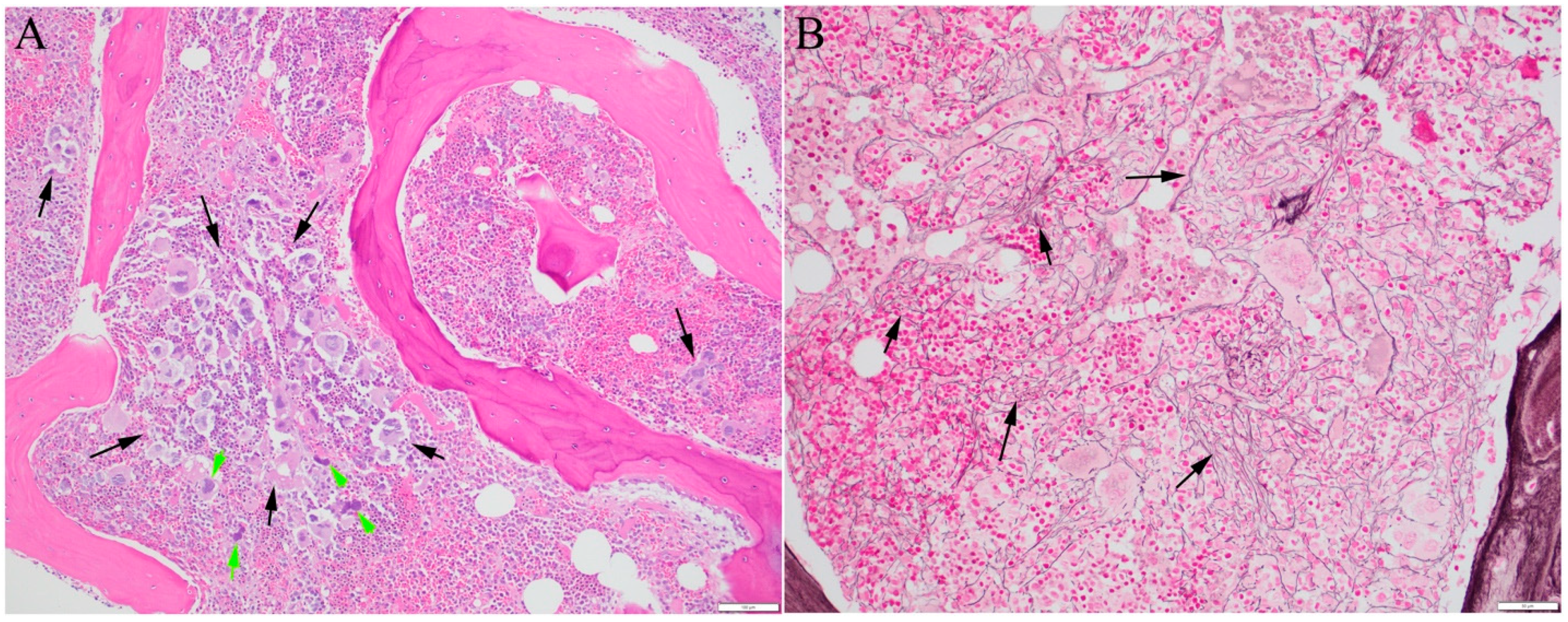{kind=link}
{kind=link}
{kind=link}
| Gene | Mutation Prevalence (%) | Most Frequent Mutations # | More Frequent in PMF Than Other MPN [34,43] | Clinical Relevance |
|---|---|---|---|---|
| Epigenetic Regulation (Chromosome Modification and DNA Methylation) | ||||
| ASXL1 | 21 | Truncation; E635Rfs | Yes | HMR Prevalence increases with age |
| DNMT3A | 12 | R882H/C | Yes | |
| EZH2 | 4 | Truncation and splice | Yes | HMR |
| IDH1/2 | 2 | IDH1 R132C/H, IDH2 R140Q/W | Yes | HMR Prevalence higher in other studies |
| TET2 | 17 | Truncation | No | The order of acquiring mutation affects phenotype |
| RNA splicing | ||||
| SF3B1 | 4 | K666N, K700E | No | Associated with ring sideroblasts |
| SRSF2 | 8 | P95 | Yes | HMR |
| U2AF1 | 5 | Q157, S34 | Yes | HMR |
| ZRSR2 | 2 | Truncation and splice | Yes | More common in SMF [41] |
| Signal transduction and transcription factors | ||||
| CBL | 6 | X366_splice, Y371H | No | Present with other additional mutations [44] Predict poor response to JAK inhibitors [45] |
| CUX1 | 3 | Truncation | Yes | |
| NFE2 | 2–5 * | E261fs | No, related to erythroid differentiation [25] | Associated with higher risk of transformation to AML, shorter OS. More common in SMF [41] |
| NRAS/KRAS | 9 | G12 | Yes | Relatively specific for MF [25,46] |
| RUNX1 | 4 | Truncation | Yes | Associated with transformation to AML [42] |
| SH2B3 | 1 | Truncation | No | May be considered a driver, or promoting JAK2 activity |
| TP53 | 2 | DNA-binding domain mutations | Yes | Relatively uncommon in MPNs. Associated with higher risk of transformation to AML [39]; however, low VAF in subclone may not increase risk [47] |
2.3. The Origin and Evolution of Neoplastic Clones

2.4. Mechanism of Fibrosis
2.5. Laboratory Test Considerations
3. Prognostic and Therapeutic Implications of Mutation Profiles
3.1. Implications in the Prognosis
3.2. Treatment Implications
| Drug | Target/Mechanism | Indications, Clinical Study Findings |
|---|---|---|
| JAK inhibitors, approved by US FDA | ||
| Ruxolitinib | JAK1/2 | Approved for intermediate and high risk PMF or SMF [121] |
| Fedratinib | JAK2 and FLT3 | Similar to ruxolitinib [122] |
| Pacritinib | JAK2, FLT3, IRAK1, CSF1R, and ACVR1 | MF with platelet count <50 K/μL [122] |
| Momelotinib | JAK1/2, and ACVR1 | Approved for intermediate- and high-risk PMF or SMF with anemia [123] |
| Drug Combinations (+/−JAKi) with promising clinical trial results | ||
| Panobinostat | HDAC | Synergistically induce apoptosis [142,143] |
| IMG7289 (Bomedemstat) | LSD1 | Synergize with ruxolitinib, selectively inhibit the ASXL1-mutant clone [144,147] |
| Pelabresib | BET | Lower inflammatory cytokines, act on megakaryocyte differentiation and proliferation [149,150,151,160] |
| C220, PRT543 | PRMT5 | Lower the mutation burden, reducing blood cell counts, and spleen size [153,154] |
| Imetelstat (monotherapy) | telomerase | Better response in patients refractory to JAKi, and harboring additional SF3B1 and U2AF1 mutations [155,156,157] |
| Navitoclax ABT-737 | BCL2 | Synergize with a JAKi, overcome acquired ruxolitinib resistance [158,159] |
| Enasidenib | IDH2 | Only for patients with IDH2 mutation [161] |
| Pegifna (+JAKi or HMA) | Inhibitor of hematopoietic cell proliferation; targeting progenitors carrying the JAK2 V617F mutation | Patients with SRSF2 or ASXL1 mutations did not respond well to IFN-α [169,170,171,172,173]. |
| Pomalidomide (monotherapy) | Immune modulator | Response not affected by additional (HMR) mutations. |
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABL1 | Abelson murine leukemia viral oncogene homolog 1 |
| ACVR1 | Activin A receptor, type I |
| ASXL1 | Additional Sex Combs Like 1 |
| BAPN | β-aminopropionitrile |
| BCR | Breakpoint cluster region |
| BET | Bromodomain and Extra-Terminal |
| CBL | Casitas B-lineage lymphoma proto-oncogene |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CRISPR/Cas9 | Clustered regularly interspaced palindromic repeats/CRISPR-associated protein 9 |
| CSF1R | Colony stimulating factor 1 receptor |
| CUX1 | Cut like homeobox 1 |
| DNMT3A | DNA (cytosine-5)-methyltransferase 3A |
| ETV6 | ETS (E-twenty-six) variant transcription factor 6 |
| EZH2 | Enhancer of zeste homolog 2 |
| FLT3 | fms-like tyrosine kinase 3 |
| G6B | Megakaryocyte and platelet inhibitory receptor G6b |
| HDAC | Histone deacetylase |
| IDH | Isocitrate dehydrogenases |
| IL | Interleukin |
| IRAK1 | Interleukin-1 receptor-associated kinase 1 |
| JAK2 | Janus Kinase 2 |
| KRAS | Kirsten rat sarcoma virus (oncogene) |
| LOX | Lysyl Oxidase |
| LSD1 | Lysine-specific demethylase-1 |
| MPL | Myeloproliferative leukemia proto-oncogene |
| NFE2 | Nuclear factor erythroid 2 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NOTCH1 | Neurogenic locus notch homolog protein 1 |
| NRAS | Neuroblastoma RAS viral oncogene homolog |
| PRMT5 | Protein arginine methyltransferase 5 |
| RUNX1 | Runt-related transcription factor 1 |
| SAP | Serum amyloid P |
| SF3B1 | Splicing Factor 3b, Subunit 1 |
| SH2B3 | Src-homology 2B adapter protein 3 |
| SRSF2 | Serine-arginine splicing factors 2 |
| STAT | Signal transducer and activator |
| TET2 | Ten-Eleven Translocation 2 |
| TP53 | Tumor protein p53 |
| U2AF1 | U2 small nuclear RNA auxiliary factor 1 |
| ZRSR2 | Zinc Finger CCCH-Type, RNA Binding Motif and Serine/Arginine Rich 2 |
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and histiocytic/Dendritic neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Passamonti, F.; Mora, B.; Barraco, D.; Maffioli, M. Post-ET and post-PV myelofibrosis: Updates on a distinct prognosis from primary myelofibrosis. Curr. Hematol. Malig. Rep. 2018, 13, 173–182. [Google Scholar] [CrossRef]
- Puglianini, O.C.; Peker, D.; Zhang, L.; Papadantonakis, N. Essential thrombocythemia and post-essential thrombocythemia myelofibrosis: Updates on diagnosis, clinical aspects, and management. Lab. Med. 2023, 54, 13–22. [Google Scholar] [CrossRef]
- Tefferi, A. How I treat myelofibrosis. Blood 2011, 117, 3494–3504. [Google Scholar] [CrossRef]
- Nangalia, J.; Grinfeld, J.; Green, A.R. Pathogenesis of myeloproliferative disorders. Annu. Rev. Pathol. 2016, 11, 101–126. [Google Scholar] [CrossRef]
