

The Role of the Complement in Clear Cell Renal Carcinoma (ccRCC)—What Future Prospects Are There for Its Use in Clinical Practice?

 , ,
, , 
Abstract
Simple Summary
Abstract
1. Introduction
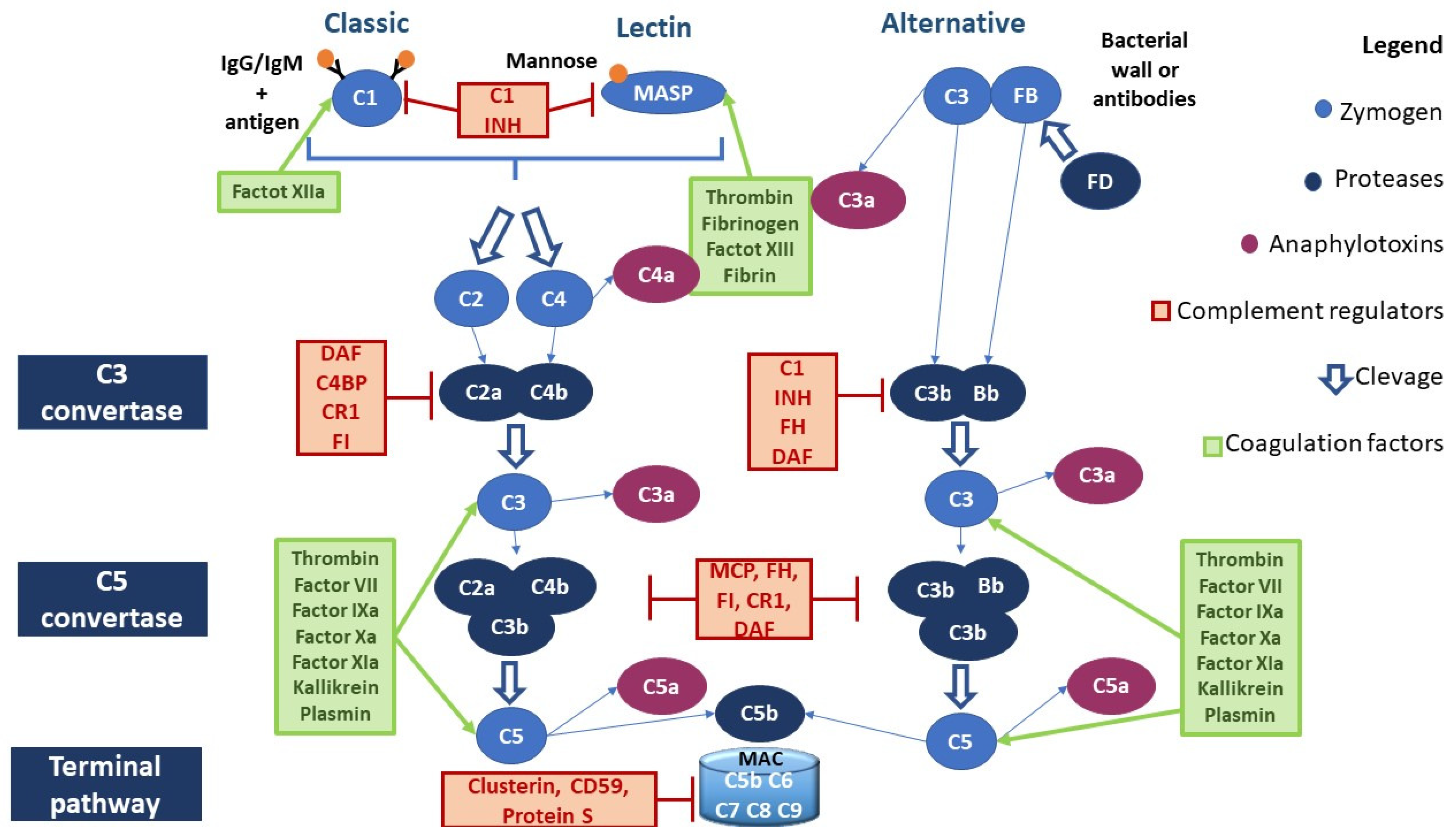
2. The Physiology of the Complement System
2.1. Canonical Pathways of Complement Activation
2.1.1. Classical Pathway
2.1.2. Lectin Pathway
2.1.3. Alternative Pathway
2.1.4. Anaphylatoxins
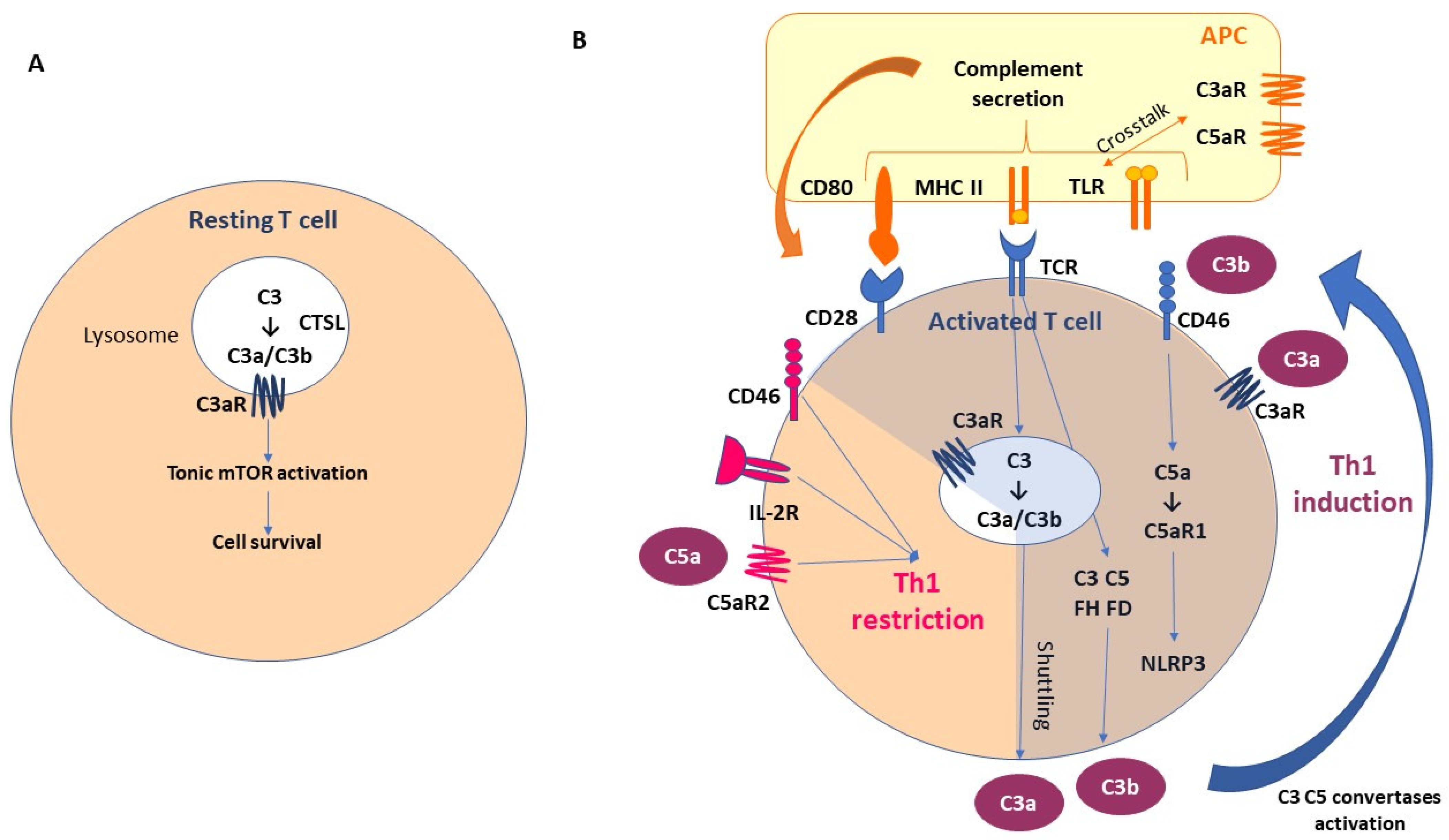
2.2. Non-Canonical Pathways of Complement Activation
2.2.1. Complement Activation through Coagulation Factors
2.2.2. Local Complement Production
2.3. Complement Negative Regulators
- (a)
- C1 inhibitor (C1 INH) is a competitive inhibitor of the C1r-C1s complex and MASP2 and interferes with the C3b–factor B interaction, blocking the activation of all three canonical pathways of CS;
- (b)
- C3 and C5 convertase inhibitors: C4b-binding protein (C4BP) controls the activation at the C4 level of the CP and LP; factor H (FH) competes with FB to C3b-bond; complement receptor 1 (CR1) is capable of binding both C3b and C4b, displacing the link with Bb and C2b, and acts as a co-factor for factor I (FI), which degrades the fragment C3b; membrane cofactor protein (MCP) is capable of binding both C3b and C4b, displacing the link with Bb and C2b, respectively; and decay-accelerating factor (DAF) accelerates the decay of C3 convertases.
- (c)
3. Cancer and Complement
4. The Prognostic Role of the Complement in Renal Cell Carcinoma
5. The Complement System as a Possible Therapeutic Target
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Albiges, L.; Tannir, N.M.; Burotto, M.; McDermott, D.; Plimack, E.R.; Barthélémy, P.; Porta, C.; Powles, T.; Donskov, F.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: Extended 4-year follow-up of the phase III CheckMate 214 trial. ESMO Open 2020, 5, e001079. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Plimack, E.R.; Soulières, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.-Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Mazumdar, M.; Bacik, J.; Russo, P.; Berg, W.J.; Metz, E.M. Effect of cytokine therapy on survival for patients with advanced renal cell carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2000, 18, 1928–1935. [Google Scholar] [CrossRef]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 3698. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Motzer, R.J.; Powles, T.; Burotto, M.; Apolo, A.B.; Escudier, B.; Tomita, Y.; McDermott, D.F.; Braun, D.A.; Han, C.; et al. Biomarker analysis from the phase 3 CheckMate 9ER trial of nivolumab + cabozantinib v sunitinib for advanced renal cell carcinoma (aRCC). J. Clin. Oncol. 2023, 41 (Suppl. S6), 608. [Google Scholar] [CrossRef]
- Motzer, R.J.; Banchereau, R.; Hamidi, H.; Powles, T.; McDermott, D.; Atkins, M.B.; Escudier, B.; Liu, L.-F.; Leng, N.; Abbas, A.R.; et al. Molecular Subsets in Renal Cancer Determine Outcome to Checkpoint and Angiogenesis Blockade. Cancer Cell 2020, 38, 803–817.e4. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Ciccarese, C.; Buti, S.; Roberto, M.; Calabro, F.; Masini, C.; Massari, F.; Cannella, M.A.; Mazzaschi, G.; Astore, S.; Di Girolamo, S.; et al. Evaluation of PBRM1, PD-L1, CD31, and CD4/CD8 ratio as a predictive signature of response to VEGFR-TKI–based therapy in patients with metastatic renal cell carcinoma (mRCC) with IMDC intermediate prognosis: Results from the APAChE-I Study. J. Clin. Oncol. 2023, 41 (Suppl. S6), 714. [Google Scholar] [CrossRef]
