
Advances in Molecular Understanding of Polycythemia Vera, Essential Thrombocythemia, and Primary Myelofibrosis: Towards Precision Medicine

Simple Summary
Abstract
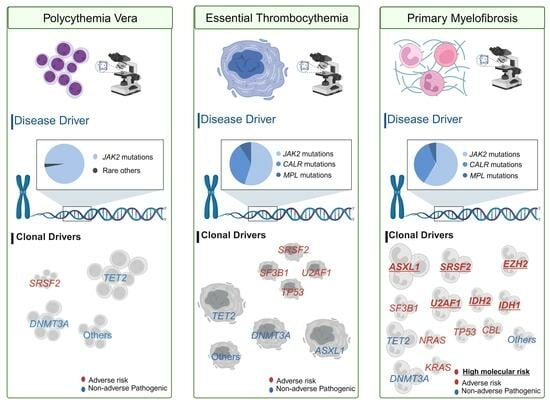
1. Introduction
2. Diagnostic Criteria for PV, ET, PMF, and MPN-NOS
3. Advances in Molecular Understanding of MPNs: Impacts on Diagnosis, Prognosis, and Treatment
3.1. Disease Drivers
3.1.1. Implications of JAK2 Mutations in MPNs
3.1.2. Implications of CALR Mutations in MPNs
3.1.3. Implications of MPL Mutations in MPNs
3.2. Clonal Drivers
3.2.1. Understanding Clonal Evolution and the Role of Additional Mutations in MPNs
3.2.2. Mutation Profiles as Prognostic Markers
Myelofibrosis Prognostic Scoring System (MIPSS)
Genetically Inspired Prognostic Scoring System (GIPSS)
3.3. Molecular Profile and Prognostic Implications in Blast Phase MPNs
3.4. Understanding Triple-Negative MPNs: Diagnostic and Molecular Challenges
3.5. Challenges Introduced by Clonal Hematopoiesis of Indeterminate Potential
4. Germline Variants in MPN Pathogenesis
5. Variant Allele Frequencies in MPNs
6. Genetic Variant Classifications in MPNs
7. Chromosomal Aberrations in MPNs
8. Implications for Treatment: A Genetic Perspective
9. Emerging Technologies and Approaches
10. Discussion
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef]
- Tefferi, A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2021, 96, 145–162. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Manshouri, T.; Patel, J.; Harris, K.; Yao, J.; Hedvat, C.; Heguy, A.; Bueso-Ramos, C.; Kantarjian, H.; Levine, R.L.; et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70, 447–452. [Google Scholar] [CrossRef]
- Network, N.C.C. Myeloproliferative Neoplasms (Version 1.2024). Available online: https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf (accessed on 29 March 2024).
- Swerdlow, S.; Campo, E.; Harris, N.L.; Jaffe, E.; Pileri, S.; Stein, H.; Thiele, J.; Arber, D.; Hasserjian, R.; Le Beau, M. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017; p. 421. [Google Scholar]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Score, J.; Mannarelli, C.; Pancrazzi, A.; Biamonte, F.; Pardanani, A.; Zoi, K.; Reiter, A.; et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: An international study of 797 patients. Leukemia 2014, 28, 1804–1810. [Google Scholar] [CrossRef]
- Li, M.M.; Cottrell, C.E.; Pullambhatla, M.; Roy, S.; Temple-Smolkin, R.L.; Turner, S.A.; Wang, K.; Zhou, Y.; Vnencak-Jones, C.L. Assessments of Somatic Variant Classification Using the Association for Molecular Pathology/American Society of Clinical Oncology/College of American Pathologists Guidelines: A Report from the Association for Molecular Pathology. J. Mol. Diagn. 2023, 25, 69–86. [Google Scholar] [CrossRef] [PubMed]
- How, J.; Garcia, J.S.; Mullally, A. Biology and therapeutic targeting of molecular mechanisms in MPNs. Blood 2023, 141, 1922–1933. [Google Scholar] [CrossRef] [PubMed]
- Luque Paz, D.; Kralovics, R.; Skoda, R.C. Genetic basis and molecular profiling in myeloproliferative neoplasms. Blood 2023, 141, 1909–1921. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martinez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G.; et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014, 124, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Rotunno, G.; Fanelli, T.; Pacilli, A.; Brogi, G.; Calabresi, L.; Pancrazzi, A.; Vannucchi, A.M. Validation of the differential prognostic impact of type 1/type 1-like versus type 2/type 2-like CALR mutations in myelofibrosis. Blood Cancer J. 2015, 5, e360. [Google Scholar] [CrossRef] [PubMed]