- Chen, M.-L.; Zhang, H.-C.; Yang, E.-P. Current status and hotspots evolution in myeloproliferative neoplasm: A bibliometric analysis from 2001 to 2022. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 4510–4519. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Barosi, G.; Massa, M.; Campanelli, R.; Fois, G.; Catarsi, P.; Viarengo, G.; Villani, L.; Poletto, V.; Bosoni, T.; Magrini, U.; et al. Primary myelofibrosis: Older age and high JAK2V617F allele burden are associated with elevated plasma high-sensitivity C-reactive protein levels and a phenotype of progressive disease. Leuk. Res. 2017, 60, 18–23. [Google Scholar] [CrossRef]
- Cazzola, M. Mutant calreticulin: When a chaperone becomes intrusive. Blood 2016, 127, 1219–1221. [Google Scholar] [CrossRef]
- Kanagal-Shamanna, R.; Kikkeri, N.N.; Sandeep, S.D.; Mesa, R.; Giraudier, S.; Harrison, C.N.; Milojkovic, D. Primary Myelofibrosis. WHO Classifications of Tumours Online. Available online: https://tumourclassification.iarc.who.int/chaptercontent/63/15 (accessed on 3 December 2023).
- Kim, S.Y.; Im, K.; Park, S.N.; Kwon, J.; Kim, J.-A.; Lee, D.S. CALR, JAK2, and MPL mutation profiles in patients with four different subtypes of myeloproliferative neoplasms: Primary myelofibrosis, essential thrombocythemia, polycythemia vera, and myeloproliferative neoplasm, unclassifiable. Am. J. Clin. Pathol. 2015, 143, 635–644. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef]
- Pecquet, C.; Papadopoulos, N.; Balligand, T.; Chachoua, I.; Tisserand, A.; Vertenoeil, G.; Nédélec, A.; Vertommen, D.; Roy, A.; Marty, C.; et al. Secreted mutant calreticulins as rogue cytokines in myeloproliferative neoplasms. Blood 2023, 141, 917–929. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Maccari, C.; Sordi, B.; Balliu, M.; Atanasio, A.; Mannarelli, C.; Capecchi, G.; Sestini, I.; Coltro, G.; Loscocco, G.G.; et al. Phenotypic correlations of CALR mutation variant allele frequency in patients with myelofibrosis. Blood Cancer J. 2023, 13, 21. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, D.; Zhang, X. Overt primary myelofibrosis with coexistence of JAK2V617F and MPLW515R driver mutations. Int. J. Lab. Hematol. 2024, 46, 180–182. [Google Scholar] [CrossRef]
- Wang, Y.; Ran, F.; Lin, J.; Zhang, J.; Ma, D. Genetic and clinical characteristics of patients with Philadelphia-negative myeloproliferative neoplasm carrying concurrent mutations in JAK2V617F, CALR, and MPL. Technol. Cancer Res. Treat. 2023, 22, 15330338231154092. [Google Scholar] [CrossRef]
- Thompson, E.R.; Nguyen, T.; Kankanige, Y.; Yeh, P.; Ingbritsen, M.; McBean, M.; Semple, T.; Mir Arnau, G.; Burbury, K.; Lee, N.; et al. Clonal independence of JAK2 and CALR or MPL mutations in comutated myeloproliferative neoplasms demonstrated by single cell DNA sequencing. Haematologica 2021, 106, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, M.S.; Di Gregorio, S.; Tirrò, E.; Romano, C.; Duminuco, A.; Garibaldi, B.; Giuffrida, G.; Manzella, L.; Vigneri, P.; Palumbo, G.A. Additional genetic alterations and clonal evolution of MPNs with double mutations on the MPL gene: Two case reports. Hematol. Rep. 2023, 15, 317–324. [Google Scholar] [CrossRef]
- Milosevic Feenstra, J.D.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef]
- Cabagnols, X.; Favale, F.; Pasquier, F.; Messaoudi, K.; Defour, J.P.; Ianotto, J.C.; Marzac, C.; Le Couédic, J.P.; Droin, N.; Chachoua, I.; et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood 2016, 127, 333–342. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Lin, H.-C.; Chiang, Y.-H.; Chen, C.G.-S.; Huang, L.; Wang, W.-T.; Cheng, C.-C.; Lin, J.; Chang, Y.-F.; Chang, M.-C.; et al. Targeted next-generation sequencing identified novel mutations in triple-negative myeloproliferative neoplasms. Med. Oncol. 2017, 34, 83. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and personalized prognosis in myeloproliferative neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Lemoine, S.; Mornet, C.; Quintin-Roue, I.; Rousselet, M.-C.; Cottin, L.; Georgeais, A.; Dubouis, L.; Boyer, F.; Orvain, C.; Caillon, C.; et al. Histological and genetic characterization and follow-up of 130 patients with chronic triple-negative thrombocytosis. Haematologica 2022, 107, 2725–2731. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.-J.; Velazquez, L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 2017, 31, 1661–1670. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Alduaij, W.; McNamara, C.J.; Schuh, A.; Arruda, A.; Sukhai, M.; Kanwar, N.; Thomas, M.; Spiegel, J.; Kennedy, J.A.; Stockley, T.; et al. Clinical utility of next-generation sequencing in the management of myeloproliferative neoplasms: A single-center experience. Hemasphere 2018, 2, e44. [Google Scholar] [CrossRef] [PubMed]
- Delic, S.; Rose, D.; Kern, W.; Nadarajah, N.; Haferlach, C.; Haferlach, T.; Meggendorfer, M. Application of an NGS-based 28-gene panel in myeloproliferative neoplasms reveals distinct mutation patterns in essential thrombocythaemia, primary myelofibrosis and polycythaemia vera. Br. J. Haematol. 2016, 175, 419–426. [Google Scholar] [CrossRef] [PubMed]
- AACR Project GENIE Consortium. AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Galán, I.; Martín, I.; Ferrer, B.; Hernández-Boluda, J.-C. Impact of molecular profiling on the management of patients with myelofibrosis. Cancer Treat. Rev. 2022, 109, 102435. [Google Scholar] [CrossRef] [PubMed]
- Mroczkowska-Bękarciak, A.; Wróbel, T. BCR::ABL1-negative myeloproliferative neoplasms in the era of next-generation sequencing. Front. Genet. 2023, 14, 1241912. [Google Scholar] [CrossRef] [PubMed]
- Hussein, K.; Van Dyke, D.L.; Tefferi, A. Conventional cytogenetics in myelofibrosis: Literature review and discussion. Eur. J. Haematol. 2009, 82, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hussaini, M.; Zhang, H.; Shao, H.; Qin, D.; Zhang, X.; Ma, Z.; Hussnain Naqvi, S.M.; Zhang, L.; Moscinski, L.C. Comparison of the mutational profiles of primary myelofibrosis, polycythemia vera, and essential thrombocytosis. Am. J. Clin. Pathol. 2017, 147, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Rolles, B.; Mullally, A. Molecular pathogenesis of myeloproliferative neoplasms. Curr. Hematol. Malig. Rep. 2022, 17, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef]
- Senín, A.; Fernández-Rodríguez, C.; Bellosillo, B.; Camacho, L.; Longarón, R.; Angona, A.; Besses, C.; Álvarez-Larrán, A. Non-driver mutations in patients with JAK2V617F-mutated polycythemia vera or essential thrombocythemia with long-term molecular follow-up. Ann. Hematol. 2018, 97, 443–451. [Google Scholar] [CrossRef]
- Courtier, F.; Garnier, S.; Carbuccia, N.; Guille, A.; Adélaide, J.; Chaffanet, M.; Hirsch, P.; Paz, D.L.; Slama, B.; Vey, N.; et al. Targeted molecular characterization shows differences between primary and secondary myelofibrosis. Genes Chromosomes Cancer 2020, 59, 30–39. [Google Scholar] [CrossRef]