- Bimbatti, D.; Ciccarese, C.; Fantinel, E.; Sava, T.; Massari, F.; Bisogno, I.; Romano, M.; Porcaro, A.; Brunelli, M.; Martignoni, G.; et al. Predictive role of changes in the tumor burden and International Metastatic Renal Cell Carcinoma Database Consortium class during active surveillance for metastatic renal cell carcinoma. Urol. Oncol. 2018, 36, e13–e526.e18. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, R.; Lanoy, E.; Albiges, L.; Escudier, B. Tumour burden is an independent prognostic factor in metastatic renal cell carcinoma. BJU Int. 2012, 110, 1747–1753. [Google Scholar] [CrossRef]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in cancer: Untangling an intricate relationship. Nat. Rev. Immunol. 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Magrini, E.; Minute, L.; Dambra, M.; Garlanda, C. Complement activation in cancer: Effects on tumor-associated myeloid cells and immunosuppression. Semin. Immunol. 2022, 60, 101642. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautès-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef]
- Reese, B.; Silwal, A.; Daugherity, E.; Daugherity, M.; Arabi, M.; Daly, P.; Paterson, Y.; Woolford, L.; Christie, A.; Elias, R.; et al. Complement as Prognostic Biomarker and Potential Therapeutic Target in Renal Cell Carcinoma. J. Immunol. 2020, 205, 3218–3229. [Google Scholar] [CrossRef]
- Ajona, D.; Ortiz-Espinosa, S.; Moreno, H.; Lozano, T.; Pajares, M.J.; Agorreta, J.; Bértolo, C.; Lasarte, J.J.; Vicent, S.; Hoehlig, K.; et al. A Combined PD-1/C5a Blockade Synergistically Protects against Lung Cancer Growth and Metastasis. Cancer Discov. 2017, 7, 694–703. [Google Scholar] [CrossRef]
- Kolev, M.; Le Friec, G.; Kemper, C. The role of complement in CD4+ T cell homeostasis and effector functions. Semin. Immunol. 2013, 25, 12–19. [Google Scholar] [CrossRef]
- Ricklin, D.; Lambris, J.D. Complement in immune and inflammatory disorders: Pathophysiological mechanisms. J. Immunol. 2013, 190, 3831–3838. [Google Scholar] [CrossRef]
- Lubbers, R.; van Essen, M.F.; van Kooten, C.; Trouw, L.A. Production of complement components by cells of the immune system. Clin. Exp. Immunol. 2017, 188, 183–194. [Google Scholar] [CrossRef]
- Ricklin, D.; Lambris, J.D. Complement-targeted therapeutics. Nat. Biotechnol. 2007, 25, 1265–1275. [Google Scholar] [CrossRef]
- Porter, R.R.; Reid, K.B. Activation of the complement system by antibody-antigen complexes: The classical pathway. Adv. Protein Chem. 1979, 33, 1–71. [Google Scholar] [CrossRef]
- Garred, P.; Genster, N.; Pilely, K.; Bayarri-Olmos, R.; Rosbjerg, A.; Ma, Y.J.; Skjoedt, M.-O. A journey through the lectin pathway of complement-MBL and beyond. Immunol. Rev. 2016, 274, 74–97. [Google Scholar] [CrossRef] [PubMed]
- Cooper, P.D. Complement and cancer: Activation of the alternative pathway as a theoretical base for immunotherapy. Adv. Immun. Cancer Ther. 1985, 1, 125–166. [Google Scholar] [CrossRef]
- Tsuruta, T.; Yamamoto, T.; Matsubara, S.; Nagasawa, S.; Tanase, S.; Tanaka, J.; Takagi, K.; Kambara, T. Novel function of C4a anaphylatoxin. Release from monocytes of protein which inhibits monocyte chemotaxis. Am. J. Pathol. 1993, 142, 1848–1857. [Google Scholar]
- Zhao, Y.; Xu, H.; Yu, W.; Xie, B.-D. Complement anaphylatoxin C4a inhibits C5a-induced neointima formation following arterial injury. Mol. Med. Rep. 2014, 10, 45–52. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barnum, S.R. C4a: An Anaphylatoxin in Name Only. J. Innate Immun. 2015, 7, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Klos, A.; Tenner, A.J.; Johswich, K.-O.; Ager, R.R.; Reis, E.S.; Köhl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef] [PubMed]
- El-Lati, S.G.; Dahinden, C.A.; Church, M.K. Complement peptides C3a- and C5a-induced mediator release from dissociated human skin mast cells. J. Investig. Dermatol. 1994, 102, 803–806. [Google Scholar] [CrossRef]
- Elsner, J.; Oppermann, M.; Czech, W.; Kapp, A. C3a activates the respiratory burst in human polymorphonuclear neutrophilic leukocytes via pertussis toxin-sensitive G-proteins. Blood 1994, 83, 3324–3331. [Google Scholar] [CrossRef] [PubMed]