- Tamari, R.; Rapaport, F.; Zhang, N.; McNamara, C.; Kuykendall, A.; Sallman, D.A.; Komrokji, R.; Arruda, A.; Najfeld, V.; Sandy, L.; et al. Impact of High-Molecular-Risk Mutations on Transplantation Outcomes in Patients with Myelofibrosis. Biol. Blood Marrow Transpl. 2019, 25, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Tenedini, E.; Bernardis, I.; Artusi, V.; Artuso, L.; Roncaglia, E.; Guglielmelli, P.; Pieri, L.; Bogani, C.; Biamonte, F.; Rotunno, G.; et al. Targeted cancer exome sequencing reveals recurrent mutations in myeloproliferative neoplasms. Leukemia 2014, 28, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Aldoss, I.; Yang, D.; Mokhtari, S.; Khaled, S.; Aribi, A.; Afkhami, M.; Al Malki, M.M.; Cao, T.; Mei, M.; et al. MIPSS70+ v2.0 predicts long-term survival in myelofibrosis after allogeneic HCT with the Flu/Mel conditioning regimen. Blood Adv. 2019, 3, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.J.; Velazquez, L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 2017, 31, 1661–1670. [Google Scholar] [CrossRef]
- Oh, S.T.; Simonds, E.F.; Jones, C.; Hale, M.B.; Goltsev, Y.; Gibbs, K.D., Jr.; Merker, J.D.; Zehnder, J.L.; Nolan, G.P.; Gotlib, J. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood 2010, 116, 988–992. [Google Scholar] [CrossRef]
- Lasho, T.L.; Mudireddy, M.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Szuber, N.; Begna, K.H.; Patnaik, M.M.; Gangat, N.; Pardanani, A.; et al. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018, 2, 370–380. [Google Scholar] [CrossRef]
- Dunbar, A.J.; Rampal, R.K.; Levine, R. Leukemia secondary to myeloproliferative neoplasms. Blood 2020, 136, 61–70. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef]
- Jamieson, C.H.; Gotlib, J.; Durocher, J.A.; Chao, M.P.; Mariappan, M.R.; Lay, M.; Jones, C.; Zehnder, J.L.; Lilleberg, S.L.; Weissman, I.L. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 6224–6229. [Google Scholar] [CrossRef]
- Maddali, M.; Kulkarni, U.P.; Ravindra, N.; Jajodia, E.; Arunachalam, A.K.; Suresh, H.; Venkatraman, A.; George, B.; Mathews, V.; Balasubramanian, P. JAK2 exon 12 mutations in cases with JAK2V617F-negative polycythemia vera and primary myelofibrosis. Ann. Hematol. 2020, 99, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Nilsri, N.; Jangprasert, P.; Pawinwongchai, J.; Israsena, N.; Rojnuckarin, P. Distinct effects of V617F and exon12-mutated JAK2 expressions on erythropoiesis in a human induced pluripotent stem cell (iPSC)-based model. Sci. Rep. 2021, 11, 5255. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Pietra, D.; Li, S.; Brisci, A.; Passamonti, F.; Rumi, E.; Theocharides, A.; Ferrari, M.; Gisslinger, H.; Kralovics, R.; Cremonesi, L.; et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood 2008, 111, 1686–1689. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, J.; Elbanna, Y.A.; Kurylowicz, K.; Ciboddo, M.; Greenbaum, H.S.; Arellano, N.S.; Rodriguez, D.; Evers, M.; Bock-Hughes, A.; Liu, C.; et al. Type I but Not Type II Calreticulin Mutations Activate the IRE1alpha/XBP1 Pathway of the Unfolded Protein Response to Drive Myeloproliferative Neoplasms. Blood Cancer Discov. 2022, 3, 298–315. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Tischer, A.; Wassie, E.A.; Finke, C.M.; Belachew, A.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.D. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood 2014, 124, 2465–2466. [Google Scholar] [CrossRef] [PubMed]
- Beer, P.A.; Campbell, P.J.; Scott, L.M.; Bench, A.J.; Erber, W.N.; Bareford, D.; Wilkins, B.S.; Reilly, J.T.; Hasselbalch, H.C.; Bowman, R.; et al. MPL mutations in myeloproliferative disorders: Analysis of the PT-1 cohort. Blood 2008, 112, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Pancrazzi, A.; Bergamaschi, G.; Rosti, V.; Villani, L.; Antonioli, E.; Bosi, A.; Barosi, G.; Vannucchi, A.M.; GIMEMA—Italian Registry of Myelofibrosis; et al. Anaemia characterises patients with myelofibrosis harbouring Mpl mutation. Br. J. Haematol. 2007, 137, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Milosevic Feenstra, J.D.