
Distinct Capabilities in NAD Metabolism Mediate Resistance to NAMPT Inhibition in Glioblastoma


Abstract
Simple Summary
Abstract
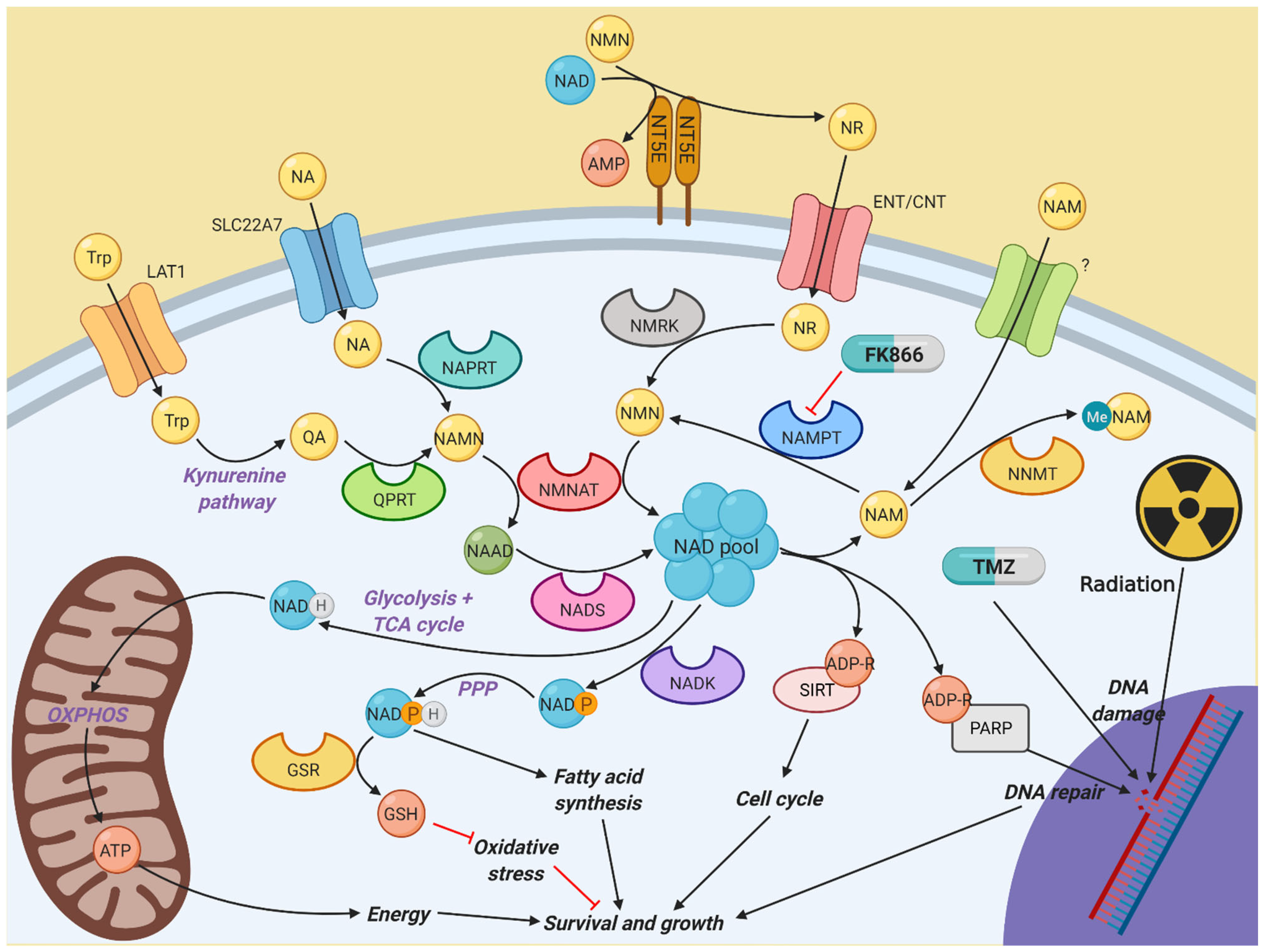
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Tissue, Cell Lines and Compounds
2.3. Reverse Transcription-Quantitative PCR (RT-qPCR)
2.4. Western Blot
2.5. Proliferation Assay
2.6. Cell Viability, Apoptosis and Cell Cycle Assays
2.7. NAD/NADH Assay
2.8. Real-Time Metabolism Assays
2.9. 3D Cell Culture Assays
2.10. In Vivo Work
2.11. Immunohistochemistry (IHC)
2.12. RNAseq
2.13. Data Analysis and Statistics
3. Results
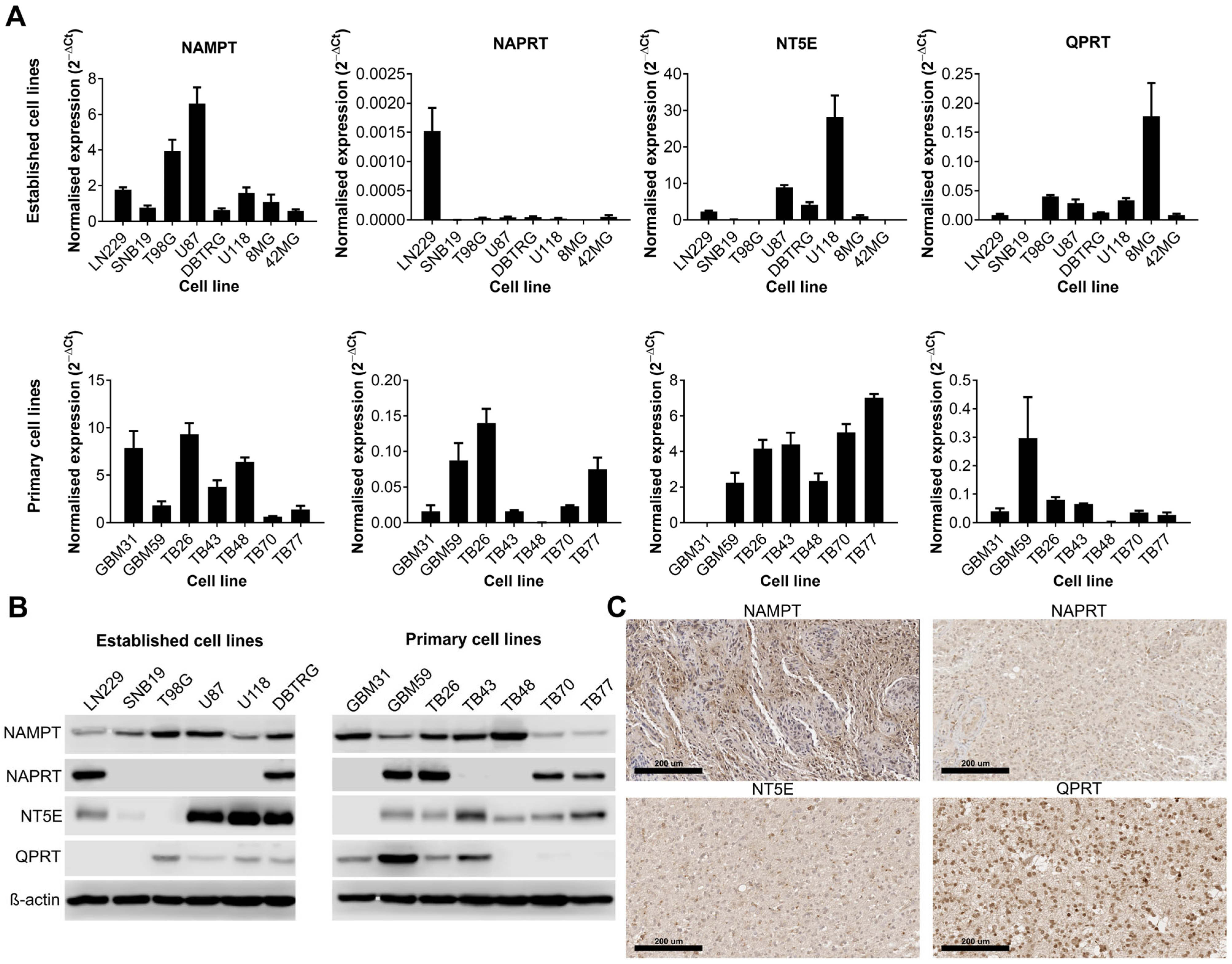
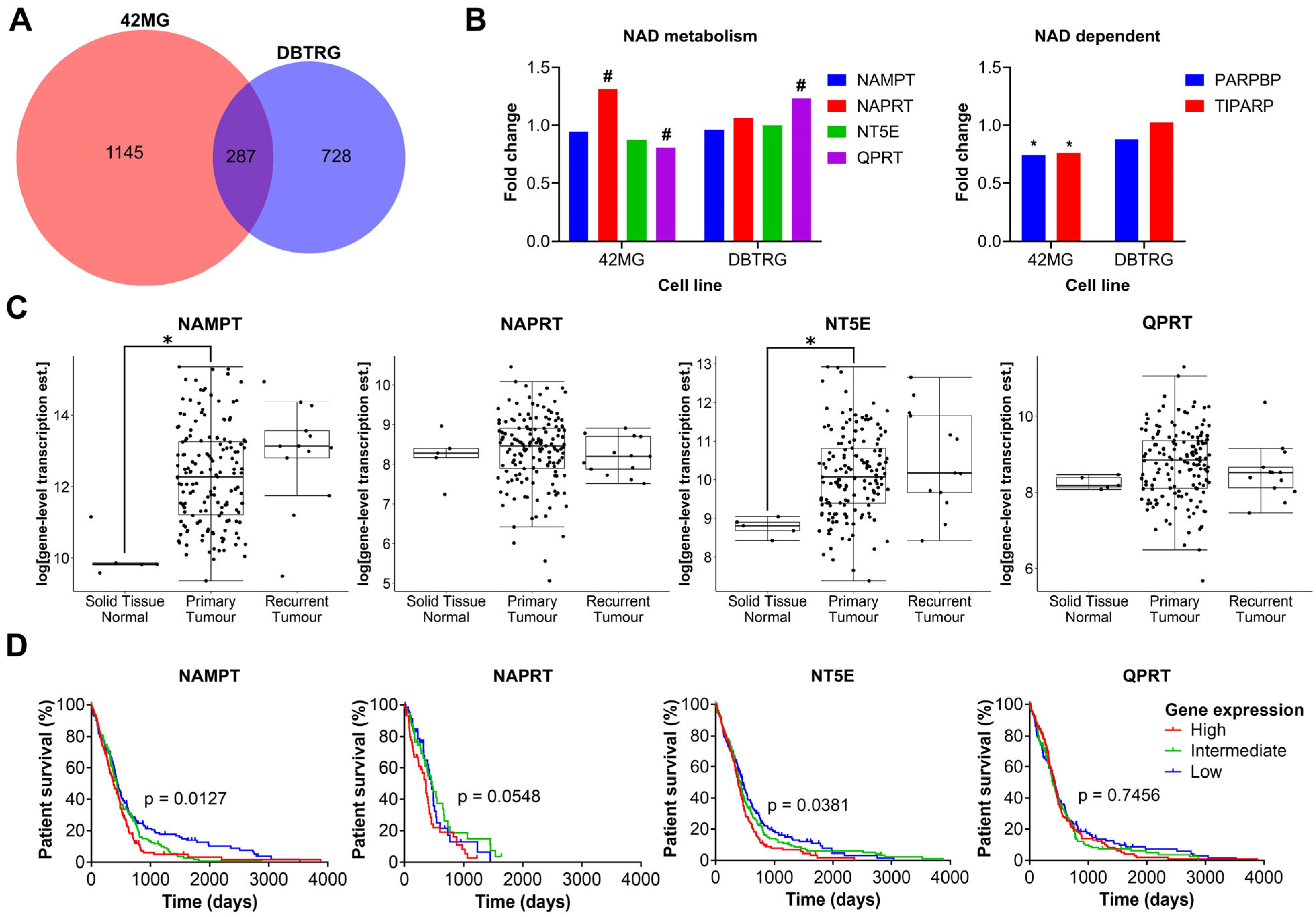
3.1. The Expression of Key Enzymes Involved in NAD Metabolism Is Highly Variable amongst GBM Cell Lines
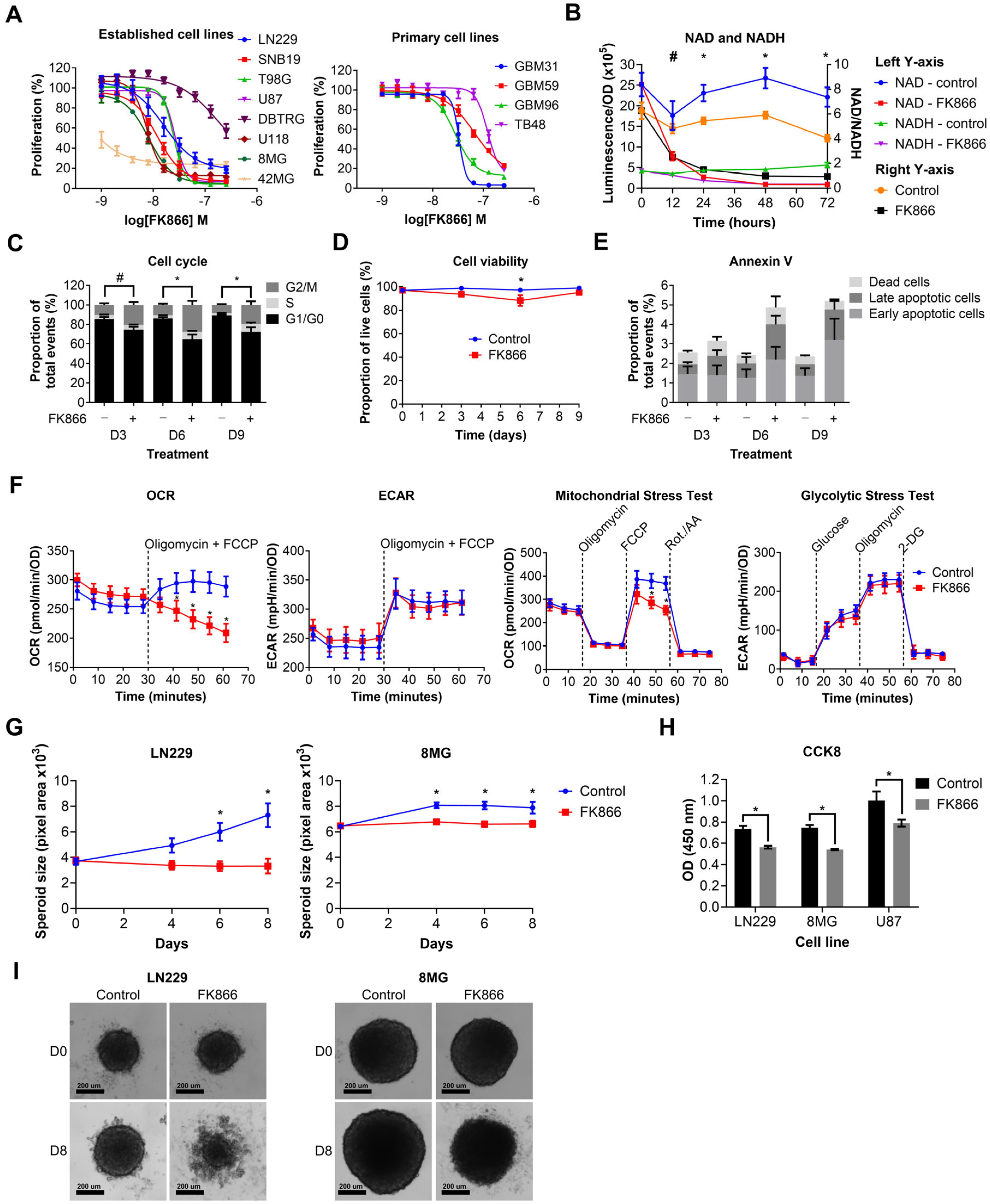
3.2. NAMPT Inhibition with FK866 Effectively Reduces NAD Levels and Inhibits Proliferation of GBM Cells by Initiating a G2/M Cell Cycle Block
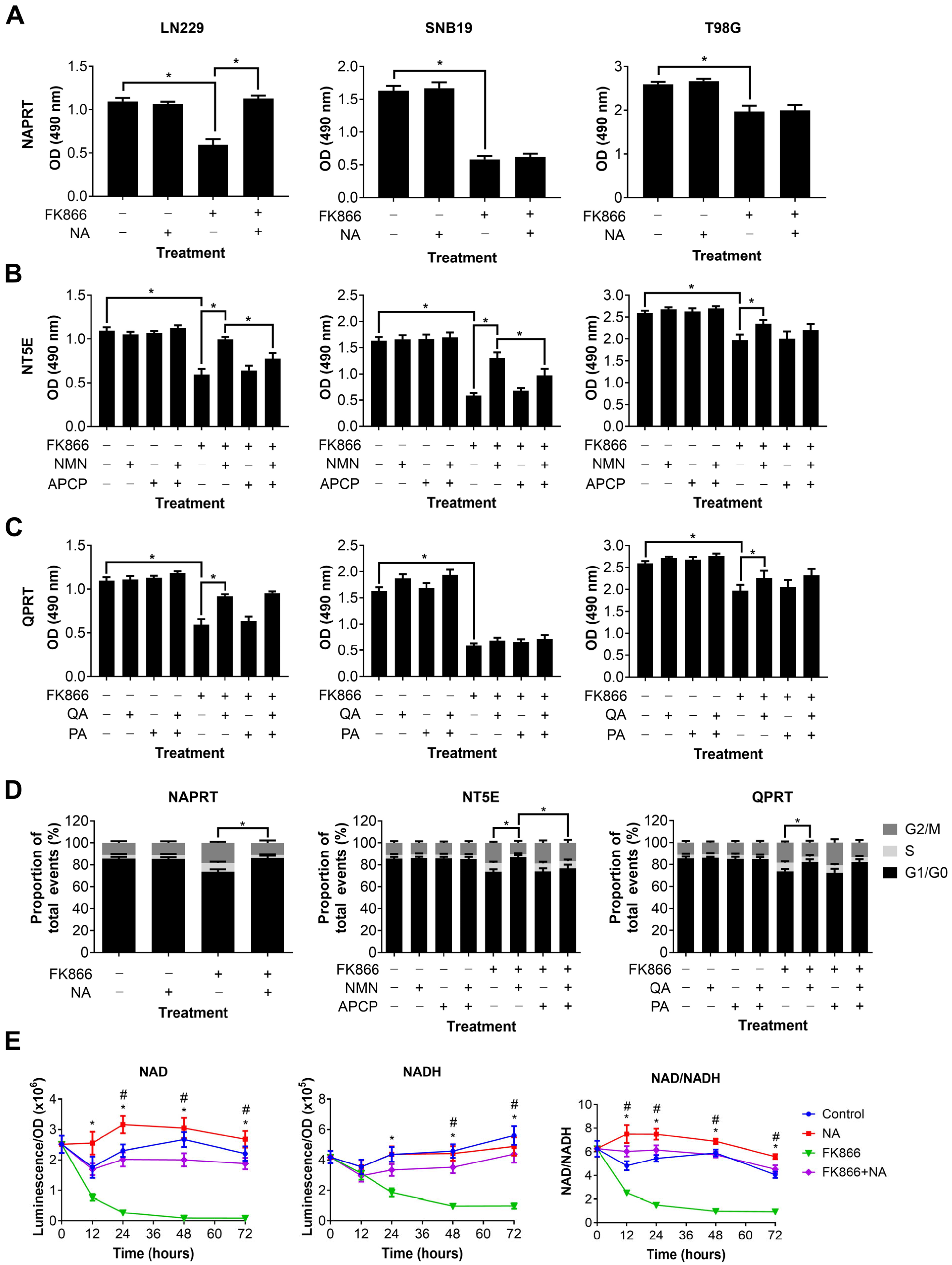
3.3. Specific NAD Precursors Can Restore Proliferation in GBM Cells Treated with FK866
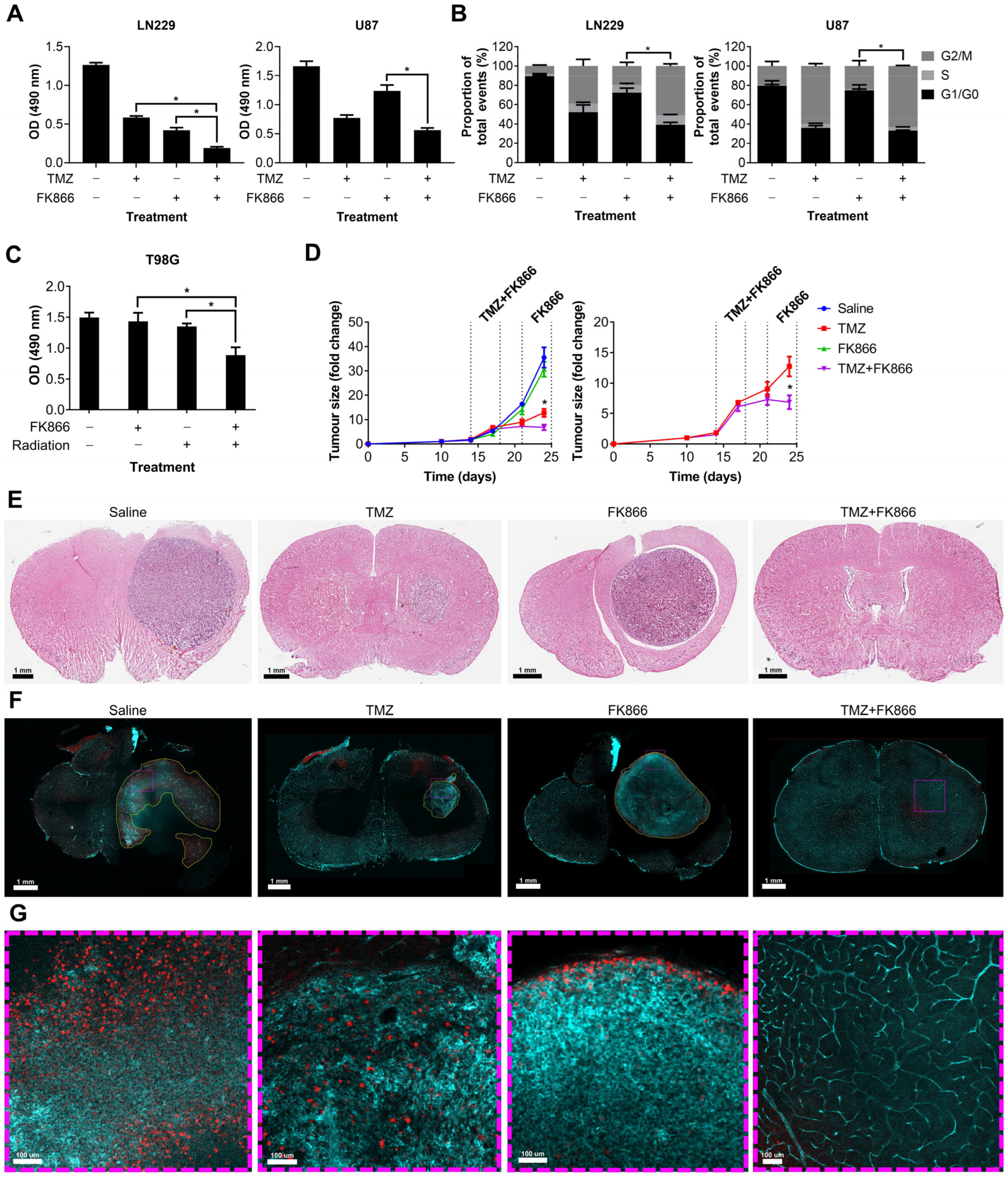
3.4. FK866 Potentiates the Effects of Radiation and TMZ In Vitro and In Vivo
3.5. GBM Cells Treated with FK866 Show Significant Dysregulation of the Cell Cycle, and Changes in the Expression of NAD Metabolism Genes Are Associated with Patient Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Weller, M.; Le Rhun, E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat. Rev. 2020, 87, 102029. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.C.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly (ADP-ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhang, Y.; Liu, Y.; Yamada, K.; Tso, J.L.; Menjivar, J.C.; Tian, J.Y.; Yong, W.H.; Schaue, D.; Mischel, P.S.; et al. Protective properties of radio-chemoresistant glioblastoma stem cell clones are associated with metabolic adaptation to reduced glucose dependence. PLoS ONE 2013, 8, e80397. [Google Scholar] [CrossRef]
- Gujar, A.D.; Le, S.; Mao, D.D.; Dadey, D.Y.A.; Turski, A.; Sasaki, Y.; Aum, D.; Luo, J.; Dahiya, S.; Yuan, L.; et al. An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc. Natl. Acad. Sci. USA 2016, 113, 8247–8256. [Google Scholar] [CrossRef]