- Luque Paz, D.; Riou, J.; Verger, E.; Cassinat, B.; Chauveau, A.; Ianotto, J.-C.; Dupriez, B.; Boyer, F.; Renard, M.; Mansier, O.; et al. Genomic analysis of primary and secondary myelofibrosis redefines the prognostic impact of ASXL1 mutations: A FIM study. Blood Adv. 2021, 5, 1442–1451. [Google Scholar] [CrossRef]
- Yan, X.; Xu, Z.; Zhang, P.; Sun, Q.; Jia, Y.; Qin, T.; Qu, S.; Pan, L.; Li, Z.; Liu, J.; et al. Non-driver mutations landscape in different stages of primary myelofibrosis determined ASXL1 mutations play a critical role in disease progression. Blood Cancer J. 2023, 13, 56. [Google Scholar] [CrossRef] [PubMed]
- Luque Paz, D.; Kralovics, R.; Skoda, R.C. Genetic basis and molecular profiling in myeloproliferative neoplasms. Blood 2023, 141, 1909–1921. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, H.; Adamsson, J.; Johansson, P.; Nilsson, S.; Palmqvist, L.; Andréasson, B.; Asp, J. The clinical relevance of broad mutational screening of myeloproliferative neoplasms at diagnosis. Front. Oncol. 2023, 13, 1190305. [Google Scholar] [CrossRef] [PubMed]
- Coltro, G.; Rotunno, G.; Mannelli, L.; Mannarelli, C.; Fiaccabrino, S.; Romagnoli, S.; Bartalucci, N.; Ravenda, E.; Gelli, E.; Sant’Antonio, E.; et al. RAS/CBL mutations predict resistance to JAK inhibitors in myelofibrosis and are associated with poor prognostic features. Blood Adv. 2020, 4, 3677–3687. [Google Scholar] [CrossRef]
- Reynolds, S.B.; Pettit, K.; Kandarpa, M.; Talpaz, M.; Li, Q. Exploring the molecular landscape of myelofibrosis, with a focus on Ras and mitogen-activated protein (MAP) kinase signaling. Cancers 2023, 15, 4654. [Google Scholar] [CrossRef]
- Rodriguez-Meira, A.; Rahman, H.; Norfo, R.; Wen, W.; Chédeville, A.; O’Sullivan, J.; Wang, G.; Paterson, A.; Louka, E.; Brierley, C.K.; et al. Single-cell multi-omics reveals the genetic, cellular and molecular landscape of TP53 mutated leukemic transformation in MPN. Blood 2021, 138, 3. [Google Scholar] [CrossRef]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and visualization of longitudinal genomic and clinical data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef]
- Marcault, C.; Zhao, L.-P.; Maslah, N.; Verger, E.; Daltro de Oliveira, R.; Soret-Dulphy, J.; Cazaux, M.; Gauthier, N.; Roux, B.; Clappier, E.; et al. Impact of NFE2 mutations on AML transformation and overall survival in patients with myeloproliferative neoplasms. Blood 2021, 138, 2142–2148. [Google Scholar] [CrossRef]
- Chifotides, H.T.; Verstovsek, S.; Bose, P. Association of myelofibrosis phenotypes with clinical manifestations, molecular profiles, and treatments. Cancers 2023, 15, 3331. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Kubovcakova, L.; Nienhold, R.; Zmajkovic, J.; Meyer, S.C.; Hao-Shen, H.; Geier, F.; Dirnhofer, S.; Guglielmelli, P.; Vannucchi, A.M.; et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J. Exp. Med. 2016, 213, 1479–1496. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.M.; Andersen, C.L.; Westman, M.; Kristensen, L.S.; Asmar, F.; Kruse, T.A.; Thomassen, M.; Larsen, T.S.; Skov, V.; Hansen, L.L.; et al. Epigenetic changes in myelofibrosis: Distinct methylation changes in the myeloid compartments and in cases with ASXL1 mutations. Sci. Rep. 2017, 7, 6774. [Google Scholar] [CrossRef]
- Nam, A.S.; Kim, K.-T.; Chaligne, R.; Izzo, F.; Ang, C.; Taylor, J.; Myers, R.M.; Abu-Zeinah, G.; Brand, R.; Omans, N.D.; et al. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature 2019, 571, 355–360. [Google Scholar] [CrossRef]
- Rodriguez-Meira, A.; Buck, G.; Clark, S.-A.; Povinelli, B.J.; Alcolea, V.; Louka, E.; McGowan, S.; Hamblin, A.; Sousos, N.; Barkas, N.; et al. Unravelling intratumoral heterogeneity through high-sensitivity single-cell mutational analysis and parallel RNA sequencing. Mol. Cell 2019, 73, 1292–1305. [Google Scholar] [CrossRef]
- Willekens, C.; Laplane, L.; Dagher, T.; Benlabiod, C.; Papadopoulos, N.; Lacout, C.; Rameau, P.; Catelain, C.; Alfaro, A.; Edmond, V.; et al. SRSF2-P95H decreases JAK/STAT signaling in hematopoietic cells and delays myelofibrosis development in mice. Leukemia 2023, 37, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Theocharides, A.; Boissinot, M.; Girodon, F.; Garand, R.; Teo, S.-S.; Lippert, E.; Talmant, P.; Tichelli, A.; Hermouet, S.; Skoda, R.C. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood 2007, 110, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Verger, E.; Giraudier, S.; Chea, M.; Hoffman, R.; Mascarenhas, J.; Cassinat, B.; Kiladjian, J.-J. Single-cell analysis reveals selection of TP53-mutated clones after MDM2 inhibition. Blood Adv. 2022, 6, 2813–2823. [Google Scholar] [CrossRef] [PubMed]
- Gagelmann, N.; Badbaran, A.; Salit, R.B.; Schroeder, T.; Gurnari, C.; Pagliuca, S.; Panagiota, V.; Rautenberg, C.; Cassinat, B.; Thol, F.; et al. Impact of TP53 on outcome of patients with myelofibrosis undergoing hematopoietic stem cell transplantation. Blood 2023, 141, 2901–2911. [Google Scholar] [CrossRef] [PubMed]
- Dilip, D.; Menghrajani, K.; Glass, J.; Rampal, R.K.; Famulare, C.; Sirenko, M.; Levine, R.L.; Koche, R. MPN transformation is characterized by heterogeneous shifts in lineage character. Blood 2023, 142 (Suppl. 1), 749. [Google Scholar] [CrossRef]
- Ishii, T.; Bruno, E.; Hoffman, R.; Xu, M. Involvement of various hematopoietic-cell lineages by the JAK2V617F mutation in polycythemia vera. Blood 2006, 108, 3128–3134. [Google Scholar] [CrossRef] [PubMed]
- Visani, G.; Etebari, M.; Fuligni, F.; Di Guardo, A.; Isidori, A.; Loscocco, F.; Paolini, S.; Navari, M.; Piccaluga, P.P. Use of next generation sequencing to define the origin of primary myelofibrosis. Cancers 2023, 15, 1785. [Google Scholar] [CrossRef]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [PubMed]