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Luque Paz, D.; Bader, M.S.; Nienhold, R.; Rai, S.; Almeida Fonseca, T.; Stetka, J.; Hao-Shen, H.; Mild-Schneider, G.; Passweg, J.R.; Skoda, R.C. Impact of Clonal Architecture on Clinical Course and Prognosis in Patients With Myeloproliferative Neoplasms. Hemasphere 2023, 7, e885. [Google Scholar] [CrossRef]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients with Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Kuykendall, A.T.; Talati, C.; Padron, E.; Sweet, K.; Sallman, D.; List, A.F.; Lancet, J.E.; Komrokji, R.S. Genetically inspired prognostic scoring system (GIPSS) outperforms dynamic international prognostic scoring system (DIPSS) in myelofibrosis patients. Am. J. Hematol. 2019, 94, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Shahin, O.A.; Chifotides, H.T.; Bose, P.; Masarova, L.; Verstovsek, S. Accelerated Phase of Myeloproliferative Neoplasms. Acta Haematol. 2021, 144, 484–499. [Google Scholar] [CrossRef] [PubMed]
- Michail, O.; McCallion, P.; McGimpsey, J.; Hindley, A.; Greenfield, G.; McAllister, R.; Feerick, J.; Arnold, C.; Cross, N.; Cuthbert, R.; et al. Mutational profiling in suspected triple-negative essential thrombocythaemia using targeted next-generation sequencing in a real-world cohort. J. Clin. Pathol. 2021, 74, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Maddali, M.; Venkatraman, A.; Kulkarni, U.P.; Mani, S.; Raj, S.; Sigamani, E.; Korula, A.; Fouzia, N.A.; Lionel, S.A.; Selvarajan, S.; et al. Molecular characterization of triple-negative myeloproliferative neoplasms by next-generation sequencing. Ann. Hematol. 2022, 101, 1987–2000. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghamdi, Y.A.; Lake, J.; Bagg, A.; Thakral, B.; Wang, S.A.; Bueso-Ramos, C.; Masarova, L.; Verstovsek, S.; Rogers, H.J.; Hsi, E.D.; et al. Triple-Negative Primary Myelofibrosis: A Bone Marrow Pathology Group Study. Mod. Pathol. 2023, 36, 100016. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Weeks, L.D.; Niroula, A.; Neuberg, D.; Wong, W.; Lindsley, R.C.; Luskin, M.; Berliner, N.; Stone, R.M.; DeAngelo, D.J.; Soiffer, R.; et al. Prediction of risk for myeloid malignancy in clonal hematopoiesis. NEJM Evid. 2023, 2, EVIDoa2200310. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaan8292. [Google Scholar] [CrossRef] [PubMed]
- Jacquelin, S.; Kramer, F.; Mullally, A.; Lane, S.W. Murine Models of Myelofibrosis. Cancers 2020, 12, 2381. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Benajiba, L.; Giraudier, S.; Kiladjian, J.J.; Cassinat, B. Clonal architecture evolution in Myeloproliferative Neoplasms: From a driver mutation to a complex heterogeneous mutational and phenotypic landscape. Leukemia 2023, 37, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef]
- Brown, D.W.; Cato, L.D.; Zhao, Y.; Nandakumar, S.K.; Bao, E.L.; Gardner, E.J.; Hubbard, A.K.; DePaulis, A.; Rehling, T.; Song, L.; et al. Shared and distinct genetic etiologies for different types of clonal hematopoiesis. Nat. Commun. 2023, 14, 5536. [Google Scholar] [CrossRef] [PubMed]
- Cordua, S.; Kjaer, L.; Skov, V.; Pallisgaard, N.; Hasselbalch, H.C.; Ellervik, C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 2019, 134, 469–479, Erratum in Blood 2019, 134, 1195. [Google Scholar] [CrossRef] [PubMed]
- McKerrell, T.; Park, N.; Chi, J.; Collord, G.; Moreno, T.; Ponstingl, H.; Dias, J.; Gerasimou, P.; Melanthiou, K.; Prokopiou, C.; et al. JAK2 V617F hematopoietic clones are present several years prior to MPN diagnosis and follow different expansion kinetics. Blood Adv. 2017, 1, 968–971. [Google Scholar] [CrossRef]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O’Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2(V617F) causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaam7729. [Google Scholar] [CrossRef]
- Olcaydu, D.; Harutyunyan, A.; Jager, R.; Berg, T.; Gisslinger, B.; Pabinger, I.; Gisslinger, H.; Kralovics, R. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat. Genet. 2009, 41, 450–454. [Google Scholar] [CrossRef]