- Garten, A.; Petzold, S.; Körner, A.; Imai, S.-i.; Kiess, W. Nampt: Linking NAD biology, metabolism and cancer. Trends Endocrinol. Metab. 2009, 20, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jiménez-García, M.P. NAMPT overexpression induces cancer stemness and defines a novel tumor signature for glioma prognosis. Oncotarget 2017, 8, 99514–99530. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Liu, L.Y.; Qie, L.L.; Ling, K.N.; Xu, L.H.; Wang, F.; Fang, S.-H.; Lu, Y.-B.; Hu, H.; Wei, E.-Q.; et al. Anti-proliferation effect of APO866 on C6 glioblastoma cells by inhibiting nicotinamide phosphoribosyltransferase. Eur. J. Pharmacol. 2012, 674, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Zhang, L.; Shi, Q.J.; Lu, Y.B.; Wu, M.; Wei, E.Q.; Zhang, W.-P. Nicotinamide phosphoribosyltransferase inhibitor APO866 induces C6 glioblastoma cell death via autophagy. Pharmazie 2015, 70, 650–655. [Google Scholar]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-Driven glycolysis is a therapeutic target in glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef]
- Goellner, E.M.; Grimme, B.; Brown, A.R.; Lin, Y.C.; Wang, X.H.; Sugrue, K.F.; Mitchell, L.; Trivedi, R.N.; Tang, J.-B.; Sobol, R.W. Overcoming temozolomide resistance in glioblastoma via dual inhibition of NAD+ biosynthesis and base excision repair. Cancer Res. 2011, 71, 2308–2317. [Google Scholar] [CrossRef] [PubMed]
- Holen, K.; Saltz, L.B.; Hollywood, E.; Burk, K.; Hanauske, A.R. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Investig. New Drugs 2008, 26, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Von Heideman, A.; Berglund, Å.; Larsson, R.; Nygren, P.; Larsson, R. Safety and efficacy of NAD depleting cancer drugs: Results of a phase i clinical trial of CHS 828 and overview of published data. Cancer Chemother. Pharmacol. 2010, 65, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Hashimoto, T.; Tsuchiya, M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J. Biol. Chem. 2007, 282, 24574–24582. [Google Scholar] [CrossRef] [PubMed]