- Mullally, A.; Lane, S.W.; Ball, B.; Megerdichian, C.; Okabe, R.; Al-Shahrour, F.; Paktinat, M.; Haydu, J.E.; Housman, E.; Lord, A.M.; et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell 2010, 17, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Prins, D.; Park, H.J.; Grinfeld, J.; Gonzalez-Arias, C.; Loughran, S.; Dovey, O.M.; Klampfl, T.; Bennett, C.; Hamilton, T.L.; et al. Mutant calreticulin knockin mice develop thrombocytosis and myelofibrosis without a stem cell self-renewal advantage. Blood 2018, 131, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Cordua, S.; Kjaer, L.; Skov, V.; Pallisgaard, N.; Hasselbalch, H.C.; Ellervik, C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 2019, 134, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.; Lee, J.; Mitchell, E.; Moore, L.; Baxter, E.J.; Hewinson, J.; Dawson, K.J.; Menzies, A.; Godfrey, A.L.; Green, A.R.; et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature 2022, 602, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.; Park, H.J.; Celik, H.; Ostrander, E.L.; Reyes, J.M.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.; Ding, L.; et al. Loss of Dnmt3a immortalizes hematopoietic stem cells in vivo. Cell Rep. 2018, 23, 1–10. [Google Scholar] [CrossRef]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, e438–e442. [Google Scholar] [CrossRef]
- Streck, A.; Kaufmann, T.L.; Schwarz, R.F. SMITH: Spatially constrained stochastic model for simulation of intra-tumour heterogeneity. Bioinformatics 2023, 39, btad102. [Google Scholar] [CrossRef]
- Sud, A.; Chattopadhyay, S.; Thomsen, H.; Sundquist, K.; Sundquist, J.; Houlston, R.S.; Hemminki, K. Familial risks of acute myeloid leukemia, myelodysplastic syndromes, and myeloproliferative neoplasms. Blood 2018, 132, 973–976. [Google Scholar] [CrossRef]
- Dunbar, A.; Bowman, R.L.; Park, Y.; Izzo, F.; Myers, R.M.; Karzai, A.; Jun Kim, W.; Fernández Maestre, I.; Waarts, M.R.; Nazir, A.; et al. Jak2V617F reversible activation shows an essential requirement for Jak2V617F in myeloproliferative neoplasms (MPNs). Blood 2022, 140 (Suppl. 1), 803–804. [Google Scholar] [CrossRef]
- Martyré, M.C. TGF-beta and megakaryocytes in the pathogenesis of myelofibrosis in myeloproliferative disorders. Leuk. Lymphoma 1995, 20, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Kramer, F.; Dernedde, J.; Mezheyeuski, A.; Tauber, R.; Micke, P.; Kappert, K. Platelet-derived growth factor receptor β activation and regulation in murine myelofibrosis. Haematologica 2020, 105, 2083–2094. [Google Scholar] [CrossRef] [PubMed]
- Bock, O.; Loch, G.; Schade, U.; von Wasielewski, R.; Schlué, J.; Kreipe, H. Aberrant expression of transforming growth factor beta-1 (TGF beta-1) per se does not discriminate fibrotic from non-fibrotic chronic myeloproliferative disorders. J. Pathol. 2005, 205, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Bock, O.; Loch, G.; Schade, U.; Büsche, G.; Wasielewski, R.; Wiese, B.; Kreipe, H. Osteosclerosis in advanced chronic idiopathic myelofibrosis is associated with endothelial overexpression of osteoprotegerin. Br. J. Haematol. 2005, 130, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell 2018, 33, 29–43.e7. [Google Scholar] [CrossRef]
- Jutzi, J.S.; Mullally, A. Two to tango! IL-13 and TGF-β drive myelofibrosis. Blood 2022, 140, 2767–2768. [Google Scholar] [CrossRef]
- Melo-Cardenas, J.; Bezavada, L.; Crawford, J.C.; Gurbuxani, S.; Cotton, A.; Kang, G.; Gossett, J.; Marinaccio, C.; Weinberg, R.; Hoffman, R.; et al. IL-13/IL-4 signaling contributes to fibrotic progression of the myeloproliferative neoplasms. Blood 2022, 140, 2805–2817. [Google Scholar] [CrossRef]
- Kumari, S.; Panda, T.K.; Pradhan, T. Lysyl oxidase: Its diversity in health and diseases. Indian J. Clin. Biochem. 2017, 32, 134–141. [Google Scholar] [CrossRef]
- Papadantonakis, N.; Matsuura, S.; Ravid, K. Megakaryocyte pathology and bone marrow fibrosis: The lysyl oxidase connection. Blood 2012, 120, 1774–1781. [Google Scholar] [CrossRef]
- Tadmor, T.; Bejar, J.; Attias, D.; Mischenko, E.; Sabo, E.; Neufeld, G.; Vadasz, Z. The expression of lysyl-oxidase gene family members in myeloproliferative neoplasms. Am. J. Hematol. 2013, 88, 355–358. [Google Scholar] [CrossRef]
- Piasecki, A.; Leiva, O.; Ravid, K. Lysyl oxidase inhibition in primary myelofibrosis: A renewed strategy. Arch. Stem Cell Ther. 2020, 1, 23–27. [Google Scholar] [CrossRef]
- Verstovsek, S.; Manshouri, T.; Pilling, D.; Bueso-Ramos, C.E.; Newberry, K.J.; Prijic, S.; Knez, L.; Bozinovic, K.; Harris, D.M.; Spaeth, E.L.; et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J. Exp. Med. 2016, 213, 1723–1740. [Google Scholar] [CrossRef]
- Ozono, Y.; Shide, K.; Kameda, T.; Kamiunten, A.; Tahira, Y.; Sekine, M.; Akizuki, K.; Nakamura, K.; Iwakiri, H.; Sueta, M.; et al. Neoplastic fibrocytes play an essential role in bone marrow fibrosis in Jak2V617F-induced primary myelofibrosis mice. Leukemia 2021, 35, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Vining, K.H.; Marneth, A.E.; Adu-Berchie, K.; Grolman, J.M.; Tringides, C.M.; Liu, Y.; Wong, W.J.; Pozdnyakova, O.; Severgnini, M.; Stafford, A.; et al. Mechanical checkpoint regulates monocyte differentiation in fibrotic niches. Nat. Mater. 2022, 21, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Shome, D.K.; Kulkarni, B.; Ghosh, M.K.; Ghosh, K. Fibrosis and bone marrow: Understanding causation and pathobiology. J. Transl. Med. 2023, 21, 703. [Google Scholar] [CrossRef]
- Hussein, K.; Bock, O.; Theophile, K.; von Neuhoff, N.; Buhr, T.; Schlué, J.; Büsche, G.; Kreipe, H. JAK2(V617F) allele burden discriminates essential thrombocythemia from a subset of prefibrotic-stage primary myelofibrosis. Exp. Hematol. 2009, 37, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.-C.; Chuang, W.-Y.; Chang, H.; Lin, T.-H.; Wu, J.-H.; Lin, T.-L.; Ou, C.-W.; Hung, Y.-S.; Huang, T.-Y.; Huang, Y.-J.; et al. Comparison of clinical and molecular features between patients with essential thrombocythemia and early/prefibrotic primary myelofibrosis presenting with thrombocytosis in Taiwan. Am. J. Clin. Pathol. 2023, 159, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.M.; Izzo, F.; Prieto, T.; Mimitou, E.; Raviram, R.; Chaligne, R.; Hoffman, R.; Stahl, M.; Abdel-Wahab, O.; Marcellino, B.; et al. High throughput single-cell simultaneous genotyping and chromatin accessibility reveals genotype to phenotype relationship in human myeloproliferation. Blood 2021, 138, 678. [Google Scholar] [CrossRef]
- Myers, R.M.; Izzo, F.; Kottapalli, S.; Prieto, T.; Dunbar, A.; Bowman, R.L.; Mimitou, E.P.; Stahl, M.; El Ghaity-Beckley, S.; Arandela, J.; et al. Integrated single-cell genotyping and chromatin accessibility charts JAK2V617F human hematopoietic differentiation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Psaila, B.; Wang, G.; Rodriguez-Meira, A.; Li, R.; Heuston, E.F.; Murphy, L.; Yee, D.; Hitchcock, I.S.; Sousos, N.; O’Sullivan, J.; et al. Single-cell analyses reveal megakaryocyte-biased hematopoiesis in myelofibrosis and identify mutant clone-specific targets. Mol. Cell 2020, 78, 477–492.e8. [Google Scholar] [CrossRef]