- Jones, A.V.; Chase, A.; Silver, R.T.; Oscier, D.; Zoi, K.; Wang, Y.L.; Cario, H.; Pahl, H.L.; Collins, A.; Reiter, A.; et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat. Genet. 2009, 41, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Kilpivaara, O.; Mukherjee, S.; Schram, A.M.; Wadleigh, M.; Mullally, A.; Ebert, B.L.; Bass, A.; Marubayashi, S.; Heguy, A.; Garcia-Manero, G.; et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat. Genet. 2009, 41, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Tapper, W.; Jones, A.V.; Kralovics, R.; Harutyunyan, A.S.; Zoi, K.; Leung, W.; Godfrey, A.L.; Guglielmelli, P.; Callaway, A.; Ward, D.; et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat. Commun. 2015, 6, 6691. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; Nienhold, R.; Ambrosetti, A.; Cervantes, F.; Perez-Encinas, M.M.; Skoda, R.C. Somatic mutations in calreticulin can be found in pedigrees with familial predisposition to myeloproliferative neoplasms. Blood 2014, 123, 2744–2745. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G.; Campanelli, R.; Catarsi, P.; Abba, C.; Carolei, A.; Massa, M.; Gale, R.P.; Rosti, V. Type 1 CALR mutation allele frequency correlates with CD34/CXCR4 expression in myelofibrosis-type megakaryocyte dysplasia: A mechanism of disease progression? Blood Cancer J. 2024, 14, 18. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Maccari, C.; Sordi, B.; Balliu, M.; Atanasio, A.; Mannarelli, C.; Capecchi, G.; Sestini, I.; Coltro, G.; Loscocco, G.G.; et al. Phenotypic correlations of CALR mutation variant allele frequency in patients with myelofibrosis. Blood Cancer J. 2023, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Grabek, J.; Straube, J.; Bywater, M.; Lane, S.W. MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and their Effect on Treatment. Cells 2020, 9, 1901. [Google Scholar] [CrossRef]
- Kiladjian, J.J.; Cassinat, B.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Menot, M.L.; Massonnet, G.; Dutel, J.L.; Ghomari, K.; et al. High molecular response rate of polycythemia vera patients treated with pegylated interferon alpha-2a. Blood 2006, 108, 2037–2040. [Google Scholar] [CrossRef] [PubMed]
- Verger, E.; Cassinat, B.; Chauveau, A.; Dosquet, C.; Giraudier, S.; Schlageter, M.H.; Ianotto, J.C.; Yassin, M.A.; Al-Dewik, N.; Carillo, S.; et al. Clinical and molecular response to interferon-alpha therapy in essential thrombocythemia patients with CALR mutations. Blood 2015, 126, 2585–2591. [Google Scholar] [CrossRef]
- Pardanani, A.; Gotlib, J.R.; Jamieson, C.; Cortes, J.E.; Talpaz, M.; Stone, R.M.; Silverman, M.H.; Gilliland, D.G.; Shorr, J.; Tefferi, A. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J. Clin. Oncol. 2011, 29, 789–796. [Google Scholar] [CrossRef]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar] [CrossRef]
- Tefferi, A.; Cervantes, F.; Mesa, R.; Passamonti, F.; Verstovsek, S.; Vannucchi, A.M.; Gotlib, J.; Dupriez, B.; Pardanani, A.; Harrison, C.; et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood 2013, 122, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Terraf, P.; Pareja, F.; Brown, D.N.; Ceyhan-Birsoy, O.; Misyura, M.; Rana, S.; O’Reilly, E.; Carlo, M.I.; Aghajanian, C.; Liu, Y.; et al. Comprehensive assessment of germline pathogenic variant detection in tumor-only sequencing. Ann. Oncol. 2022, 33, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Kuzbari, Z.; Bandlamudi, C.; Loveday, C.; Garrett, A.; Mehine, M.; George, A.; Hanson, H.; Snape, K.; Kulkarni, A.; Allen, S.; et al. Germline-focused analysis of tumour-detected variants in 49,264 cancer patients: ESMO Precision Medicine Working Group recommendations. Ann. Oncol. 2023, 34, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Horak, P.; Griffith, M.; Danos, A.M.; Pitel, B.A.; Madhavan, S.; Liu, X.; Chow, C.; Williams, H.; Carmody, L.; Barrow-Laing, L.; et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genet. Med. 2022, 24, 986–998. [Google Scholar] [CrossRef]
- Schaafsma, E.; Takacs, E.M.; Kaur, S.; Cheng, C.; Kurokawa, M. Predicting clinical outcomes of cancer patients with a p53 deficiency gene signature. Sci. Rep. 2022, 12, 1317. [Google Scholar] [CrossRef] [PubMed]