- Heyes, M.P.; Chen, C.Y.; Major, E.O.; Saito, K. Different kynurenine pathway enzymes limit quinolinic acid formation by various human cell types. Biochem. J. 1997, 326, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Beavis, P.A.; Darcy, P.K.; Stagg, J. Immunosuppressive activities of adenosine in cancer. Curr. Opin. Pharmacol. 2016, 29, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Grozio, A.; Sociali, G.; Sturla, L.; Caffa, I.; Soncini, D.; Salis, A.; Raffaelli, N.; De Flora, A.; Nencioni, A.; Bruzzone, S. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J. Biol. Chem. 2013, 288, 25938–25949. [Google Scholar] [CrossRef] [PubMed]
- Perbellini, F.; Liu, A.K.L.; Watson, S.A.; Bardi, I.; Rothery, S.M.; Terracciano, C.M. Free-of-Acrylamide SDS-based Tissue Clearing (FASTClear) for three dimensional visualization of myocardial tissue. Sci. Rep. 2017, 7, 5188. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Pan, J.H.; Zhou, H.; Zhu SBin Huang, J.L.; Zhao, X.X.; Ding, H.; Qin, L.; Pan, Y.-L. Nicotinamide phosphoribosyl transferase regulates cell growth via the Sirt1/P53 signaling pathway and is a prognosis marker in colorectal cancer. J. Cell Physiol. 2018, 234, 4385–4395. [Google Scholar] [CrossRef]
- Paz, M.F.; Yaya-tur, R.; Rojas-marcos, I.; Reynes, G.; Pollan, M.; Aguirrecruz, L.; García-Lopez, J.L.; Piquer, J.; Safont, M.-J.; Balaña, C.; et al. CpG Island Hypermethylation of the DNA Repair Enzyme Methyltransferase Predicts Response to Temozolomide in Primary Gliomas. Clin. Cancer Res. 2004, 10, 4933–4938. [Google Scholar] [CrossRef] [PubMed]
- Nahimana, A.; Attinger, A.; Aubry, D.; Greaney, P.; Ireson, C.; Thougaard, A.V.; Tjørnelund, J.; Dawson, K.M.; Dupuis, M.; Duchosal, M.A. The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic malignancies. Blood 2009, 113, 3276–3286. [Google Scholar] [CrossRef] [PubMed]
- Hjarnaa, P.; Julia, V.; Jonsson, E.; Latini, S.; Dhar, S.; Larsson, R.; Bramm, E.; Binderup, L.; Madsen, M.W. CHS 828, a Novel Pyridyl Cyanoguanidine with Potent Antitumor Activity in Vitro and in Vivo CHS 828, a Novel Pyridyl Cyanoguanidine with Potent Antitumor Activity in Vitro and in Vivo. Cancer Res. 1999, 59, 5751–5757. [Google Scholar] [PubMed]
