
Current and Future Trends of Colorectal Cancer Treatment: Exploring Advances in Immunotherapy

 ,
, 
 , , and
, , and 
Abstract
Simple Summary
Abstract
1. Introduction
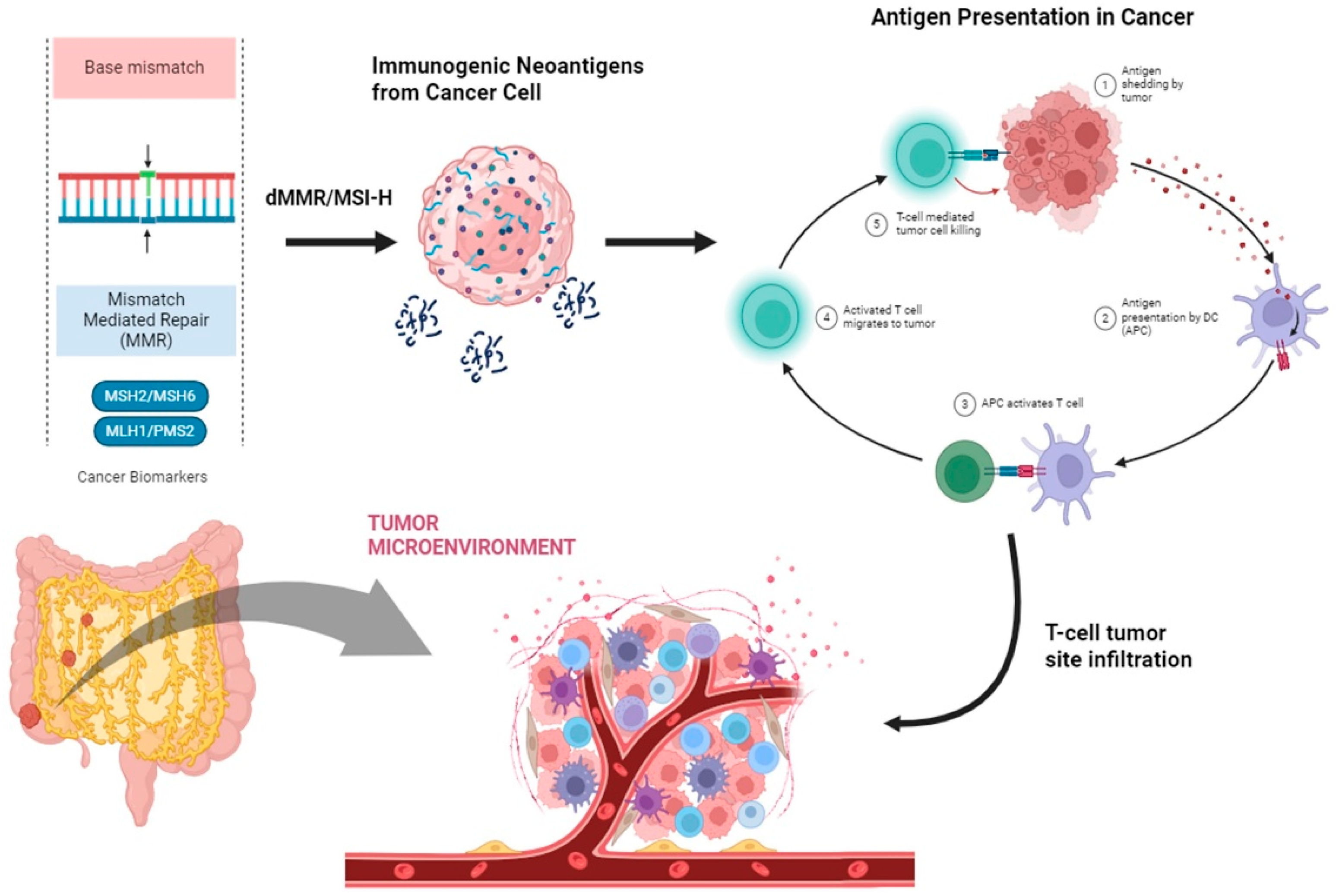
2. Genetic Determinants of Immune Response
2.1. Mismatch Repair Complex and Microsatellite Status in CRC
2.2. BRAFV600E
3. Immune Response in CRC
4. Immune Evasion of CRC
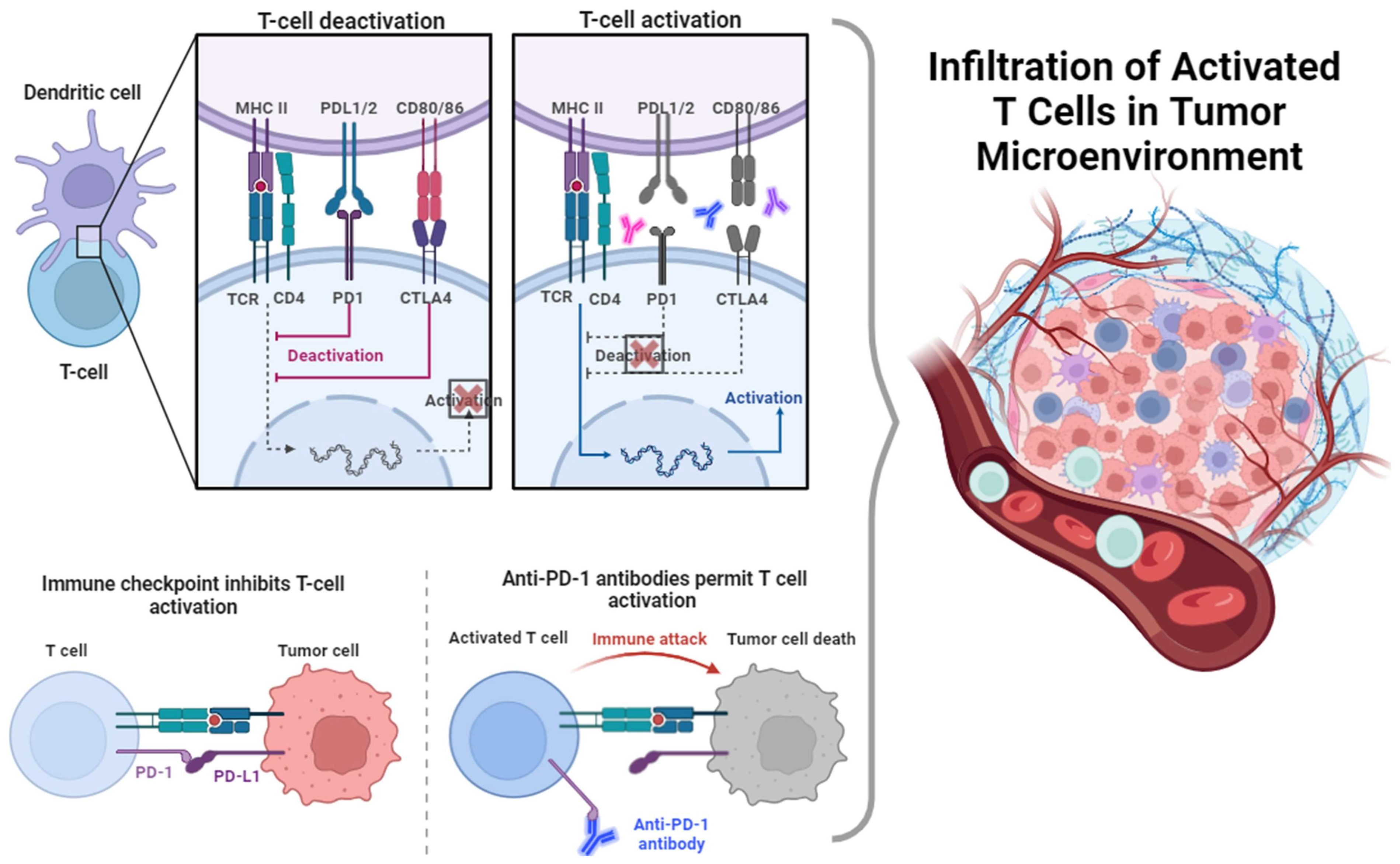
5. Immune Checkpoints
5.1. Programmed Death Ligand 1 (PD L1) and Programmed Death 1 (PD-1)
5.2. Cytotoxic T-Lymphocyte Antigen-4 (CTLA-4)
6. Immune Checkpoint Inhibitors (ICIs)
6.1. Anti-PD1/PDL-1 Abs
6.2. Anti-CTLA-4 Abs
7. Adoptive Cell Therapy (ACT)
7.1. Chimeric Antigen Receptor (CAR) T-Cell Therapy
7.2. Tumor-Infiltrating Lymphocyte (TIL) Therapy
8. Anti-Tumor Vaccine Therapy
9. Emerging Biomarkers of Clinical Response
10. Dynamics of Telomere Length
11. Telomere Length and CRC
12. Telomerase Activity and CRC
13. Therapeutic Implications of Telomerase Activity Inhibition
14. Potential Interplay between Telomere Length and Immune Therapies
15. Conclusions
- Pearls:
- (1)
- In cases of early-stage CRC, the dMMR/MSI-H profile is more common (10–18%) than that of mCRC (about 3–5%).