- Kepp, O.; Liu, P.; Zhao, L.; Plo, I.; Kroemer, G. Surface-exposed and soluble calreticulin: Conflicting biomarkers for cancer prognosis. Oncoimmunology 2020, 9, 1792037. [Google Scholar] [CrossRef]
- Foßelteder, J.; Pabst, G.; Sconocchia, T.; Schlacher, A.; Auinger, L.; Kashofer, K.; Beham-Schmid, C.; Trajanoski, S.; Waskow, C.; Schöll, W.; et al. Human gene-engineered calreticulin mutant stem cells recapitulate MPN hallmarks and identify targetable vulnerabilities. Leukemia 2023, 37, 843–853. [Google Scholar] [CrossRef]
- Abdulkarim, K.; Ridell, B.; Johansson, P.; Kutti, J.; Safai-Kutti, S.; Andréasson, B. The impact of peripheral blood values and bone marrow findings on prognosis for patients with essential thrombocythemia and polycythemia vera. Eur. J. Haematol. 2011, 86, 148–155. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Passamonti, F.; Rumi, E.; Boveri, E.; Randi, M.L.; Bertozzi, I.; Marino, F.; Vannucchi, A.M.; Pieri, L.; et al. Initial bone marrow reticulin fibrosis in polycythemia vera exerts an impact on clinical outcome. Blood 2012, 119, 2239–2241. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, C.; Hu, Z.; Li, Z.; Li, M.; Wu, L.; Zhou, M.; Liang, D. CRISPR/Cas12a-based ultrasensitive and rapid detection of JAK2 V617F somatic mutation in myeloproliferative neoplasms. Biosensors 2021, 11, 247. [Google Scholar] [CrossRef] [PubMed]
- Hill, M. Conformation sensitive gel electrophoresis. Methods Mol. Biol. 2011, 688, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, N.A.; Rosle, N.A.; Siti Asmaa, M.J.; Aziee, S.; Haiyuni, M.Y.; Samat, N.A.; Husin, A.; Hassan, R.; Ramli, M.; Mohamed Yusoff, S.; et al. Conformation sensitive gel electrophoresis for the detection of calreticulin mutations in BCR-ABL1-negative myeloproliferative neoplasms. Int. J. Lab. Hematol. 2021, 43, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, K.; Chai, J.; Wang, Y.; Naeem, R.; Goldstein, D.Y. Pitfalls of using polymerase chain reaction-based assays for JAK2 and CALR exon 9 variant testing in myeloproliferative neoplasms: Knowing when to go the extra mile! Am. J. Clin. Pathol. 2023, aqad122. [Google Scholar] [CrossRef] [PubMed]
- Verger, E.; Maslah, N.; Schlageter, M.-H.; Chomienne, C.; Kiladjian, J.-J.; Giraudier, S.; Cassinat, B. Pitfalls in CALR exon 9 mutation detection: A single-center experience in 571 positive patients. Int. J. Lab. Hematol. 2020, 42, 827–832. [Google Scholar] [CrossRef]
- Nienhold, R.; Ashcroft, P.; Zmajkovic, J.; Rai, S.; Rao, T.N.; Drexler, B.; Meyer, S.C.; Lundberg, P.; Passweg, J.R.; Leković, D.; et al. MPN patients with low mutant JAK2 allele burden show late expansion restricted to erythroid and megakaryocytic lineages. Blood 2020, 136, 2591–2595. [Google Scholar] [CrossRef]
- Craven, K.E.; Fischer, C.G.; Jiang, L.; Pallavajjala, A.; Lin, M.-T.; Eshleman, J.R. Optimizing insertion and deletion detection using next-generation sequencing in the clinical laboratory. J. Mol. Diagn. 2022, 24, 1217–1231. [Google Scholar] [CrossRef]
- Khurana, H.; Muthusamy, B.; Yanamandra, U.; Garapati, K.; Premdeep, H.; Subramanian, S.; Pandey, A. Whole exome sequencing reveals novel variants in unexplained erythrocytosis. OMICS J. Integr. Biol. 2023, 27, 299–304. [Google Scholar] [CrossRef]
- Sakuma, M.; Blombery, P.; Meggendorfer, M.; Haferlach, C.; Lindauer, M.; Martens, U.M.; Kern, W.; Haferlach, T.; Walter, W. Novel causative variants of VEXAS in UBA1 detected through whole genome transcriptome sequencing in a large cohort of hematological malignancies. Leukemia 2023, 37, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.-L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009, 113, 2895–2901. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and karyotype-enhanced international prognostic scoring system for primary myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-enhanced international prognostic score system for transplantation-age patients with primary myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef]
- Loscocco, G.G.; Rotunno, G.; Mannelli, F.; Coltro, G.; Gesullo, F.; Pancani, F.; Signori, L.; Maccari, C.; Esposito, M.; Paoli, C.; et al. The prognostic contribution of CBL, NRAS, KRAS, RUNX1, and TP53 mutations to mutation-enhanced international prognostic score systems (MIPSS70/plus/plus v2.0) for primary myelofibrosis. Am. J. Hematol. 2023, 99, 68–78. [Google Scholar] [CrossRef]
- Sobieralski, P.; Wasąg, B.; Leszczyńska, A.; Żuk, M.; Bieniaszewska, M. The molecular profile in patients with polycythemia vera and essential thrombocythemia is dynamic and correlates with disease’s phenotype. Front. Oncol. 2023, 13, 1224590. [Google Scholar] [CrossRef]
- Passamonti, F.; Giorgino, T.; Mora, B.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2726–2731. [Google Scholar] [CrossRef]
- Tefferi, A. Primary myelofibrosis: 2023 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2023, 98, 801–821. [Google Scholar] [CrossRef]
- Gagelmann, N.; Ditschkowski, M.; Bogdanov, R.; Bredin, S.; Robin, M.; Cassinat, B.; Shahswar, R.; Thol, F.; Heuser, M.; Socié, G.; et al. Comprehensive clinical-molecular transplant scoring system for myelofibrosis undergoing stem cell transplantation. Blood 2019, 133, 2233–2242. [Google Scholar] [CrossRef]
- Asp, J.; Andréasson, B.; Hansson, U.; Wasslavik, C.; Abelsson, J.; Johansson, P.; Palmqvist, L. Mutation status of essential thrombocythemia and primary myelofibrosis defines clinical outcome. Haematologica 2016, 101, e129–e132. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Nicolosi, M.; Mudireddy, M.; Szuber, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N.; et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1095 patients. Am. J. Hematol. 2018, 93, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Ersal, T.; Özkocaman, V.; Pınar, İ.E.; Yalçın, C.; Orhan, B.; Candar, Ö.; Çubukçu, S.; Koca, T.G.; Hunutlu, F.Ç.; Yavuz, Ş.; et al. Systemic inflammatory indices for predicting prognosis of myelofibrosis. Sci. Rep. 2023, 13, 12539. [Google Scholar] [CrossRef] [PubMed]
- Kröger, N.; Wolschke, C.; Gagelmann, N. How I treat transplant-eligible patients with myelofibrosis. Blood 2023, 142, 1683–1696. [Google Scholar] [CrossRef]