- Olesen, U.H.; Christensen, M.K.; Björkling, F.; Jäättelä, M.; Jensen, P.B.; Sehested, M.; Nielsen, S.J. Anticancer agent CHS-828 inhibits cellular synthesis of NAD. Biochem. Biophys. Res. Commun. 2008, 367, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Hasmann, M.; Schemainda, I. FK866, a Highly Specific Noncompetitive Inhibitor of Nicotinamide Phosphoribosyltransferase, Represents a Novel Mechanism for Induction of Tumor Cell Apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar] [PubMed]
- Cerna, D.; Li, H.; Flaherty, S.; Takebe, N.; Coleman, C.N.; Yoo, S.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates Reactive Oxygen Species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribosyltransferase1 (NAPRT1)-dependent manner. J. Biol. Chem. 2012, 287, 22408–22417. [Google Scholar] [CrossRef]
- Shames, D.S.; Elkins, K.; Walter, K.; Holcomb, T.; Du, P.; Mohl, D.; Xiao, Y.; Pham, T.; Haverty, P.M.; Liederer, B.; et al. Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin. Cancer Res. 2013, 19, 6912–6923. [Google Scholar] [CrossRef]
- Jin, D.; Fan, J.; Wang, L.; Thompson, L.F.; Liu, A.; Daniel, B.J.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 on tumor cells impairs antitumor T-cell responses: A novel mechanism of tumor-induced immune suppression. Cancer Res. 2010, 70, 2245–2255. [Google Scholar] [CrossRef]
- Bavaresco, L.; Bernardi, A.; Braganhol, E.; Cappellari, A.R.; Rockenbach, L.; Farias, P.F.; Wink, M.R.; Delgado-Cañedo, A.; Battastini, A.M.O. The role of ecto-5′-nucleotidase/CD73 in glioma cell line proliferation. Mol. Cell Biochem. 2008, 319, 61–68. [Google Scholar] [CrossRef]
- Krüger, K.H.; Thompson, L.F.; Kaufmann, M.; Möller, P. Expression of ecto-5′-nucleotidase (CD73) in normal mammary gland and in breast carcinoma. Br. J. Cancer 1991, 63, 114–118. [Google Scholar] [CrossRef]
- Sociali, G.; Raffaghello, L.; Magnone, M.; Zamporlini, F.; Emionite, L.; Sturla, L.; Bianchi, G.; Vigliarolo, T.; Nahimana, A.; Nencioni, A.; et al. Antitumor effect of combined NAMPT and CD73 inhibition in an ovarian cancer model. Oncotarget 2015, 7, 2968–2984. [Google Scholar] [CrossRef] [PubMed]