- (2)
- Activation and inhibitory signals of T cells are mediated through CTLA-4 [229].
- (3)
- A suppression of immune responses is exhibited by the interplay of PD-1 and PD-L1/PD-L2, inducing a minimized function of T effector cells over immune responses.
- (4)
- PD-L1 is highly expressed in inflamed cells and PD-L2 is expressed only in antigen-presenting cells [230].
Author Contributions
Funding
Conflicts of Interest
References
- Hedrick, T.L. Colorectal Cancer. Surg. Oncol. Clin. N. Am. 2022, 31, xv–xvi. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef] [PubMed]
- Li, L.B.; Wang, L.Y.; Chen, D.M.; Liu, Y.X.; Zhang, Y.H.; Song, W.X.; Shen, X.B.; Fang, S.Q.; Ma, Z.Y. A systematic analysis of the global and regional burden of colon and rectum cancer and the difference between early- and late-onset CRC from 1990 to 2019. Front. Oncol. 2023, 13, 1102673. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Azad, N.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Garrido-Laguna, I.; et al. Rectal Cancer, Version 2.2022. JNCCN J. Natl. Compr. Cancer Netw. 2022, 20, 1139–1167. [Google Scholar] [CrossRef]
- Nikolouzakis, T.K.; Stivaktakis, P.D.; Apalaki, P.; Kalliantasi, K.; Sapsakos, T.M.; Spandidos, D.A.; Tsatsakis, A.; Souglakos, J.; Tsiaoussis, J. Effect of systemic treatment on the micronuclei frequency in the peripheral blood of patients with metastatic colorectal cancer. Oncol. Lett. 2019, 17, 2703–2712. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Borelli, B.; Antoniotti, C.; Carullo, M.; Germani, M.M.; Conca, V.; Masi, G. Immune-Checkpoint Inhibitors (ICIs) in Metastatic Colorectal Cancer (mCRC) Patients beyond Microsatellite Instability. Cancers 2022, 14, 4974. [Google Scholar] [CrossRef] [PubMed]
- Benatti, P.; Gafà, R.; Barana, D.; Marino, M.; Scarselli, A.; Pedroni, M.; Maestri, I.; Guerzoni, L.; Roncucci, L.; Menigatti, M.; et al. Microsatellite instability and colorectal cancer prognosis. Clin. Cancer Res. 2005, 11, 8332–8340. [Google Scholar] [CrossRef] [PubMed]
- André, T.; De Gramont, A.; Vernerey, D.; Chibaudel, B.; Bonnetain, F.; Tijeras-Raballand, A.; Scriva, A.; Hickish, T.; Tabernero, J.; Van Laethem, J.L.; et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in stage II to III colon cancer: Updated 10-year survival and outcomes according to BRAF mutation and mismatch repair status of the MOSAIC study. J. Clin. Oncol. 2015, 33, 4176–4187. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Lenz, H.J.; Van Cutsem, E.; Limon, M.L.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; García-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.B.; et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J. Clin. Oncol. 2022, 40, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Gaissmaier, L.; Elshiaty, M.; Christopoulos, P. Breaking Bottlenecks for the TCR Therapy of Cancer. Cells 2020, 9, 2095. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef]
- Yi, X.; Hu, W. Advances in adoptive cellular therapy for colorectal cancer: A narrative review. Ann. Transl. Med. 2022, 10, 1404. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular Basis of Colorectal Cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Nikolouzakis, T.K.; Vassilopoulou, L.; Fragkiadaki, P.; Sapsakos, T.M.; Papadakis, G.Z.; Spandidos, D.A.; Tsatsakis, A.M.; Tsiaoussis, J. Improving diagnosis, prognosis and prediction by using biomarkers in CRC patients (Review). Oncol. Rep. 2018, 39, 2455–2472. [Google Scholar] [CrossRef] [PubMed]
- Piñol-Felis, C.; Fernández-Marcelo, T.; Viñas-Salas, J.; Valls-Bautista, C. Telomeres and telomerase in the clinical management of colorectal cancer. Clin. Transl. Oncol. 2017, 19, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Marcelo, T.; Sánchez-Pernaute, A.; Pascua, I.; De Juan, C.; Head, J.; Torres-García, A.J.; Iniesta, P.I. Clinical Relevance of Telomere Status and Telomerase Activity in Colorectal Cancer. PLoS ONE 2016, 11, e0149626. [Google Scholar] [CrossRef] [PubMed]