- Elsner, J.; Oppermann, M.; Czech, W.; Dobos, G.; Schöpf, E.; Norgauer, J.; Kapp, A. C3a activates reactive oxygen radical species production and intracellular calcium transients in human eosinophils. Eur. J. Immunol. 1994, 24, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Imamichi, T.; Nagasawa, S. Characterization of C3a anaphylatoxin receptor on guinea-pig macrophages. Immunology 1993, 79, 633–638. [Google Scholar] [PubMed]
- Chen, N.-J.; Mirtsos, C.; Suh, D.; Lu, Y.-C.; Lin, W.-J.; McKerlie, C.; Lee, T.; Baribault, H.; Tian, H.; Yeh, W.-C. C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature 2007, 446, 203–207. [Google Scholar] [CrossRef]
- Klaus, G.G.; Humphrey, J.H. A re-evaluation of the role of C3 in B-cell activation. Immunol. Today 1986, 7, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, P.W.; Allison, M.E.; Akkaraju, S.; Goodnow, C.C.; Fearon, D.T. C3d of complement as a molecular adjuvant: Bridging innate and acquired immunity. Science 1996, 271, 348–350. [Google Scholar] [CrossRef]
- Fang, Y.; Xu, C.; Fu, Y.X.; Holers, V.M.; Molina, H. Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J. Immunol. 1998, 160, 5273–5279. [Google Scholar] [CrossRef]
- Strainic, M.G.; Liu, J.; Huang, D.; An, F.; Lalli, P.N.; Muqim, N.; Shapiro, V.S.; Dubyak, G.R.; Heeger, P.S.; Medof, M.E. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 2008, 28, 425–435. [Google Scholar] [CrossRef]
- Peng, Q.; Li, K.; Patel, H.; Sacks, S.H.; Zhou, W. Dendritic cell synthesis of C3 is required for full T cell activation and development of a Th1 phenotype. J. Immunol. 2006, 176, 3330–3341. [Google Scholar] [CrossRef]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Brückner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular intercommunication between the complement and coagulation systems. J. Immunol. 2010, 185, 5628–5636. [Google Scholar] [CrossRef]
- Ghebrehiwet, B.; Silverberg, M.; Kaplan, A.P. Activation of the classical pathway of complement by Hageman factor fragment. J. Exp. Med. 1981, 153, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Gulla, K.C.; Gupta, K.; Krarup, A.; Gal, P.; Schwaeble, W.J.; Sim, R.B.; O’Connor, C.D.; Hajela, K. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology 2010, 129, 482–495. [Google Scholar] [CrossRef]
- Krarup, A.; Wallis, R.; Presanis, J.S.; Gál, P.; Sim, R.B. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS ONE 2007, 2, e623. [Google Scholar] [CrossRef]
- Hess, K.; Ajjan, R.; Phoenix, F.; Dobó, J.; Gál, P.; Schroeder, V. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PLoS ONE 2012, 7, e35690. [Google Scholar] [CrossRef] [PubMed]
- Lidington, E.A.; Haskard, D.O.; Mason, J.C. Induction of decay-accelerating factor by thrombin through a protease-activated receptor 1 and protein kinase C-dependent pathway protects vascular endothelial cells from complement-mediated injury. Blood 2000, 96, 2784–2792. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Kato, H.; Iwanaga, S.; Takada, K.; Kimura, T. New fluorogenic substrates for alpha-thrombin, factor Xa, kallikreins, and urokinase. J. Biochem. 1977, 82, 1495–1498. [Google Scholar] [CrossRef] [PubMed]
- Presanis, J.S.; Hajela, K.; Ambrus, G.; Gál, P.; Sim, R.B. Differential substrate and inhibitor profiles for human MASP-1 and MASP-2. Mol. Immunol. 2004, 40, 921–929. [Google Scholar] [CrossRef]
- Nangaku, M.; Couser, W.G. Mechanisms of immune-deposit formation and the mediation of immune renal injury. Clin. Exp. Nephrol. 2005, 9, 183–191. [Google Scholar] [CrossRef]
- Markiewski, M.M.; Nilsson, B.; Ekdahl, K.N.; Mollnes, T.E.; Lambris, J.D. Complement and coagulation: Strangers or partners in crime? Trends Immunol. 2007, 28, 184–192. [Google Scholar] [CrossRef]
- Amara, U.; Rittirsch, D.; Flierl, M.; Bruckner, U.; Klos, A.; Gebhard, F.; Lambris, J.D.; Huber-Lang, M. Interaction between the coagulation and complement system. Adv. Exp. Med. Biol. 2008, 632, 71–79. [Google Scholar] [CrossRef]