- Wolschke, C.; Badbaran, A.; Zabelina, T.; Christopeit, M.; Ayuk, F.; Triviai, I.; Zander, A.; Alchalby, H.; Bacher, U.; Fehse, B.; et al. Impact of molecular residual disease post allografting in myelofibrosis patients. Bone Marrow Transplant. 2017, 52, 1526–1529. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Livingston, R.A.; Hu, W.; Mascarenhas, J. Ten years of treatment with ruxolitinib for myelofibrosis: A review of safety. J. Hematol. Oncol. 2023, 16, 82. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Bose, P.; Rampal, R.; Gerds, A.T.; Fleischman, A.; Verstovsek, S. Ten years after ruxolitinib approval for myelofibrosis: A review of clinical efficacy. Leuk. Lymphoma 2023, 64, 1063–1081. [Google Scholar] [CrossRef]
- Caduc, M.J.; Koschmieder, S. Is treatment for cytopenic myelofibrosis still an unmet clinical need? Hemasphere 2023, 7, e982. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves fourth JAK inhibitor for myelofibrosis. Nat. Rev. Drug Discov. 2023, 22, 862. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G.; Klersy, C.; Villani, L.; Bonetti, E.; Catarsi, P.; Poletto, V.; Campanelli, R.; Impera, S.; Latagliata, R.; Viarengo, G.; et al. JAK2(V617F) allele burden ≥50% is associated with response to ruxolitinib in persons with MPN-associated myelofibrosis and splenomegaly requiring therapy. Leukemia 2016, 30, 1772–1775. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Garcia, J.S.; Perkins, A.; Harb, J.G.; Souers, A.J.; Werner, M.E.; Brown, C.M.; Passamonti, F. New era for myelofibrosis treatment with novel agents beyond Janus kinase-inhibitor monotherapy: Focus on clinical development of BCL-XL/BCL-2 inhibition with navitoclax. Cancer 2023, 129, 3535–3545. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G.; Gale, R.P. Does ruxolitinib really prolong survival in individuals with myelofibrosis? The never-ending story. Blood Adv. 2022, 6, 2331–2333. [Google Scholar] [CrossRef] [PubMed]
- Palandri, F.; Breccia, M.; Bonifacio, M.; Polverelli, N.; Elli, E.M.; Benevolo, G.; Tiribelli, M.; Abruzzese, E.; Iurlo, A.; Heidel, F.H.; et al. Life after ruxolitinib: Reasons for discontinuation, impact of disease phase, and outcomes in 218 patients with myelofibrosis. Cancer 2020, 126, 1243–1252. [Google Scholar] [CrossRef]
- Harrison, C.N.; Schaap, N.; Mesa, R.A. Management of myelofibrosis after ruxolitinib failure. Ann. Hematol. 2020, 99, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Talpaz, M.; Kiladjian, J.-J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 2021, 35, 1–17. [Google Scholar] [CrossRef]
- Tefferi, A.; Pardanani, A.; Gangat, N. Momelotinib (JAK1/JAK2/ACVR1 inhibitor): Mechanism of action, clinical trial reports, and therapeutic prospects beyond myelofibrosis. Haematologica 2023, 108, 2919–2932. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.T.; Mesa, R.A.; Harrison, C.N.; Bose, P.; Gerds, A.T.; Gupta, V.; Scott, B.L.; Kiladjian, J.-J.; Lucchesi, A.; Kong, T.; et al. Pacritinib is a potent ACVR1 inhibitor with significant anemia benefit in patients with myelofibrosis. Blood Adv. 2023, 7, 5835–5842. [Google Scholar] [CrossRef]
- Tremblay, D.; Mesa, R.; Scott, B.; Buckley, S.; Roman-Torres, K.; Verstovsek, S.; Mascarenhas, J. Pacritinib demonstrates spleen volume reduction in patients with myelofibrosis independent of JAK2V617F allele burden. Blood Adv. 2020, 4, 5929–5935. [Google Scholar] [CrossRef]
- Gupta, V.; Mesa, R.A.; Deininger, M.W.N.; Rivera, C.E.; Sirhan, S.; Brachmann, C.B.; Collins, H.; Kawashima, J.; Xin, Y.; Verstovsek, S. A phase 1/2, open-label study evaluating twice-daily administration of momelotinib in myelofibrosis. Haematologica 2017, 102, 94–102. [Google Scholar] [CrossRef]
- Patel, K.P.; Newberry, K.J.; Luthra, R.; Jabbour, E.; Pierce, S.; Cortes, J.; Singh, R.; Mehrotra, M.; Routbort, M.J.; Luthra, M.; et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood 2015, 126, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.Y.; McNamara, C.; Kennedy, J.A.; Panzarella, T.; Arruda, A.; Stockley, T.; Sukhai, M.; Thomas, M.; Bartoszko, J.; Ho, J.; et al. Impact of genomic alterations on outcomes in myelofibrosis patients undergoing JAK1/2 inhibitor therapy. Blood Adv. 2017, 1, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.P.S.; Getta, B.; Masarova, L.; Famulare, C.; Schulman, J.; Datoguia, T.S.; Puga, R.D.; Alves Paiva, R.d.M.; Arcila, M.E.; Hamerschlak, N.; et al. Prognostic impact of RAS-pathway mutations in patients with myelofibrosis. Leukemia 2020, 34, 799–810. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.M.; Taylor, J.; Gerds, A.; Buckley, S.; Harrison, C.N.; Oh, S.; List, A.F.; Howard, K.; Dreau, H.; Hamblin, A.; et al. RAS-pathway mutations are common in patients with ruxolitinib refractory/intolerant myelofibrosis: Molecular analysis of the PAC203 cohort. Leukemia 2023, 37, 2497–2501. [Google Scholar] [CrossRef] [PubMed]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, E.; Yoshida, K.; Frick, M.; Hoyer, K.; Christen, F.; Kaeda, J.; Obenaus, M.; Noerenberg, D.; Hennch, C.; Chan, W.; et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat. Commun. 2020, 11, 73. [Google Scholar] [CrossRef]
- Tefferi, A.; Barraco, D.; Lasho, T.L.; Shah, S.; Begna, K.H.; Al-Kali, A.; Hogan, W.J.; Litzow, M.R.; Hanson, C.A.; Ketterling, R.P.; et al. Momelotinib therapy for myelofibrosis: A 7-year follow-up. Blood Cancer J. 2018, 8, 29. [Google Scholar] [CrossRef]
- Wang, Y.; Fiskus, W.; Chong, D.G.; Buckley, K.M.; Natarajan, K.; Rao, R.; Joshi, A.; Balusu, R.; Koul, S.; Chen, J.; et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 2009, 114, 5024–5033. [Google Scholar] [CrossRef]
- Harrison, C.; Heidel, F.H.; Vannucchi, A.M.; Kiladjian, J.-J.; Hayat, A.; Passamonti, F.; Conneally, E.; Kindler, T.; Martino, B.; Lipka, D.B.; et al. A phase 1b dose-finding study of panobinostat and ruxolitinib in myelofibrosis. Hemasphere 2022, 6, e757. [Google Scholar] [CrossRef]
- Jutzi, J.S.; Kleppe, M.; Dias, J.; Staehle, H.F.; Shank, K.; Teruya-Feldstein, J.; Gambheer, S.M.M.; Dierks, C.; Rienhoff, H.Y.; Levine, R.L.; et al. LSD1 inhibition prolongs survival in mouse models of MPN by selectively targeting the disease clone. Hemasphere 2018, 2, e54. [Google Scholar] [CrossRef]
- Wang, A.; Liu, J.; Pu, J.J. Novel agents and evolving strategies in myelofibrotive neoplasm: An update from 2022 ASH annual conference. J. Hematol. Oncol. 2023, 16, 53. [Google Scholar] [CrossRef]
- Gill, H. Lysine-Specific Demethylase 1 (LSD1/KDM1A) Inhibition as a Target for Disease Modification in Myelofibrosis. Cells 2022, 11, 2107. [Google Scholar] [CrossRef]
- Pettit, K. A Phase 2 Study of the LSD1 Inhibitor IMG7289 (Bomedemstat) for the Treatment of Advanced Myelofibrosis. ASH. 2020. Available online: https://ash.confex.com/ash/2020/webprogram/Paper136001.html (accessed on 10 December 2023).