- Kroupa, M.; Kubecek, O.; Tomasova, K.; Hanak, P.; Krupova, M.; Cervena, K.; Siskova, A.; Rosendorf, J.; Hosek, P.; Vodickova, L.; et al. The dynamics of telomere length in primary and metastatic colorectal cancer lesions. Sci. Rep. 2023, 13, 9097. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Li, J.; Song, C.; Chen, W. Telomere in colorectal cancer associated with distant metastases and predicted a poor prognosis. Transl. Cancer Res. 2021, 10, 2906–2917. [Google Scholar] [CrossRef] [PubMed]
- Tsatsakis, A.; Oikonomopoulou, T.; Nikolouzakis, T.K.; Vakonaki, E.; Tzatzarakis, M.; Flamourakis, M.; Renieri, E.; Fragkiadaki, P.; Iliaki, E.; Bachlitzanaki, M.; et al. Role of telomere length in human carcinogenesis (Review). Int. J. Oncol. 2023, 63, 78. [Google Scholar] [CrossRef] [PubMed]
- Nikolouzakis, T.K.; Vakonaki, E.; Stivaktakis, P.D.; Alegakis, A.; Berdiaki, A.; Razos, N.; Souglakos, J.; Tsatsakis, A.; Tsiaoussis, J. Novel Prognostic Biomarkers in Metastatic and Locally Advanced Colorectal Cancer: Micronuclei Frequency and Telomerase Activity in Peripheral Blood Lymphocytes. Front. Oncol. 2021, 11, 683605. [Google Scholar] [CrossRef] [PubMed]
- Weng, N.P. Telomeres and immune competency. Curr. Opin. Immunol. 2012, 24, 470–475. [Google Scholar] [CrossRef]
- Nikolouzakis, T.K.; Vakonaki, E.; Alegakis, A.; Rakitskii, V.; Svistunov, A.; Chrysos, E.; Souglakos, J.; Tsatsakis, A.M.; Tsiaoussis, J. P02-06: Effect of systemic treatment on telomere length of patients with metastatic colorectal cancer. Toxicol. Lett. 2023, 384, S83. [Google Scholar] [CrossRef]
- Rolles, B.; Gorgulho, J.; Tometten, M.; Roderburg, C.; Vieri, M.; Abels, A.; Vucur, M.; Heymann, F.; Tacke, F.; Brümmendorf, T.H.; et al. Telomere Shortening in Peripheral Leukocytes Is Associated with Poor Survival in Cancer Patients Treated with Immune Checkpoint Inhibitor Therapy. Front. Oncol. 2021, 11, 729207. [Google Scholar] [CrossRef]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar]
- Losso, G.M.; Moraes, R.d.S.; Gentili, A.C.; Messias-Reason, I.T. Microsatellite instability—MSI markers (BAT26, BAT25, D2S123, D5S346, D17S250) in rectal cancer. Arq. Bras. Cir. Dig. 2012, 25, 240–244. [Google Scholar] [CrossRef]
- De La Chapelle, A.; Hampel, H. Clinical relevance of microsatellite instability in colorectal cancer. J. Clin. Oncol. 2010, 28, 3380–3387. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zheng, R.-Y.; Jin, Z.-S. Correlations between microsatellite instability and the biological behaviour of tumours. J. Cancer Res. Clin. Oncol. 2019, 145, 2891–2899. [Google Scholar] [CrossRef] [PubMed]
- Gatalica, Z.; Vranic, S.; Xiu, J.; Swensen, J.; Reddy, S. High microsatellite instability (MSI-H) colorectal carcinoma: A brief review of predictive biomarkers in the era of personalized medicine. Fam. Cancer 2016, 15, 405–412. [Google Scholar] [CrossRef]
- Li, X.; Yao, X.; Wang, Y.; Hu, F.; Wang, F.; Jiang, L.; Liu, Y.; Wang, D.; Sun, G.; Zhao, Y. MLH1 Promoter Methylation Frequency in Colorectal Cancer Patients and Related Clinicopathological and Molecular Features. PLoS ONE 2013, 8, e59064. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Laghi, L.; Bianchi, P.; Malesci, A. Differences and evolution of the methods for the assessment of microsatellite instability. Oncogene 2008, 27, 6313–6321. [Google Scholar] [CrossRef] [PubMed]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.-Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 2017, PO.17.00073. [Google Scholar] [CrossRef] [PubMed]
- Venderbosch, S.; Nagtegaal, I.D.; Maughan, T.S.; Smith, C.G.; Cheadle, J.P.; Fisher, D.; Kaplan, R.; Quirke, P.; Seymour, M.T.; Richman, S.D.; et al. Mismatch Repair Status and BRAF Mutation Status in Metastatic Colorectal Cancer Patients: A Pooled Analysis of the CAIRO, CAIRO2, COIN, and FOCUS Studies. Clin. Cancer Res. 2014, 20, 5322–5330. [Google Scholar] [CrossRef]
- Goldstein, J.; Tran, B.; Ensor, J.; Gibbs, P.; Wong, H.L.; Wong, S.F.; Vilar, E.; Tie, J.; Broaddus, R.; Kopetz, S.; et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H). Ann. Oncol. 2014, 25, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, N.; Liu, C.; Zhang, Q.; Li, J.; Zhang, X.; Gong, J.; Lu, M.; Peng, Z.; Zhou, J.; Wang, X.; et al. Characteristics and Prognosis of Acquired Resistance to Immune Checkpoint Inhibitors in Gastrointestinal Cancer. JAMA Netw. Open 2022, 5, e224637. [Google Scholar] [CrossRef] [PubMed]