- Hassin, O.; Nataraj, N.B.; Shreberk-Shaked, M.; Aylon, Y.; Yaeger, R.; Fontemaggi, G.; Mukherjee, S.; Maddalena, M.; Avioz, A.; Iancu, O.; et al. Different hotspot p53 mutants exert distinct phenotypes and predict outcome of colorectal cancer patients. Nat. Commun. 2022, 13, 2800. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Kantarjian, H.; Pierce, S.; Cortes, J.; Verstovsek, S. Prognostic model to identify patients with myelofibrosis at the highest risk of transformation to acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2013, 13, 315–318.e312. [Google Scholar] [CrossRef][Green Version]
- Mora, B.; Giorgino, T.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; Kiladjian, J.J.; et al. Value of cytogenetic abnormalities in post-polycythemia vera and post-essential thrombocythemia myelofibrosis: A study of the MYSEC project. Haematologica 2018, 103, e392–e394. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; Caramazza, D.; Vaidya, R.; George, G.; Begna, K.; Schwager, S.; Van Dyke, D.; Hanson, C.; Wu, W.; Pardanani, A.; et al. DIPSS plus: A refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J. Clin. Oncol. 2011, 29, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Neveling, K.; Kanagal-Shamanna, R. Optical genome mapping for structural variation analysis in hematologic malignancies. Am. J. Hematol. 2022, 97, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Mondal, A.K.; Tvrdik, T.; Hauenstein, J.; Shi, H.; Deeb, K.K.; Saxe, D.; Hastie, A.R.; Chaubey, A.; Savage, N.M.; et al. Clinical Validation and Diagnostic Utility of Optical Genome Mapping for Enhanced Cytogenomic Analysis of Hematological Neoplasms. J. Mol. Diagn. 2022, 24, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Mondal, A.K.; Singh, H.; Vashisht, A.; Ananth, S.; Saul, D.; Hastie, A.R.; Hilton, B.; DuPont, B.R.; Savage, N.M.; et al. Clinical Utility of Optical Genome Mapping and 523-Gene Next Generation Sequencing Panel for Comprehensive Evaluation of Myeloid Cancers. Cancers 2023, 15, 3214. [Google Scholar] [CrossRef] [PubMed]
- Downes, C.E.J.; McClure, B.J.; Bruning, J.B.; Page, E.; Breen, J.; Rehn, J.; Yeung, D.T.; White, D.L. Acquired JAK2 mutations confer resistance to JAK inhibitors in cell models of acute lymphoblastic leukemia. NPJ Precis. Oncol. 2021, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Reddy, M.M.; Schade, G.O.; Ray, A.; Chowdary, T.K.; Griffin, J.D.; Sattler, M. Kinase domain mutations confer resistance to novel inhibitors targeting JAK2V617F in myeloproliferative neoplasms. Leukemia 2012, 26, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Marit, M.R.; Chohan, M.; Matthew, N.; Huang, K.; Kuntz, D.A.; Rose, D.R.; Barber, D.L. Random mutagenesis reveals residues of JAK2 critical in evading inhibition by a tyrosine kinase inhibitor. PLoS ONE 2012, 7, e43437. [Google Scholar] [CrossRef]
- Pandey, G.; Kuykendall, A.T.; Reuther, G.W. JAK2 inhibitor persistence in MPN: Uncovering a central role of ERK activation. Blood Cancer J. 2022, 12, 13. [Google Scholar] [CrossRef]
- Spivak, J.L.; Moliterno, A.R. The Thrombopoietin Receptor, MPL, Is a Therapeutic Target of Opportunity in the MPN. Front. Oncol. 2021, 11, 641613. [Google Scholar] [CrossRef]
- Vainchenker, W.; Plo, I.; Marty, C.; Varghese, L.N.; Constantinescu, S.N. The role of the thrombopoietin receptor MPL in myeloproliferative neoplasms: Recent findings and potential therapeutic applications. Expert. Rev. Hematol. 2019, 12, 437–448. [Google Scholar] [CrossRef] [PubMed]
- McLornan, D.P.; Hargreaves, R.; Hernandez-Boluda, J.C.; Harrison, C.N. How I manage myeloproliferative neoplasm-unclassifiable: Practical approaches for 2022 and beyond. Br. J. Haematol. 2022, 197, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Verstovsek, S.; Pemmaraju, N. Novel Therapies in Myeloproliferative Neoplasms: Beyond JAK Inhibitor Monotherapy. J. Immunother. Precis. Oncol. 2021, 4, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Robinson, T.M.; Bowman, R.L.; Persaud, S.; Liu, Y.; Neigenfind, R.; Gao, Q.; Zhang, J.; Sun, X.; Miles, L.A.; Cai, S.F.; et al. Single-cell genotypic and phenotypic analysis of measurable residual disease in acute myeloid leukemia. Sci. Adv. 2023, 9, eadg0488. [Google Scholar] [CrossRef]