- Palumbo, G.A.; Duminuco, A. Myelofibrosis: In search for BETter targeted therapies. J. Clin. Oncol. 2023, 41, 5044–5048. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Gerds, A.; Verstovsek, S. Paradigm shift: Combination BET and JAK inhibition in myelofibrosis. Leukemia 2021, 35, 3361–3363. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Kremyanskaya, M.; Patriarca, A.; Palandri, F.; Devos, T.; Passamonti, F.; Rampal, R.K.; Mead, A.J.; Hobbs, G.; Scandura, J.M.; et al. MANIFEST: Pelabresib in combination with ruxolitinib for Janus kinase inhibitor treatment-naïve myelofibrosis. J. Clin. Oncol. 2023, 41, 4993–5004. [Google Scholar] [CrossRef]
- Harrison, C.N.; Gupta, V.K.; Gerds, A.T.; Rampal, R.; Verstovsek, S.; Talpaz, M.; Kiladjian, J.-J.; Mesa, R.; Kuykendall, A.T.; Vannucchi, A.M.; et al. Phase III MANIFEST-2: Pelabresib + ruxolitinib vs placebo + ruxolitinib in JAK inhibitor treatment-naive myelofibrosis. Future Oncol. 2022, 18, 2987–2997. [Google Scholar] [CrossRef] [PubMed]
- Wildschut, M.H.E.; Mena, J.; Dördelmann, C.; van Oostrum, M.; Hale, B.D.; Settelmeier, J.; Festl, Y.; Lysenko, V.; Schürch, P.M.; Ring, A.; et al. Proteogenetic drug response profiling elucidates targetable vulnerabilities of myelofibrosis. Nat. Commun. 2023, 14, 6414. [Google Scholar] [CrossRef]
- Pastore, F.; Bhagwat, N.; Pastore, A.; Radzisheuskaya, A.; Karzai, A.; Krishnan, A.; Li, B.; Bowman, R.L.; Xiao, W.; Viny, A.D.; et al. PRMT5 inhibition modulates E2F1 methylation and gene-regulatory networks leading to therapeutic efficacy in JAK2V617F-mutant MPN. Cancer Discov. 2020, 10, 1742–1757. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Monga, V.; Jauhari, S.; Stevens, D.; Masarova, L.; McKean, M.; Mauro, D.; Viscusi, J.; Scherle, P.; Bhagwat, N.; et al. A phase 1 dose escalation study of protein arginine methyltransferase 5 (PRMT5) inhibitor PRT543 in patients with myeloid malignancies. Blood 2021, 138 (Suppl. 1), 2609. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Harrison, C.N.; Kiladjian, J.-J.; Komrokji, R.S.; Koschmieder, S.; Vannucchi, A.M.; Berry, T.; Redding, D.; Sherman, L.; Dougherty, S.; et al. Imetelstat in intermediate-2 or high-risk myelofibrosis refractory to JAK inhibitor: IMpactMF phase III study design. Future Oncol. 2022, 18, 2393–2402. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Begna, K.H.; Patnaik, M.M.; Zblewski, D.L.; Finke, C.M.; Laborde, R.R.; Wassie, E.; Schimek, L.; Hanson, C.A.; et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N. Engl. J. Med. 2015, 373, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Olschok, K.; Altenburg, B.; de Toledo, M.A.S.; Maurer, A.; Abels, A.; Beier, F.; Gezer, D.; Isfort, S.; Paeschke, K.; Brümmendorf, T.H.; et al. The telomerase inhibitor imetelstat differentially targets JAK2V617F versus CALR mutant myeloproliferative neoplasm cells and inhibits JAK-STAT signaling. Front. Oncol. 2023, 13, 1277453. [Google Scholar] [CrossRef] [PubMed]
- Waibel, M.; Solomon, V.S.; Knight, D.A.; Ralli, R.A.; Kim, S.-K.; Banks, K.-M.; Vidacs, E.; Virely, C.; Sia, K.C.S.; Bracken, L.S.; et al. Combined targeting of JAK2 and Bcl-2/Bcl-xL to cure mutant JAK2-driven malignancies and overcome acquired resistance to JAK2 inhibitors. Cell Rep. 2013, 5, 1047–1059. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Mead, A.J.; Somervaille, T.C.; McCloskey, J.K.; Palandri, F.; Koschmieder, S.; Lavie, D.; Leber, B.; Yeh, S.-P.; Gómez-Casares, M.T.; et al. Transform-1: A randomized, double-blind, placebo-controlled, multicenter, international phase 3 study of navitoclax in combination with ruxolitinib versus ruxolitinib plus placebo in patients with untreated myelofibrosis. Blood 2023, 142 (Suppl. 1), 620. [Google Scholar] [CrossRef]
- Rampal, R.K.; Grosicki, S.; Chraniuk, D.; Abruzzese, E.; Bose, P.; Gerds, A.T.; Vannucchi, A.M.; Palandri, F.; Lee, S.-E.; Gupta, V.; et al. Pelabresib in combination with ruxolitinib for Janus kinase inhibitor treatment-naïve patients with myelofibrosis: Results of the MANIFEST-2 randomized, double-blind, phase 3 study. Blood 2023, 142 (Suppl. 1), 628. [Google Scholar] [CrossRef]
- Bar-Natan, M.; Mascarenhas, J.; Gerds, A.T.; Mesa, R.; Gupta, V.; Kremyanskaya, M.; Dougherty, M.; Fabris, F.; Johnson, K.; Yu, A.; et al. Molecularly targeted combination therapy for advanced phase myeloproliferative neoplasm: MPN-RC 119. Blood 2022, 140, 3988–3990. [Google Scholar] [CrossRef]
- Bonelli, M.; Kerschbaumer, A.; Kastrati, K.; Ghoreschi, K.; Gadina, M.; Heinz, L.X.; Smolen, J.S.; Aletaha, D.; O’Shea, J.; Laurence, A. Selectivity, efficacy and safety of JAKinibs: New evidence for a still evolving story. Ann. Rheum. Dis. 2024, 83, 139–160. [Google Scholar] [CrossRef] [PubMed]
- Zahr, A.A.; Salama, M.E.; Carreau, N.; Tremblay, D.; Verstovsek, S.; Mesa, R.; Hoffman, R.; Mascarenhas, J. Bone marrow fibrosis in myelofibrosis: Pathogenesis, prognosis and targeted strategies. Haematologica 2016, 101, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.A.C.; Fowles, J.S.; Zhou, A.; Oh, S.T. Inflammatory pathophysiology as a contributor to myeloproliferative neoplasms. Front. Immunol. 2021, 12, 683401. [Google Scholar] [CrossRef]