- Alemohammad, H.; Najafzadeh, B.; Asadzadeh, Z.; Baghbanzadeh, A.; Ghorbaninezhad, F.; Najafzadeh, A.; Safarpour, H.; Bernardini, R.; Brunetti, O.; Sonnessa, M.; et al. The importance of immune checkpoints in immune monitoring: A future paradigm shift in the treatment of cancer. Biomed. Pharmacother. 2022, 146, 112516. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- AVELUMAB and CETUXIMAB and mFOLFOXIRI as Initial Therapy for Unresectable Metastatic Colorectal Cancer Patients—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT04513951 (accessed on 10 January 2024).
- Antoniotti, C.; Rossini, D.; Pietrantonio, F.; Catteau, A.; Salvatore, L.; Lonardi, S.; Boquet, I.; Tamberi, S.; Marmorino, F.; Moretto, R.; et al. Upfront FOLFOXIRI plus bevacizumab with or without atezolizumab in the treatment of patients with metastatic colorectal cancer (AtezoTRIBE): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2022, 23, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Svrcek, M.; Lascols, O.; Cohen, R.; Collura, A.; Jonchère, V.; Fléjou, J.F.; Buhard, O.; Duval, A. MSI/MMR-deficient tumor diagnosis: Which standard for screening and for diagnosis? Diagnostic modalities for the colon and other sites: Differences between tumors. Bull. Cancer 2019, 106, 119–128. [Google Scholar] [CrossRef]
- French, A.J.; Sargent, D.J.; Burgart, L.J.; Foster, N.R.; Kabat, B.F.; Goldberg, R.; Shepherd, L.; Windschitl, H.E.; Thibodeau, S.N. Prognostic significance of defective mismatch repair and BRAF V600E in patients with colon cancer. Clin. Cancer Res. 2008, 14, 3408–3415. [Google Scholar] [CrossRef] [PubMed]
- Toon, C.W.; Chou, A.; Desilva, K.; Chan, J.; Patterson, J.; Clarkson, A.; Sioson, L.; Jankova, L.; Gill, A.J. BRAFV600E immunohistochemistry in conjunction with mismatch repair status predicts survival in patients with colorectal cancer. Mod. Pathol. 2014, 27, 644–650. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Cohen, R.; Salem, M.E. Immune Checkpoint Blockade Therapy in Patients with Colorectal Cancer Harboring Microsatellite Instability/Mismatch Repair Deficiency in 2022. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Toon, C.W.; Walsh, M.D.; Chou, A.; Capper, D.; Clarkson, A.; Sioson, L.; Clarke, S.; Mead, S.; Walters, R.J.; Clendenning, M.; et al. BRAFV600E immunohistochemistry facilitates universal screening of colorectal cancers for lynch syndrome. Am. J. Surg. Pathol. 2013, 37, 1592–1602. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.A.; Smith, D.M.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab versus chemotherapy for microsatellite instability-high/mismatch repair deficient metastatic colorectal cancer: The phase 3 KEYNOTE-177 Study. J. Clin. Oncol. 2020, 38, LBA4. [Google Scholar] [CrossRef]
- Cohen, R.; Colle, R.; Pudlarz, T.; Heran, M.; Duval, A.; Svrcek, M.; André, T. Immune checkpoint inhibition in metastatic colorectal cancer harboring microsatellite instability or mismatch repair deficiency. Cancers 2021, 13, 1149. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Kim, T.W.; van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability–high/mismatch repair–deficient metastatic colorectal cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Groot, H.J.; Lubberts, S.; de Wit, R.; Witjes, J.A.; Martijn Kerst, J.; de Jong, I.J.; Groenewegen, G.; van den Eertwegh, A.J.; Poortmans, P.M.; Klümpen, H.-J.; et al. Risk of Solid Cancer after Treatment of Testicular Germ Cell Cancer in the Platinum Era. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Karhemo, P.R.; Hyvönen, M.; Laakkonen, P. Metastasis-associated cell surface oncoproteomics. Front. Pharmacol. 2012, 3, 192. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Sokol, E.S.; Frampton, G.M.; Lippman, S.M.; Kurzrock, R. Microsatellite-Stable Tumors with High Mutational Burden Benefit from Immunotherapy. Cancer Immunol. Res. 2019, 7, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lanier, L.L. Natural Killer Cells and Cancer. Adv. Cancer Res. 2003, 90, 127–156. [Google Scholar]