- Wiggins, R.C.; Giclas, P.C.; Henson, P.M. Chemotactic activity generated from the fifth component of complement by plasma kallikrein of the rabbit. J. Exp. Med. 1981, 153, 1391–1404. [Google Scholar] [CrossRef]
- Frade, R.; Rodrigues-Lima, F.; Huang, S.; Xie, K.; Guillaume, N.; Bar-Eli, M. Procathepsin-L, a proteinase that cleaves human C3 (the third component of complement), confers high tumorigenic and metastatic properties to human melanoma cells. Cancer Res. 1998, 58, 2733–2736. [Google Scholar] [CrossRef] [PubMed]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Nilsson-Ekdahl, K.; Nilsson, B. Phosphorylation of C3 by a casein kinase released from activated human platelets increases opsonization of immune complexes and binding to complement receptor type 1. Eur. J. Immunol. 2001, 31, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Ekdahl, K.N.; Nilsson, B. Phosphorylation of complement component C3 and C3 fragments by a human platelet protein kinase. Inhibition of factor I-mediated cleavage of C3b. J. Immunol. 1995, 154, 6502–6510. [Google Scholar] [CrossRef]
- Tedesco, F.; Pausa, M.; Nardon, E.; Introna, M.; Mantovani, A.; Dobrina, A. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J. Exp. Med. 1997, 185, 1619–1627. [Google Scholar] [CrossRef]
- Ikeda, K.; Nagasawa, K.; Horiuchi, T.; Tsuru, T.; Nishizaka, H.; Niho, Y. C5a induces tissue factor activity on endothelial cells. Thromb. Haemost. 1997, 77, 394–398. [Google Scholar] [CrossRef]
- Weiler, J.M.; Edens, R.E.; Linhardt, R.J.; Kapelanski, D.P. Heparin and modified heparin inhibit complement activation in vivo. J. Immunol. 1992, 148, 3210–3215. [Google Scholar] [CrossRef]
- Xi, W.; Liu, L.; Wang, J.; Xia, Y.; Bai, Q.; Long, Q.; Wang, Y.; Xu, J.; Guo, J. High Level of Anaphylatoxin C5a Predicts Poor Clinical Outcome in Patients with Clear Cell Renal Cell Carcinoma. Sci. Rep. 2016, 6, 29177. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S.; et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 2013, 39, 1143–1157. [Google Scholar] [CrossRef]
- Arbore, G.; West, E.E.; Spolski, R.; Robertson, A.A.B.; Klos, A.; Rheinheimer, C.; Dutow, P.; Woodruff, T.M.; Yu, Z.X.; O’Neill, L.A.; et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science 2016, 352, aad1210. [Google Scholar] [CrossRef] [PubMed]
- Kunz, N.; Kemper, C. Complement Has Brains-Do Intracellular Complement and Immunometabolism Cooperate in Tissue Homeostasis and Behavior? Front. Immunol. 2021, 12, 629986. [Google Scholar] [CrossRef] [PubMed]
- Cardone, J.; Le Friec, G.; Vantourout, P.; Roberts, A.; Fuchs, A.; Jackson, I.; Suddason, T.; Lord, G.; Atkinson, J.P.; Cope, A.; et al. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat. Immunol. 2010, 11, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Song, W.-C. Crosstalk between complement and toll-like receptors. Toxicol. Pathol. 2012, 40, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Sarma, J.V.; Ward, P.A. The complement system. Cell Tissue Res. 2011, 343, 227–235. [Google Scholar] [CrossRef]
- Poppelaars, F.; Faria, B.; Gaya da Costa, M.; Franssen, C.F.M.; van Son, W.J.; Berger, S.P.; Daha, M.R.; Seelen, M.A. The Complement System in Dialysis: A Forgotten Story? Front. Immunol. 2018, 9, 71. [Google Scholar] [CrossRef]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef]
- Smith, M.R. Rituximab (monoclonal anti-CD20 antibody): Mechanisms of action and resistance. Oncogene 2003, 22, 7359–7368. [Google Scholar] [CrossRef]
- Gunn, L.; Ding, C.; Liu, M.; Ma, Y.; Qi, C.; Cai, Y.; Hu, X.; Aggarwal, D.; Zhang, H.-G.; Yan, J. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J. Immunol. 2012, 189, 2985–2994. [Google Scholar] [CrossRef]