- O’Sullivan, J.M.; Mead, A.J.; Psaila, B. Single-cell methods in myeloproliferative neoplasms: Old questions, new technologies. Blood 2023, 141, 380–390. [Google Scholar] [CrossRef]
- Royston, D.; Mead, A.J.; Psaila, B. Application of Single-Cell Approaches to Study Myeloproliferative Neoplasm Biology. Hematol. Oncol. Clin. N. Am. 2021, 35, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, E.; Yoshida, K.; Frick, M.; Hoyer, K.; Christen, F.; Kaeda, J.; Obenaus, M.; Noerenberg, D.; Hennch, C.; Chan, W.; et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat. Commun. 2020, 11, 73. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Kosiorek, H.E.; Prchal, J.T.; Rambaldi, A.; Berenzon, D.; Yacoub, A.; Harrison, C.N.; McMullin, M.F.; Vannucchi, A.M.; Ewing, J.; et al. A randomized phase 3 trial of interferon-alpha vs hydroxyurea in polycythemia vera and essential thrombocythemia. Blood 2022, 139, 2931–2941. [Google Scholar] [CrossRef]

{kind=link}
| Disease | WHO Criteria | Diagnosis Requirements: WHO | ICC 2022 Classification |
|---|---|---|---|
| PV * | Major: 1. Elevated hemoglobin (>16.5 g/dL in men, >16.0 g/dL in women) or hematocrit (>49% in men, >48% in women). 2. Bone marrow biopsy showing trilineage proliferation (panmyelosis) with pleomorphic, mature megakaryocytes. 3. Presence of JAK2 V617F or exon 12 mutation. Minor: Subnormal serum erythropoietin level. | Option 1: All three criteria must be met. Option 2: The first two major criteria, plus the minor criterion. | Diagnostic Criteria: Option 1: All three criteria must be met. Option 2: First and third major criteria, plus the minor criterion. Note: Increased red blood cell mass is included in the diagnostic criteria. |
| Post-PV myelofibrosis | Major Criteria: 1. Established diagnosis of PV. 2. Bone marrow fibrosis (Grade 2 or 3). Additional Criteria: 1. Anemia (below reference range considering age, sex, altitude, or sustained loss of phlebotomy or cytoreductive treatment requirement). 2. Leukoerythroblastosis. 3. Increasing splenomegaly (increase in palpable splenomegaly >50 mm from baseline or development of newly palpable splenomegaly). 4. Development of at least two of the following symptoms: weight loss (>10% in 6 months), night sweats, unexplained fever (>37.5 °C). | Major criteria plus two additional criteria. | No significant differences noted |
| ET | Major Criteria: 1. Platelet count ≥ 450 × 109/L. 2. Bone marrow biopsy showing:
Minor Criteria: 1. Presence of a clonal marker. 2. Exclusion of reactive thrombocytosis. | Option 1: All major criteria must be met. Option 2: First three major criteria plus one minor criterion. | No significant differences noted |
| Post-ET myelofibrosis | Required Criteria: 1. Previous diagnosis of WHO-defined ET. 2. Bone marrow fibrosis grade 2–3 (on a scale of 0–3). Additional Criteria: 1. Anemia (below reference range for age, sex, and altitude) with >2 g/dL decrease from baseline hemoglobin. 2. Leukoerythroblastosis. 3. Increasing splenomegaly, defined as:
5. Development of any two (or all three) of the following symptoms:
| Option 1: All required criteria must be met. Option 2: At least two additional criteria. | No significant differences noted |
| PMF, Prefibrotic Stage | Major Criteria: 1. Megakaryocytic proliferation and atypia (without reticulin fibrosis grade > 1), accompanied by:
Minor Criteria: 1. Anemia not attributed to a comorbid condition. 2. Leukocytosis ≥ 11 × 109/L. 3. Clinically and/or imaging-detected splenomegaly. 4. LDH level above the upper limit of the institutional reference range. | Required: All three major criteria. Additional: At least one minor criterion. Confirmation: Minor criteria must be confirmed in two consecutive determinations. | No significant differences noted |