- Markman, J.L.; Shiao, S.L. Impact of the immune system and immunotherapy in colorectal cancer. J. Gastrointest. Oncol. 2015, 6, 208–223. [Google Scholar]
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, A.; Ishida, T.; Ishii, T.; Komatsu, H.; Iida, S.; Ding, J.; Yonekura, K.; Takeuchi, S.; Takatsuka, Y.; Utsunomiya, A.; et al. Clinical significance of serum Th1-, Th2- and regulatory T cells-associated cytokines in adult T-cell leukemia/lymphoma: High interleukin-5 and -10 levels are significant unfavorable prognostic factors. Int. J. Cancer 2006, 118, 3054–3061. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Fu, Y.X. Tumor-infiltrating T lymphocytes: Friends or foes? Lab. Investig. 2006, 86, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Sutmuller, R.P.M.; Van Duivenvoorde, L.M.; Van Elsas, A.; Schumacher, T.N.M.; Wildenberg, M.E.; Allison, J.P.; Toes, R.E.M.; Offringa, R.; Melief, C.J.M. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25+ regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J. Exp. Med. 2001, 194, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Waldner, M.; Schimanski, C.C.; Neurath, M.F. Colon cancer and the immune system: The role of tumor invading T cells. World J. Gastroenterol. 2006, 12, 7233–7238. [Google Scholar] [CrossRef] [PubMed]
- Guidoboni, M.; Gafà, R.; Viel, A.; Doglioni, C.; Russo, A.; Santini, A.; Del Tin, L.; Macrì, E.; Lanza, G.; Boiocchi, M.; et al. Microsatellite instability and high content of activated cytotoxic lymphocytes identify colon cancer patients with a favorable prognosis. Am. J. Pathol. 2001, 159, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Prall, F.; Dührkop, T.; Weirich, V.; Ostwald, C.; Lenz, P.; Nizze, H.; Barten, M. Prognostic role of CD8+ tumor-infiltrating lymphocytes in stage III colorectal cancer with and without microsatellite instability. Hum. Pathol. 2004, 35, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Cornista, A.M.; Giolito, M.V.; Baker, K.; Hazime, H.; Dufait, I.; Datta, J.; Khumukcham, S.S.; De Ridder, M.; Roper, J.; Abreu, M.T.; et al. Colorectal Cancer Immunotherapy: State of the Art and Future Directions. Gastro Hep Adv. 2023, 2, 1103–1119. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef]
- Mezheyeuski, A.; Micke, P.; Martín-bernabé, A.; Backman, M.; Hrynchyk, I.; Hammarström, K.; Ström, S.; Ekström, J.; Edqvist, P.H.; Sundström, M.; et al. The immune landscape of colorectal cancer. Cancers 2021, 13, 5545. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T. Cancer metastasis: Characterization and identification of the behavior of metastatic tumor cells and the cell adhesion molecules, including carbohydrates. Curr. Drug Targets-Cardiovasc. Haematol. Disord. 2005, 5, 39–64. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Walsh, N.; Clynes, M. Membrane and membrane-associated proteins involved in the aggressive phenotype displayed by highly invasive cancer cells. Proteomics 2008, 8, 4054–4065. [Google Scholar] [CrossRef]
- Khong, H.T.; Restifo, N.P. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat. Immunol. 2002, 3, 999–1005. [Google Scholar] [CrossRef]
- O’Connell, J.; Bennett, M.W.; O’Sullivan, G.C.; Collins, J.K.; Shanahan, F. The Fas counterattack: Cancer as a site of immune privilege. Immunol. Today 1999, 20, 46–52. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Tumor counterattack: Fact or fiction? Cancer Immunol. Immunother. 2005, 54, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M.; Topalian, S.L. The role of CD4+ T cell responses in antitumor immunity. Curr. Opin. Immunol. 1998, 10, 588–594. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Schroeder, C.P.; Yang, P.; Newman, R.A.; Lotan, R. Eicosanoid metabolism in squamous cell carcinoma cell lines derived from primary and metastatic head and neck cancer and its modulation by celecoxib. Cancer Biol. Ther. 2004, 3, 847–852. [Google Scholar] [CrossRef]