- Nevedomskaya, E.; Perryman, R.; Solanki, S.; Syed, N.; Mayboroda, O.A.; Keun, H.C. A Systems Oncology Approach Identifies NT5E as a Key Metabolic Regulator in Tumor Cells and Modulator of Platinum Sensitivity. J. Proteome Res. 2016, 15, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Cheng, S.P.; Chen, M.J.; Lin, C.H.; Chen, S.N.; Kuo, Y.H.; Chang, Y.-C. Quinolinate Phosphoribosyltransferase Promotes Invasiveness of Breast Cancer Through Myosin Light Chain Phosphorylation. Front. Endocrinol. 2021, 11, 621944. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Guillemin, G.J.; Grant, R. Effects of kynurenine pathway inhibition on NAD+ metabolism and cell viability in human primary astrocytes and neurons. Int. J. Tryptophan Res. 2011, 4, 29–37. [Google Scholar] [CrossRef]
- Xiao, Y.; Elkins, K.; Durieux, J.K.; Lee, L.; Oeh, J.; Yang, L.X.; Liang, X.; DelNagro, C.; Tremayne, J.; Kwong, M.; et al. Dependence of tumor cell lines and patient-derived tumors on the NAD salvage pathway renders them sensitive to NAMPT inhibition with GNE-618. Neoplasia 2013, 15, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Oezen, I.; Opitz, C.A.; Radlwimmer, B.; Von Deimling, A.; Ahrendt, T.; Adams, S.; Bode, H.B.; Guillemin, G.J.; Wick, W.; et al. The endogenous tryptophan metabolite and NAD+ precursor quinolinic acid confers resistance of gliomas to oxidative stress. Cancer Res. 2013, 73, 3225–3234. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Nakagawa, H.; Ueda, K.; Chung, S.; Kashiwaya, K.; Eguchi, H.; Ohigashi, H.; Ishikawa, O.; Daigo, Y.; Matsuda, K.; et al. C12orf48, termed PARP-1 binding protein, enhances poly(ADP-ribose) polymerase-1 (PARP-1) activity and protects pancreatic cancer cells from DNA damage. Genes Chromosomes Cancer 2011, 50, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Manetsch, P.; Böhi, F.; Nowak, K.; Pedrioli, D.M.L.; Hottiger, M.O. PARP7-mediated ADP-ribosylation of FRA1 promotes cancer cell growth by repressing IRF1-and IRF3-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2023, 120, 47120. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Yan, P.F.; Zhao, H.Y.; Zhang, F.C.; Zhao, W.H.; Feng, M. Inhibitor of Nicotinamide Phosphoribosyltransferase Sensitizes Glioblastoma Cells to Temozolomide via Activating ROS/JNK Signaling Pathway. BioMed Res. Int. 2016, 2016, 1450843. [Google Scholar] [CrossRef]