- Gorter, A.; Meri, S. Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol. Today 1999, 20, 576–582. [Google Scholar] [CrossRef]
- Schmidt, C.Q.; Lambris, J.D.; Ricklin, D. Protection of host cells by complement regulators. Immunol. Rev. 2016, 274, 152–171. [Google Scholar] [CrossRef]
- Piao, C.; Cai, L.; Qiu, S.; Jia, L.; Song, W.; Du, J. Complement 5a Enhances Hepatic Metastases of Colon Cancer via Monocyte Chemoattractant Protein-1-mediated Inflammatory Cell Infiltration. J. Biol. Chem. 2015, 290, 10667–10676. [Google Scholar] [CrossRef]
- Cho, M.S.; Vasquez, H.G.; Rupaimoole, R.; Pradeep, S.; Wu, S.; Zand, B.; Han, H.-D.; Rodriguez-Aguayo, C.; Bottsford-Miller, J.; Huang, J.; et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014, 6, 1085–1095. [Google Scholar] [CrossRef]
- Köhl, J. Self, non-self, and danger: A complementary view. Adv. Exp. Med. Biol. 2006, 586, 71–94. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; DeAngelis, R.A.; Benencia, F.; Ricklin-Lichtsteiner, S.K.; Koutoulaki, A.; Gerard, C.; Coukos, G.; Lambris, J.D. Modulation of the antitumor immune response by complement. Nat. Immunol. 2008, 9, 1225–1235. [Google Scholar] [CrossRef]
- Corrales, L.; Ajona, D.; Rafail, S.; Lasarte, J.J.; Riezu-Boj, J.I.; Lambris, J.D.; Rouzaut, A.; Pajares, M.J.; Montuenga, L.M.; Pio, R. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J. Immunol. 2012, 189, 4674–4683. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Espinosa, S.; Morales, X.; Senent, Y.; Alignani, D.; Tavira, B.; Macaya, I.; Ruiz, B.; Moreno, H.; Remírez, A.; Sainz, C.; et al. Complement C5a induces the formation of neutrophil extracellular traps by myeloid-derived suppressor cells to promote metastasis. Cancer Lett. 2022, 529, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Nabizadeh, J.A.; Manthey, H.D.; Steyn, F.J.; Chen, W.; Widiapradja, A.; Md Akhir, F.N.; Boyle, G.M.; Taylor, S.M.; Woodruff, T.M.; Rolfe, B.E. The Complement C3a Receptor Contributes to Melanoma Tumorigenesis by Inhibiting Neutrophil and CD4+ T Cell Responses. J. Immunol. 2016, 196, 4783–4792. [Google Scholar] [CrossRef]
- Guglietta, S.; Chiavelli, A.; Zagato, E.; Krieg, C.; Gandini, S.; Ravenda, P.S.; Bazolli, B.; Lu, B.; Penna, G.; Rescigno, M. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat. Commun. 2016, 7, 11037. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Assiri, A.M.; Broering, D.C. Complement and macrophage crosstalk during process of angiogenesis in tumor progression. J. Biomed. Sci. 2015, 22, 58. [Google Scholar] [CrossRef]
- Piao, C.; Zhang, W.-M.; Li, T.-T.; Zhang, C.-C.; Qiu, S.; Liu, Y.; Liu, S.; Jin, M.; Jia, L.-X.; Song, W.-C.; et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp. Cell Res. 2018, 366, 127–138. [Google Scholar] [CrossRef]
- Fu, L.-Q.; Du, W.-L.; Cai, M.-H.; Yao, J.-Y.; Zhao, Y.-Y.; Mou, X.-Z. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell. Immunol. 2020, 353, 104119. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Crivellato, E.; Vacca, A. Macrophages and tumor angiogenesis. Leukemia 2007, 21, 2085–2089. [Google Scholar] [CrossRef] [PubMed]
- Lamagna, C.; Aurrand-Lions, M.; Imhof, B.A. Dual role of macrophages in tumor growth and angiogenesis. J. Leukoc. Biol. 2006, 80, 705–713. [Google Scholar] [CrossRef]
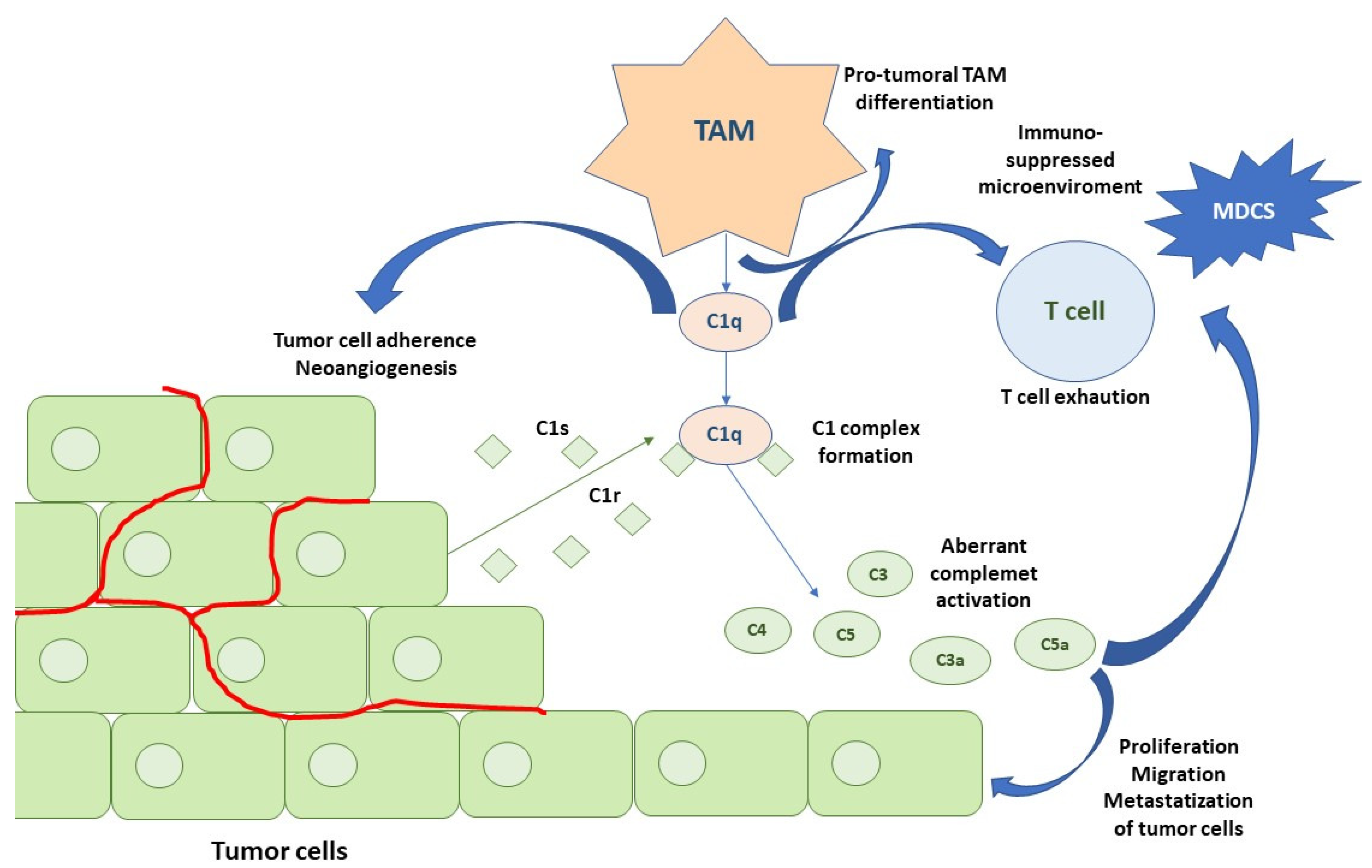
- Revel, M.; Sautès-Fridman, C.; Fridman, W.-H.; Roumenina, L.T. C1q+ macrophages: Passengers or drivers of cancer progression. Trends Cancer 2022, 8, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Towner, L.D.; Wheat, R.A.; Hughes, T.R.; Morgan, B.P. Complement Membrane Attack and Tumorigenesis: A Systems Biology Approach. J. Biol. Chem. 2016, 291, 14927–14938. [Google Scholar] [CrossRef]
- Vlaicu, S.I.; Tegla, C.A.; Cudrici, C.D.; Danoff, J.; Madani, H.; Sugarman, A.; Niculescu, F.; Mircea, P.A.; Rus, V.; Rus, H. Role of C5b-9 complement complex and response gene to complement-32 (RGC-32) in cancer. Immunol. Res. 2013, 56, 109–121. [Google Scholar] [CrossRef]