- Qin, H.; Valentino, J.; Manna, S.; Tripathi, P.K.; Bhattacharya-Chatterjee, M.; Foon, K.A.; O’Malley, B.W.; Chatterjee, S.K. Gene therapy for head and neck cancer using vaccinia virus expressing IL-2 in a murine model, with evidence of immune suppression. Mol. Ther. 2001, 4, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Dennis, K.L.; Blatner, N.R.; Gounari, F.; Khazaie, K. Current status of interleukin-10 and regulatory T-cells in cancer. Curr. Opin. Oncol. 2013, 25, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Golshani, G.; Zhang, Y. Advances in immunotherapy for colorectal cancer: A review. Therap. Adv. Gastroenterol. 2020, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Sadeghi Rad, H.; Monkman, J.; Warkiani, M.E.; Ladwa, R.; O’Byrne, K.; Rezaei, N.; Kulasinghe, A. Understanding the tumor microenvironment for effective immunotherapy. Med. Res. Rev. 2021, 41, 1474–1498. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Collins, M. The B7 family of ligands and its receptors: New pathways for costimulation and inhibition of immune responses. Annu. Rev. Immunol. 2002, 20, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Staron, M.M.; Gray, S.M.; Marshall, H.D.; Parish, I.A.; Chen, J.H.; Perry, C.J.; Cui, G.; Li, M.O.; Kaech, S.M. The Transcription Factor FoxO1 Sustains Expression of the Inhibitory Receptor PD-1 and Survival of Antiviral CD8+ T Cells during Chronic Infection. Immunity 2014, 41, 802–814. [Google Scholar] [CrossRef]
- Li, C.; Li, W.; Xiao, J.; Jiao, S.; Teng, F.; Xue, S.; Zhang, C.; Sheng, C.; Leng, Q.; Rudd, C.E.; et al. ADAP and SKAP 55 deficiency suppresses PD -1 expression in CD 8 + cytotoxic T lymphocytes for enhanced anti-tumor immunotherapy. EMBO Mol. Med. 2015, 7, 754–769. [Google Scholar] [CrossRef]
- Xiao, G.; Deng, A.; Liu, H.; Ge, G.; Liu, X. Activator protein 1 suppresses antitumor T-cell function via the induction of programmed death 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15419–15424. [Google Scholar] [CrossRef] [PubMed]
- Salmaninejad, A.; Khoramshahi, V.; Azani, A.; Soltaninejad, E.; Aslani, S.; Zamani, M.R.; Zal, M.; Nesaei, A.; Hosseini, S.M. PD-1 and cancer: Molecular mechanisms and polymorphisms. Immunogenetics 2018, 70, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Chen, L. Inducible expression of B7-H1 (PD-L1) and its selective role in tumor site immune modulation. Cancer J. 2014, 20, 256–261. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef]
- Abiko, K.; Matsumura, N.; Hamanishi, J.; Horikawa, N.; Murakami, R.; Yamaguchi, K.; Yoshioka, Y.; Baba, T.; Konishi, I.; Mandai, M. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br. J. Cancer 2015, 112, 1501–1509. [Google Scholar] [CrossRef]
- Bellucci, R.; Martin, A.; Bommarito, D.; Wang, K.; Hansen, S.H.; Freeman, G.J.; Ritz, J. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology 2015, 4, e1008824. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-intrinsic PD-L1 signaling in cancer initiation, development and treatment: Beyond immune evasion. Front. Oncol. 2018, 8, 386. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Song, Y.; Wang, Y.; Huang, Y.; Li, Z.; Cui, Y.; Yi, M.; Xia, L.; Zhuang, W.; Wu, X.; et al. PD-1/PD-L1 blockade rescue exhausted CD8+ T cells in gastrointestinal stromal tumours via the PI3K/Akt/mTOR signalling pathway. Cell Prolif. 2019, 52, e12571. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, K.; Gunatilake, D.; Gallagher, S.J.; Tiffen, J.; Rizos, H.; Hersey, P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-κB. PLoS ONE 2015, 10, e0123410. [Google Scholar] [CrossRef] [PubMed]