- Pogrebniak, A.; Schemainda, I.; Azzam, K.; Pelka Fleischer, R.; Nüssler, V.; Hasmann, M. Chemopotentiating effects of a novel NAD biosynthesis inhibitor, FK866, in combination with antineoplastic agents. Eur. J. Med. Res. 2006, 11, 313–321. [Google Scholar]
- Wang, K.; Ye, K.; Zhang, X.; Wang, T.; Qi, Z.; Wang, Y.; Jiang, S.; Zhang, K. Dual Nicotinamide Phosphoribosyltransferase (NAMPT) and Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors for the Treatment of Drug-Resistant Nonsmall-Cell Lung Cancer. J. Med. Chem. 2023, 66, 1027–1047. [Google Scholar] [CrossRef]
- Abu Aboud, O.; Chen, C.H.; Senapedis, W.; Baloglu, E.; Argueta, C.; Weiss, R.H. Dual and Specific Inhibition of NAMPT and PAK4 By KPT-9274 Decreases Kidney Cancer Growth. Mol. Cancer Ther. 2016, 15, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dong, G.; Wu, Y.; Zhang, W.; Miao, C.; Sheng, C. Dual NAMPT/HDAC Inhibitors as a New Strategy for Multitargeting Antitumor Drug Discovery. ACS Med. Chem. Lett. 2018, 9, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.A.; Noronha, K.J.; Wang, Y.; Friedman, A.P.; Paradkar, S.; Suh, H.W.; Sundaram, R.K.; Brenner, C.; Saltzman, W.M.; Bindra, R.S. Exploiting Metabolic Defects in Glioma with Nanoparticle Encapsulated NAMPT Inhibitors. Mol. Cancer Ther. 2024. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kirtane, A.R.; Kiyokawa, J.; Nagashima, H.; Lopes, A.; Tirmizi, Z.A.; Lee, C.K.; Traverso, G.; Cahill, D.P.; Wakimoto, H. Local targeting of NAD+ salvage pathway alters the immune tumor microenvironment and enhances checkpoint immunotherapy in glioblastoma. Cancer Res. 2020, 80, 5024–5034. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, Z.; Wang, Y.; Gao, J.; Meng, Y.; Wang, S.; Zhao, X.; Tang, C.; Yang, W.; Li, Y.; et al. The regulatory relationship between NAMPT and PD-L1 in cancer and identification of a dual-targeting inhibitor. EMBO Mol. Med. 2024, 16, 885–903. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Jill, P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Set | Pathway | Normalized Enrichment Score | FDR | Size | Leading Edge Number |
|---|---|---|---|---|---|
| R-HSA-69278 | Cell Cycle, Mitotic | −2.00 | 0 | 455 | 177 |
| R-HSA-2467813 | Separation of Sister Chromatids | −1.91 | 1.03 × 10−4 | 174 | 73 |
| R-HSA-68886 | M Phase | −1.91 | 1.15 × 10−4 | 320 | 123 |