- Nitta, H.; Murakami, Y.; Wada, Y.; Eto, M.; Baba, H.; Imamura, T. Cancer cells release anaphylatoxin C5a from C5 by serine protease to enhance invasiveness. Oncol. Rep. 2014, 32, 1715–1719. [Google Scholar] [CrossRef]
- Cho, M.S.; Rupaimoole, R.; Choi, H.-J.; Noh, K.; Chen, J.; Hu, Q.; Sood, A.K.; Afshar-Kharghan, V. Complement Component 3 Is Regulated by TWIST1 and Mediates Epithelial-Mesenchymal Transition. J. Immunol. 2016, 196, 1412–1418. [Google Scholar] [CrossRef]
- Fan, Z.; Qin, J.; Wang, D.; Geng, S. Complement C3a promotes proliferation, migration and stemness in cutaneous squamous cell carcinoma. J. Cell. Mol. Med. 2019, 23, 3097–3107. [Google Scholar] [CrossRef]
- Casula, M.; Montecucco, F.; Bonaventura, A.; Liberale, L.; Vecchié, A.; Dallegri, F.; Carbone, F. Update on the role of Pentraxin 3 in atherosclerosis and cardiovascular diseases. Vascul. Pharmacol. 2017, 99, 1–12. [Google Scholar] [CrossRef]
- Presta, M.; Foglio, E.; Churruca Schuind, A.; Ronca, R. Long Pentraxin-3 Modulates the Angiogenic Activity of Fibroblast Growth Factor-2. Front. Immunol. 2018, 9, 2327. [Google Scholar] [CrossRef] [PubMed]
- Daigo, K.; Inforzato, A.; Barajon, I.; Garlanda, C.; Bottazzi, B.; Meri, S.; Mantovani, A. Pentraxins in the activation and regulation of innate immunity. Immunol. Rev. 2016, 274, 202–217. [Google Scholar] [CrossRef]
- Bottazzi, B.; Garlanda, C.; Cotena, A.; Moalli, F.; Jaillon, S.; Deban, L.; Mantovani, A. The long pentraxin PTX3 as a prototypic humoral pattern recognition receptor: Interplay with cellular innate immunity. Immunol. Rev. 2009, 227, 9–18. [Google Scholar] [CrossRef]
- Souza, D.G.; Amaral, F.A.; Fagundes, C.T.; Coelho, F.M.; Arantes, R.M.E.; Sousa, L.P.; Matzuk, M.M.; Garlanda, C.; Mantovani, A.; Dias, A.A.; et al. The long pentraxin PTX3 is crucial for tissue inflammation after intestinal ischemia and reperfusion in mice. Am. J. Pathol. 2009, 174, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Inforzato, A.; Bottazzi, B.; Garlanda, C.; Valentino, S.; Mantovani, A. Pentraxins in humoral innate immunity. Adv. Exp. Med. Biol. 2012, 946, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Daigo, K.; Hamakubo, T. Host-protective effect of circulating pentraxin 3 (PTX3) and complex formation with neutrophil extracellular traps. Front. Immunol. 2012, 3, 378. [Google Scholar] [CrossRef]
- Gout, E.; Moriscot, C.; Doni, A.; Dumestre-Pérard, C.; Lacroix, M.; Pérard, J.; Schoehn, G.; Mantovani, A.; Arlaud, G.J.; Thielens, N.M. M-ficolin interacts with the long pentraxin PTX3: A novel case of cross-talk between soluble pattern-recognition molecules. J. Immunol. 2011, 186, 5815–5822. [Google Scholar] [CrossRef]
- Deban, L.; Jarva, H.; Lehtinen, M.J.; Bottazzi, B.; Bastone, A.; Doni, A.; Jokiranta, T.S.; Mantovani, A.; Meri, S. Binding of the long pentraxin PTX3 to factor H: Interacting domains and function in the regulation of complement activation. J. Immunol. 2008, 181, 8433–8440. [Google Scholar] [CrossRef]
- Bonavita, E.; Gentile, S.; Rubino, M.; Maina, V.; Papait, R.; Kunderfranco, P.; Greco, C.; Feruglio, F.; Molgora, M.; Laface, I.; et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell 2015, 160, 700–714. [Google Scholar] [CrossRef]
- Olivieri, F.; Albertini, M.C.; Orciani, M.; Ceka, A.; Cricca, M.; Procopio, A.D.; Bonafè, M. DNA damage response (DDR) and senescence: Shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget 2015, 6, 35509–35521. [Google Scholar] [CrossRef] [PubMed]