- Calledda, F.R.; Malara, A.; Balduini, A. Inflammation and bone marrow fibrosis: Novel immunotherapeutic targets. Curr. Opin. Hematol. 2023, 30, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.-J.; Giraudier, S.; Cassinat, B. Interferon-alpha for the therapy of myeloproliferative neoplasms: Targeting the malignant clone. Leukemia 2016, 30, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Mosca, M.; Hermange, G.; Tisserand, A.; Noble, R.; Marzac, C.; Marty, C.; Le Sueur, C.; Campario, H.; Vertenoeil, G.; El-Khoury, M.; et al. Inferring the dynamics of mutated hematopoietic stem and progenitor cells induced by IFNα in myeloproliferative neoplasms. Blood 2021, 138, 2231–2243. [Google Scholar] [CrossRef]
- Knudsen, T.A.; Skov, V.; Stevenson, K.; Werner, L.; Duke, W.; Laurore, C.; Gibson, C.J.; Nag, A.; Thorner, A.R.; Wollison, B.; et al. Genomic profiling of a randomized trial of interferon-α vs hydroxyurea in MPN reveals mutation-specific responses. Blood Adv. 2022, 6, 2107–2119. [Google Scholar] [CrossRef]
- How, J.; Garcia, J.S.; Mullally, A. Biology and therapeutic targeting of molecular mechanisms in MPNs. Blood 2023, 141, 1922–1933. [Google Scholar] [CrossRef]
- Rao, T.N.; Hansen, N.; Stetka, J.; Luque Paz, D.; Kalmer, M.; Hilfiker, J.; Endele, M.; Ahmed, N.; Kubovcakova, L.; Rybarikova, M.; et al. JAK2-V617F and interferon-α induce megakaryocyte-biased stem cells characterized by decreased long-term functionality. Blood 2021, 137, 2139–2151. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.T.; Barel, A.C.; Lascu, E.; Ritchie, E.K.; Roboz, G.J.; Christos, P.J.; Orazi, A.; Hassane, D.C.; Tam, W.; Cross, N.C.P. The effect of initial molecular profile on response to recombinant interferon-α (rIFNα) treatment in early myelofibrosis. Cancer 2017, 123, 2680–2687. [Google Scholar] [CrossRef]
- Lock, M.; Luque Paz, D.; Hansen, N.; Almeida Fonseca, T.; Usart, M.; Rai, S.; Hao-Shen, H.; Mild, G.; Dirnhofer, S.; Skoda, R.C.; et al. Combination of 5-Azacytidine and Pegifna is able to overcome resistance in JAK2-V617F positive MPN with loss of Dnmt3a. Blood 2022, 140 (Suppl. 1), 3876–3877. [Google Scholar] [CrossRef]
- Kiladjian, J.-J.; Ianotto, J.-C.; Soret, J.; Maslah, N.; Chaffaut, C.; Boyer Perrard, F.; Barraco, F.; Dubruille, V.; Capron, C.; Tisserand, A.; et al. Final results of Ruxopeg, a phase 1/2 adaptive randomized trial of ruxolitinib (rux) and pegylated interferon alpha (ifna) 2a in patients with myelofibrosis (MF). Blood 2022, 140 (Suppl. 1), 577–578. [Google Scholar] [CrossRef]
- Castillo-Tokumori, F.; Talati, C.; Al Ali, N.; Sallman, D.; Yun, S.; Sweet, K.; Padron, E.; Lancet, J.; Komrokji, R.; Kuykendall, A.T. Retrospective analysis of the clinical use and benefit of lenalidomide and thalidomide in myelofibrosis. Clin. Lymphoma Myeloma Leuk. 2020, 20, e956–e960. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M.; Müllauer, L.; Buxhofer-Ausch, V.; Gisslinger, B.; Gisslinger, H. Essential thrombocythemia versus early primary myelofibrosis: A multicenter study to validate the WHO classification. Blood 2011, 117, 5710–5718. [Google Scholar] [CrossRef]
- Barosi, G.; Rosti, V.; Gale, R.P. Myelofibrosis-type megakaryocyte dysplasia (MTMD) as a distinct category of BCR::ABL-negative myeloproliferative neoplasms. Challenges and perspectives. Leukemia 2023, 37, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Carobbio, A.; Guglielmelli, P.; Rumi, E.; Cavalloni, C.; De Stefano, V.; Betti, S.; Rambaldi, A.; Finazzi, M.C.; Thiele, J.; Vannucchi, A.M.; et al. A multistate model of survival prediction and event monitoring in prefibrotic myelofibrosis. Blood Cancer J. 2020, 10, 100. [Google Scholar] [CrossRef] [PubMed]


| Prognostic Model | Karyotype or Mutations Included in the Score Calculation (Score points) | Risk Groups (OS) |
|---|---|---|
| MIPSS70 + v2.0 [107] | Non-CALR type 1 (2) HMR = 1 (2), HMR ≥ 2 (3), HR karyotype (3), VHR karyotype (4) | (Median OS) Very low (not reached) Low (16.4 year) Intermediate (7.7 year) High (4.1 year) Very high (1.8) |
| MPN Personalized Risk Calculator [25] (not for MF only) | Mutations in 33 genes Cytogenetic abnormalities | Individualized risk calculator |
| Myelofibrosis Secondary to PV and ET- Prognostic Model (MYSEC-PM) [112] | CALR-unmutated genotype | (Median OS) Low risk (not reached) Intermediate-1 (9.3 year) Intermediate-2 (4.4 year) high risk (2 year) |
| MTSS [114] | Non CALR/MPL (2) ASXL1 (1) | (5-year OS) Low (83%) Intermediate (64%) High (37%) Very high (22%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verma, T.; Papadantonakis, N.; Peker Barclift, D.; Zhang, L. Molecular Genetic Profile of Myelofibrosis: Implications in the Diagnosis, Prognosis, and Treatment Advancements. Cancers 2024, 16, 514. https://doi.org/10.3390/cancers16030514
Verma T, Papadantonakis N, Peker Barclift D, Zhang L. Molecular Genetic Profile of Myelofibrosis: Implications in the Diagnosis, Prognosis, and Treatment Advancements. Cancers. 2024; 16(3):514. https://doi.org/10.3390/cancers16030514
Chicago/Turabian StyleVerma, Tanvi, Nikolaos Papadantonakis, Deniz Peker Barclift, and Linsheng Zhang. 2024. "Molecular Genetic Profile of Myelofibrosis: Implications in the Diagnosis, Prognosis, and Treatment Advancements" Cancers 16, no. 3: 514. https://doi.org/10.3390/cancers16030514
APA StyleVerma, T., Papadantonakis, N., Peker Barclift, D., & Zhang, L. (2024). Molecular Genetic Profile of Myelofibrosis: Implications in the Diagnosis, Prognosis, and Treatment Advancements. Cancers, 16(3), 514. https://doi.org/10.3390/cancers16030514