| R-HSA-141424 | Amplification of signal from the kinetochores | −1.92 | 1.29 × 10−4 | 92 | 44 |
| R-HSA-141444 | Amplification of signal from unattached kinetochores via a MAD2 inhibitory signal | −1.92 | 1.29 × 10−4 | 92 | 44 |
| R-HSA-68882 | Mitotic Anaphase | −1.92 | 1.72 × 10−4 | 184 | 76 |
| R-HSA-69618 | Mitotic Spindle Checkpoint | −1.88 | 1.88 × 10−4 | 108 | 53 |
| R-HSA-2555396 | Mitotic Metaphase and Anaphase | −1.92 | 2.07 × 10−4 | 185 | 77 |
| R-HSA-69620 | Cell Cycle Checkpoints | −1.93 | 2.58 × 10−4 | 252 | 105 |
| R-HSA-2500257 | Resolution of Sister Chromatid Cohesion | −1.95 | 3.44 × 10−4 | 113 | 59 |
| Gene Set | Pathway | Normalized Enrichment Score | FDR | Size | Leading Edge Number |
|---|---|---|---|---|---|
| R-HSA-72163 | mRNA Splicing—Major Pathway | −1.92 | 0 | 174 | 56 |
| R-HSA-390466 | Chaperonin-mediated protein folding | −1.92 | 0 | 67 | 28 |
| R-HSA-162909 | Host Interactions of HIV factors | −1.93 | 0 | 113 | 52 |
| R-HSA-72172 | mRNA Splicing | −1.93 | 0 | 182 | 57 |
| R-HSA-168255 | Influenza Life Cycle | −1.93 | 0 | 135 | 71 |
| R-HSA-69275 | G2/M Transition | −1.93 | 0 | 176 | 62 |
| R-HSA-391251 | Protein folding | −1.93 | 0 | 72 | 29 |
| R-HSA-2500257 | Resolution of Sister Chromatid Cohesion | −1.94 | 0 | 112 | 62 |
| R-HSA-168254 | Influenza Infection | −1.95 | 0 | 145 | 69 |
| R-HSA-162906 | HIV Infection | −1.95 | 0 | 201 | 83 |
| ENSEMBL ID | Gene | Gene Name | Fold Change | Adjusted p Value | Poorest Survival Group |
|---|---|---|---|---|---|
| ENSG00000044574 | HSPA5 | Heat shock protein family A (Hsp70) member 5 | −1.297 | 1.94 × 10−6 | High (p = 0.008) |
| ENSG00000123485 | HJURP | Holliday junction recognition protein | −1.235 | 0.001 | None |
| ENSG00000166598 | HSP90B1 | Heat shock protein 90 beta family member 1 | −1.249 | 0.001 | High (p = 0.02) |
| ENSG00000168010 | ATG16L2 | Autophagy-related 16 like 2 | 1.346 | 0.004 | None |
| ENSG00000163659 | TIPARP | TCDD-inducible poly(ADP-ribose) polymerase | −1.311 | 0.004 | None |
| ENSG00000128965 | CHAC1 | ChaC glutathione specific gamma-glutamylcyclotransferase 1 | 1.478 | 0.006 | None |
| ENSG00000145050 | MANF | Mesencephalic astrocyte-derived neurotrophic factor | −1.234 | 0.006 | None |