- Roumenina, L.T.; Daugan, M.V.; Noé, R.; Petitprez, F.; Vano, Y.A.; Sanchez-Salas, R.; Becht, E.; Meilleroux, J.; Le Clec’h, B.; Giraldo, N.A.; et al. Tumor Cells Hijack Macrophage-Produced Complement C1q to Promote Tumor Growth. Cancer Immunol. Res. 2019, 7, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Daugan, M.V.; Revel, M.; Russick, J.; Dragon-Durey, M.-A.; Gaboriaud, C.; Robe-Rybkine, T.; Poillerat, V.; Grunenwald, A.; Lacroix, G.; Bougouin, A.; et al. Complement C1s and C4d as Prognostic Biomarkers in Renal Cancer: Emergence of Noncanonical Functions of C1s. Cancer Immunol. Res. 2021, 9, 891–908. [Google Scholar] [CrossRef] [PubMed]
- Magrini, E.; Garlanda, C. Noncanonical Functions of C1s Complement Its Canonical Functions in Renal Cancer. Cancer Immunol. Res. 2021, 9, 855. [Google Scholar] [CrossRef]
- Yang, C.; Yang, F.; Chen, X.; Li, Y.; Hu, X.; Guo, J.; Yao, J. Overexpression of complement C5a indicates poor survival and therapeutic response in metastatic renal cell carcinoma. Int. J. Biol. Markers 2023, 38, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Ma, W.-M.; Yang, W.; Hao, L.; Zhang, S.-Q.; Fang, K.; Hu, C.-H.; Zhang, Q.-J.; Shi, Z.-D.; Zhang, W.; et al. Identification of C3 and FN1 as potential biomarkers associated with progression and prognosis for clear cell renal cell carcinoma. BMC Cancer 2021, 21, 1135. [Google Scholar] [CrossRef] [PubMed]
- Daugan, M.V.; Revel, M.; Thouenon, R.; Dragon-Durey, M.-A.; Robe-Rybkine, T.; Torset, C.; Merle, N.S.; Noé, R.; Verkarre, V.; Oudard, S.M.; et al. Intracellular Factor H Drives Tumor Progression Independently of the Complement Cascade. Cancer Immunol. Res. 2021, 9, 909–925. [Google Scholar] [CrossRef]
- Netti, G.S.; Lucarelli, G.; Spadaccino, F.; Castellano, G.; Gigante, M.; Divella, C.; Rocchetti, M.T.; Rascio, F.; Mancini, V.; Stallone, G.; et al. PTX3 modulates the immunoflogosis in tumor microenvironment and is a prognostic factor for patients with clear cell renal cell carcinoma. Aging 2020, 12, 7585–7602. [Google Scholar] [CrossRef]
- Ajona, D.; Ortiz-Espinosa, S.; Pio, R. Complement anaphylatoxins C3a and C5a: Emerging roles in cancer progression and treatment. Semin. Cell Dev. Biol. 2019, 85, 153–163. [Google Scholar] [CrossRef]
- Lin, E.; Liu, X.; Liu, Y.; Zhang, Z.; Xie, L.; Tian, K.; Liu, J.; Yu, Y. Roles of the Dynamic Tumor Immune Microenvironment in the Individualized Treatment of Advanced Clear Cell Renal Cell Carcinoma. Front. Immunol. 2021, 12, 653358. [Google Scholar] [CrossRef]
- Lasorsa, F.; di Meo, N.A.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Tataru, O.S.; Autorino, R.; Battaglia, M.; et al. Immune Checkpoint Inhibitors in Renal Cell Carcinoma: Molecular Basis and Rationale for Their Use in Clinical Practice. Biomedicines 2023, 11, 1071. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.L.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Tataru, O.S.; Autorino, R.; Battaglia, M.; Ditonno, P.; et al. Cellular and Molecular Players in the Tumor Microenvironment of Renal Cell Carcinoma. J. Clin. Med. 2023, 12, 3888. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Robbins, P.B.; Powles, T.; Albiges, L.; Haanen, J.B.; Larkin, J.; Mu, X.J.; Ching, K.A.; Uemura, M.; Pal, S.K.; et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: Biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 2020, 26, 1733–1741. [Google Scholar] [CrossRef]
- Iacovelli, R.; Atzori, F.; Basso, U.; Bersanelli, M.; Bonomi, L.; Bracarda, S.; Buttigliero, C.; Fantinel, E.; Calabro, F.; Chiuri, V.E.; et al. Phase II study of avelumab plus intermittent axitinib in previously untreated patients with metastatic renal cell carcinoma (Tide-A study). J. Clin. Oncol. 2020, 38 (Suppl. S6), TPS762. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Factor | Substrate | Action of the Complement/Coagulation Factor on the Substrate: Activated (+)/Inactivated (−) | References |
|---|---|---|---|
| Factor XIIa | C1r | + | [37] |
| Factor XIII, Fibrinogen, Fibrin, Thrombin | MASP1 | + | [38,40,42,43] |