| PMF, Fibrotic Stage | Major Criteria: 1. Megakaryocytic proliferation and atypia, with reticulin and/or collagen fibrosis grade 2 or 3. 2. Does not meet diagnostic criteria for:
Minor Criteria: 1. Anemia not attributed to a comorbid condition. 2. Leukocytosis ≥ 11 × 109/L. 3. Clinically and/or imaging-detected splenomegaly. 4. LDH level above the upper limit of the institutional reference range. 5. Leukoerythroblastosis. | Required: All three major criteria. Additional: At least one minor criterion. Confirmation: Minor criteria must be confirmed in two consecutive determinations. | No significant differences noted |
| MPN-NOS/MPN-U | Required Criteria: 1. Presence of any one of the following features:
Exclusion Criteria: 1. Insufficient clinical data or inadequate bone marrow specimen for accurate evaluation and classification. 2. Recent history of cytotoxic or growth factor therapy, especially when dysplastic features are present. | Required: Presence of all required criteria and absence of all exclusion criteria | Similar diagnostic criteria |
| Gene | Frequency in MPN [12] | Reported Oncogenic/Likely Oncogenic Mutations and (Reference Transcripts) [3] | Significance and Impact on Prognosis [6] |
|---|---|---|---|
| Disease Drivers | |||
| JAK2 | PV *: 98% (~95% V617, ~4% exon 12) ET: 55% PMF: 60% | V617F; Missense/indel in aa range: pp. 536–547 (NM_004972) | WHO/ICC criterion for diagnosis; intermediate prognosis with a heightened risk of thrombosis relative to CALR type 1 mutation carriers [6,13] |
| MPL | PV: 0% ET: 5–7%, PMF 7–10% | S505G, S505N, S505C, L510P, del513, W515A, W515R, W515K, W515S, W515L, A519T, A519V, Y591D, W515-518KT. (NM_005373) | WHO/ICC criterion for diagnosis; intermediate prognosis with a heightened risk of thrombosis relative to CALR type 1 mutation carriers [6,13] |
| CALR | PV: 0% ET: 25–30% PMF: 20–30% | Frameshift in exon 9 (NM_004343) | WHO/ICC criterion for diagnosis; CALR 1: enhanced OS and reduced thrombosis risk in comparison to those with JAK2 mutations and TN-PMF, as well as better OS than CALR type 2 mutation carriers [14,15,16]; CALR 2: lower OS than CALR 1 [17] |
| Clonal Drivers | |||
| DNMT3A | PV: 5–10% ET: 1–5% PMF: 8–12% | Frameshift/nonsense/splice-site; missense in aa range: pp. 292–350, 482–614, 634–912 (NM_022552) | Inferior OS post-HCT [18] |
| IDH1 | PV: 1–2% ET: 1–2% PMF: 5–6% | Frameshift/nonsense/splice-site in exon 11–12 (NM_015338) | HMR Inferior OS and reduced PFS post-HCT [8] |
| IDH2 | PV: 1–2% ET: 1–2% PMF: 5–6% | Missense at R132 (NM_005896) | HMR Inferior OS and reduced PFS post-HCT [8] |
| ASXL1 | PV: 2–7% ET: 5–10% PMF: 15–35% | Frameshift/nonsense/splice-site in exon 11–12 (NM_015338) | HMR Adverse impact, particularly in PMF; marked by poorer OS and LFS, including post-HCT [8] |
| EZH2 | PV: 1–2% ET: 1–2% PMF: 7–10% | Frameshift/nonsense/splice-site; missense in SET domain (pp. 617–732) (NM_001203247) | HMR Inferior OS [8] |
| NRAS | PV: <2% ET: <2% PMF: 2–4% | Missense at G12/G13/Q61 (NM_002524) | Inferior OS [19] |
| KRAS | PV: <2% ET: <2% PMF: 2% | Missense at G12/G13/Q61 (NM_033360) | Similar to NRAS |
| CBL | PV: <2% ET: <2% PMF: 4% | Missense in Linker/RING finger domains (pp. 345–434) (NM_005188) | Inferior OS post-HCT [18] |
| SRSF2 | PV: <2% ET: <2% PMF: 6–14% | Missense/in-frame deletion involving P95 (NM_003016) | HMR in all MPNs Inferior OS and LFS; adverse prognosis in transformation [8] |
| U2AF1 | PV: <2% ET: <2% PMF: 7–10% | Missense at S34/Q157 (NM_006758) | HMR Adverse prognosis in PMF and secondary AML; diminished OS post-HCT, with U2AF1 Q157 mutation associated with worse outcomes compared to U2AF1 S34 mutations or unmutated MF [18] |
| TP53 | PV: <2% ET: <2% PMF: 2–5% Increased frequency in advanced stages/post-MPN AML | Frameshift/nonsense/splice-site; missense in aa range: pp. 72, 95–288, 337 (NM_001126112) | Higher likelihood of leukemic transformation [20] |