| ENSG00000178913 | TAF7 | TATA-box-binding-protein-associated factor 7 | −1.239 | 0.006 | None |
| ENSG00000100321 | SYNGR1 | Synaptogyrin 1 | 1.237 | 0.007 | None |
| ENSG00000136541 | ERMN | Ermin | −1.375 | 0.011 | None |
| ENSEMBL ID | Gene | Gene Name | Fold Change | Adjusted p Value | Poorest Survival Group |
|---|---|---|---|---|---|
| ENSG00000162496 | DHRS3 | Dehydrogenase/reductase 3 | 2.005 | 4.32 × 10−14 | None |
| ENSG00000107731 | UNC5B | Unc-5 netrin receptor B | 1.378 | 2.29 × 10−9 | None |
| ENSG00000159399 | HK2 | Hexokinase 2 | 1.392 | 2.43 × 10−8 | None |
| ENSG00000104081 | BMF | Bcl2 modifying factor | 1.361 | 8.08 × 10−8 | None |
| ENSG00000175727 | MLXIP | MLX interacting protein | 1.263 | 1.42 × 10−7 | None |
| ENSG00000095752 | IL11 | Interleukin 11 | −1.514 | 1.69 × 10−6 | None |
| ENSG00000138316 | ADAMTS14 | ADAM metallopeptidase with thrombospondin type 1 motif 14 | 1.665 | 1.78 × 10−6 | High (p = 0.031) |
| ENSG00000179981 | TSHZ1 | Teashirt zinc finger homeobox 1 | 1.274 | 1.78 × 10−6 | None |
| ENSG00000120093 | HOXB3 | Homeobox B3 | 1.332 | 1.67 × 10−5 | None |
| ENSG00000112715 | VEGFA | Vascular endothelial growth factor A | 1.215 | 2.00 × 10−5 | High (p = 0.00027) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perryman, R.; Chau, T.W.; De-Felice, J.; O’Neill, K.; Syed, N. Distinct Capabilities in NAD Metabolism Mediate Resistance to NAMPT Inhibition in Glioblastoma. Cancers 2024, 16, 2054. https://doi.org/10.3390/cancers16112054
Perryman R, Chau TW, De-Felice J, O’Neill K, Syed N. Distinct Capabilities in NAD Metabolism Mediate Resistance to NAMPT Inhibition in Glioblastoma. Cancers. 2024; 16(11):2054. https://doi.org/10.3390/cancers16112054
Chicago/Turabian StylePerryman, Richard, Tsz Wing Chau, John De-Felice, Kevin O’Neill, and Nelofer Syed. 2024. "Distinct Capabilities in NAD Metabolism Mediate Resistance to NAMPT Inhibition in Glioblastoma" Cancers 16, no. 11: 2054. https://doi.org/10.3390/cancers16112054
APA StylePerryman, R., Chau, T. W., De-Felice, J., O’Neill, K., & Syed, N. (2024). Distinct Capabilities in NAD Metabolism Mediate Resistance to NAMPT Inhibition in Glioblastoma. Cancers, 16(11), 2054. https://doi.org/10.3390/cancers16112054