| Fibrin, Thrombin | MASP2 | + | [43,46,47] |
| Thrombin, Factor VII, Factor IXa, Factor Xa, Factor Xia, Kallikrein, Plasmin | C3 and C5 | + | [40,48,49,50,51,52,53] |
| Platelets | C3 | + | [50,51,54,55] |
| Thrombin | DAF | + (− complement activity) | [41] |
| MAC, C5a | Tissue Factor | + | [56,57] |
| Complement | Heparin | - | [58] |
| Study | Negative Prognostic Complement Factor Evaluated | Laboratory Investigation | Patients (n) | Type of Study [R: Retrospective; P: Prospective] | Stage (%) | Survival Outcome |
|---|---|---|---|---|---|---|
| L.T. Roumenina et al., Cancer Immunol. Res., 2019 [102] | C1q C3 C4 C4d | IHC | Cohort 1: 106 | R | I (40) II (6) III (41) IV (14) | C1q OP: PFS p = 0.008, OS p = 0.0016 Stage I–II: PFS p = 0.711, OS p = 0.256 Stage III–IV: PFS p = 0.00356, OS p = 0.00198 |
| C4c OP: PFS p = 0.0235, OS p = 0.0299 | ||||||
| C4d OP: PFS p = 0.013, OS p = 0.007 | ||||||
| C3 OP: PFS p = 0.0349, OS p = 0.07 | ||||||
| Cohort 2: 154 | R | I (40) II (5) III (54) IV (2) | C1q: Stage I–II: PFS p = 0.527, Stage III–IV: PFS p = 0.0109 | |||
| Cohort 3: 43 | R | IV (100) | C1q: PFS p = 0.00276, OS p = 0.00126 C4d: PFS p = 0.0176 | |||
| M.V. Daugan et al. Cancer Immunol. Res., 2021 [103] | C1s C4d deposits Plasma C4d | IHC IHC Plasma | Cohort 1: 82 | R | I (40) II (6) III (39) IV (15) | C4d deposits OP: PFS p = 0.00176 |
| Cohort 2: 26 | P | / | Plasma C4d OP: PFS p = 0.09 | |||
| Cohort 3:92 (longer FU) | P | I (54) II (8) III (17) IV (18) | Plasma C4d OP: PFS p = 0.0125 | |||
| Wei Xi et al. Scientific Reports, 2016 [59] | C5a | IHC | 272 | R | I (62) II (8) III (24) IV (7) | OP: OS p = 0.011, DFS p = 0.079 Stage I–II: OS p = 0.845 Stage III–IV: OS p < 0.001 |
| C. Yang et al. IJBM, 2023 [105] | C5a | IHC | 231 | IV | OS p = 0.0199, PFS p = 0.0138 | |
| Dong et al., BMC 2021 [106] | C3 C3AR1 | Transcriptomics analysis | 532 (TCGA-KIRC dataset) | R | C3 OP: OS p = 0.0004, RFS p = 0.007 C3AR1 OP: OS p = 0.204, RFS p = 0.323 | |
| Daugan et al. Cancer Immunol. Res., 2021 [107] | mb-FH int-FH | IHC | Cohort 1: 133 | R | I (0) II (24) III (64) IV (9) | OP mb-FH: DFS p = 0.14 OP int-FH: DFS p = 0.004 |
| Cohort 2:91 | R | I (5) II (22) III (48) IV (8) | OP mb-FH: PFS p = 0.226, OS p = 0.627 OP int-FH: PFS p = 0.0274, OS p = 0.0727 | |||
| Netti et al. Aging (Albany NY). 2020 [108] | PTX3 | Plasma | Cohort 1: 168 | R | pT1 (62) pT2 (14) pT3 (21) pT4 (3) pN+ (20) cM+ (18) | 10-yr OS rate: 73.7% ↓PTX3 vs. 48.4% ↑PTX3, p = 0.002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panebianco, M.; Ciccarese, C.; Strusi, A.; Beccia, V.; Carbone, C.; Agostini, A.; Piro, G.; Tortora, G.; Iacovelli, R. The Role of the Complement in Clear Cell Renal Carcinoma (ccRCC)—What Future Prospects Are There for Its Use in Clinical Practice? Cancers 2024, 16, 490. https://doi.org/10.3390/cancers16030490
Panebianco M, Ciccarese C, Strusi A, Beccia V, Carbone C, Agostini A, Piro G, Tortora G, Iacovelli R. The Role of the Complement in Clear Cell Renal Carcinoma (ccRCC)—What Future Prospects Are There for Its Use in Clinical Practice? Cancers. 2024; 16(3):490. https://doi.org/10.3390/cancers16030490
Chicago/Turabian StylePanebianco, Martina, Chiara Ciccarese, Alessandro Strusi, Viria Beccia, Carmine Carbone, Antonio Agostini, Geny Piro, Giampaolo Tortora, and Roberto Iacovelli. 2024. "The Role of the Complement in Clear Cell Renal Carcinoma (ccRCC)—What Future Prospects Are There for Its Use in Clinical Practice?" Cancers 16, no. 3: 490. https://doi.org/10.3390/cancers16030490
APA StylePanebianco, M., Ciccarese, C., Strusi, A., Beccia, V., Carbone, C., Agostini, A., Piro, G., Tortora, G., & Iacovelli, R. (2024). The Role of the Complement in Clear Cell Renal Carcinoma (ccRCC)—What Future Prospects Are There for Its Use in Clinical Practice? Cancers, 16(3), 490. https://doi.org/10.3390/cancers16030490