| TET2 | PV: 10–20% ET: 3–10% PMF: 10–20% | Frameshift/nonsense/splice-site; aa range: pp. 1104–1481, 1843–2002 (NM_001127208) | No consensus impact on prognosis |
| SH2B3 (LNK) | PV: 2–9% ET: 1–3% PMF: 2–4% | Frameshift/nonsense/splice-site; Missense at E208Q (NM_005475) [21] | Reported as potential driver in JAK2 negative MPN [22] |
| RUNX1 | PV: <2% ET: <2% PMF: 2–3% | Frameshift/nonsense/splice-site, S73F, H78Q, H78L, R80C, R80P, R80H, L85Q, P86L, P86H, S114L, D133Y, L134P, R135G, R135K, R135S, R139Q, R142S, A165V, R174Q, R177L, R177Q, A224T, D171G, D171V, D171N, R205W, R223C (NM_001001890) | Frequent in leukemic transformation [23,24] |
| SF3B1 | PV: 2–3% ET: 2–5% PMF: 5–7% The possibility of mixed myelodysplastic component should be considered [3,4] | Missense in terminal HEAT domains (pp. 529–1201) (NM_012433) | Adverse impact in ET [12] |
| Mutation | Targeted Therapy Options | Clinical Trial Evidence | FDA Approval Status |
|---|---|---|---|
| JAK2 V617F | Ruxolitinib, Pacritinib, Fedratinib | Ruxolitinib showed significant spleen volume reduction and improved quality of life in MF and PV patients | Ruxolitinib, Pacritinib, and Fedratinib are FDA-approved |
| CALR Mutations | Immunological therapies, CALR-targeted, mutant CALR peptide vaccine | Investigations on disrupting CALRdel52-MPL signaling complexes in CALR-mutated cells | No specific FDA approvals for CALR-targeted therapies yet |
| Telomerase Activity | Imetelstat (Telomerase inhibitor) | A phase 2 trial showed clinical improvements in intermediate-2/high-risk MF patients relapsed or refractory to ruxolitinib | Not yet FDA-approved; phase 3 trial ongoing |
| HSP90 * | PU-H71 (HSP90 inhibitor) | Early phase clinical trials ongoing for safety, tolerability, and pharmacokinetic profile in MPN patients | Not yet FDA-approved |
| MDM2/TP53 Pathway | Idasanutlin, KRT232 (MDM2 antagonists) | Phase I/Ib study in AML patients with idasanutlin showed durable responses; ongoing studies in MF. Idasanutlin in PV showed rapidly reduced JAK2 allele burden in PV patients [101] | Not yet FDA-approved for MPNs |
| Hepcidin Mimetics in PV | Rusfertide (PTG-300) | Phase 2 trials showed reduced hematocrit levels and therapeutic phlebotomy needs in PV patients | Phase 3 trial underway; not yet FDA-approved |
| Bcl-2/Bcl-xL Inhibition | Navitoclax | Phase 2 trial showed safety and efficacy in MF patients, with ongoing phase 3 trials | Not yet FDA-approved; phase 3 trial ongoing |
| Interferons | Pegylated interferons | Induce durable molecular responses and preferentially deplete JAK2-mutated HSCs, showing efficacy in ET and PV | Used in clinical practice but specific FDA approval varies |
| CD123 Targeted Therapy | Tagraxofusp (SL-401) | Phase I/II clinical trial ongoing in intermediate- or high-risk and relapsed/refractory MF patients | FDA-approved for BPDCN, not specifically for MPNs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tashkandi, H.; Younes, I.E. Advances in Molecular Understanding of Polycythemia Vera, Essential Thrombocythemia, and Primary Myelofibrosis: Towards Precision Medicine. Cancers 2024, 16, 1679. https://doi.org/10.3390/cancers16091679
Tashkandi H, Younes IE. Advances in Molecular Understanding of Polycythemia Vera, Essential Thrombocythemia, and Primary Myelofibrosis: Towards Precision Medicine. Cancers. 2024; 16(9):1679. https://doi.org/10.3390/cancers16091679
Chicago/Turabian StyleTashkandi, Hammad, and Ismail Elbaz Younes. 2024. "Advances in Molecular Understanding of Polycythemia Vera, Essential Thrombocythemia, and Primary Myelofibrosis: Towards Precision Medicine" Cancers 16, no. 9: 1679. https://doi.org/10.3390/cancers16091679
APA StyleTashkandi, H., & Younes, I. E. (2024). Advances in Molecular Understanding of Polycythemia Vera, Essential Thrombocythemia, and Primary Myelofibrosis: Towards Precision Medicine. Cancers, 16(9), 1679. https://doi.org/10.3390/cancers16091679