- Stutvoet, T.S.; Kol, A.; de Vries, E.G.E.; de Bruyn, M.; Fehrmann, R.S.N.; Terwisscha van Scheltinga, A.G.T.; de Jong, S. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J. Pathol. 2019, 249, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Ishikawa, T.; Okayama, T.; Oka, K.; Mizushima, K.; Yasuda, T.; Sakamoto, N.; Katada, K.; Kamada, K.; Uchiyama, K.; et al. The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines. Oncol. Rep. 2017, 37, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Rothstein, D.; Rudd, C.E. Small-Molecule Inhibition of PD-1 Transcription Is an Effective Alternative to Antibody Blockade in Cancer Therapy. Cancer Res. 2018, 78, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.W.; Wang, H.; Zhang, W.W.; Wang, J.H.; Liu, W.J.; Xia, Z.J.; Huang, H.Q.; Jiang, W.Q.; Zhang, Y.J.; Wang, L. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 2016, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhang, Y.; Sun, B.; McMahon, A.P.; Wang, Y. Hedgehog Signaling: From Basic Biology to Cancer Therapy. Cell Chem. Biol. 2017, 24, 252–280. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Yao, H.; Li, C.S.; Liang, L.X.; Zhang, Y.; Chen, Y.X.; Fang, J.Y.; Xu, J. Rise of PD-L1 expression during metastasis of colorectal cancer: Implications for immunotherapy. J. Dig. Dis. 2017, 18, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.H.; Cavalcanti, M.S.; Segal, N.H.; Hechtman, J.F.; Weiser, M.R.; Smith, J.J.; Garcia-Aguilar, J.; Sadot, E.; Ntiamoah, P.; Markowitz, A.J.; et al. Patterns and prognostic relevance of PD-1 and PD-L1 expression in colorectal carcinoma. Mod. Pathol. 2016, 29, 1433–1442. [Google Scholar] [CrossRef]
- Scalapino, K.J.; Daikh, D.I. CTLA-4: A key regulatory point in the control of autoimmune disease. Immunol. Rev. 2008, 223, 143–155. [Google Scholar] [CrossRef]
- Valk, E.; Rudd, C.E.; Schneider, H. CTLA-4 trafficking and surface expression. Trends Immunol. 2008, 29, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Bradshaw, J.; Greene, J.A.; Peach, R.; Bennett, K.L.; Mittler, R.S. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 1996, 4, 535–543. [Google Scholar] [CrossRef]
- Egen, J.G.; Allison, J.P. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 2002, 16, 23–35. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.A.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Read, S.; Malmström, V.; Powrie, F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 2000, 192, 295–302. [Google Scholar] [CrossRef]
- Schneider, H.; Smith, X.; Liu, H.; Bismuth, G.; Rudd, C.E. CTLA-4 disrupts ZAP70 microcluster formation with reduced T cell/APC dwell times and calcium mobilization. Eur. J. Immunol. 2008, 38, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, N.; Derakhshani, A.; Kooshkaki, O.; Shadbad, M.A.; Hajiasgharzadeh, K.; Baghbanzadeh, A.; Safarpour, H.; Mokhtarzadeh, A.; Brunetti, O.; Yue, S.C.; et al. Immune checkpoints and car-t cells: The pioneers in future cancer therapies? Int. J. Mol. Sci. 2020, 21, 8305. [Google Scholar] [CrossRef]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: Mechanisms of action, efficacy, and limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Pernot, S.; Terme, M.; Voron, T.; Colussi, O.; Marcheteau, E.; Tartour, E.; Taieb, J. Colorectal cancer and immunity: What we know and perspectives. World J. Gastroenterol. 2014, 20, 3738–3750. [Google Scholar] [CrossRef]
- Andre, T.; Amonkar, M.; Norquist, J.M.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.A.; Smith, D.; Garcia-Carbonero, R.; et al. Health-related quality of life in patients with microsatellite instability-high or mismatch repair deficient metastatic colorectal cancer treated with first-line pembrolizumab versus chemotherapy (KEYNOTE-177): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 665–677. [Google Scholar] [CrossRef]
- US National Institutes of Health. A Study to Investigate Efficacy and Safety of Cobimetinib Plus Atezolizumab and Atezolizumab Monotherapy versus Regorafenib in Participants with Metastatic Colorectal Adenocarcinoma; NCT02788279; US National Institutes of Health: Bethesda, MD, USA, 2016.
- Study Details|Atezolizumab in Patients with MSI-h/MMR-D Stage III Colorectal Cancer Ineligible for Oxaliplatin|ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05118724 (accessed on 13 January 2024).
- Study of Induction PD-1 Blockade in Subjects with Locally Advanced Mismatch Repair Deficient Solid Tumors—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT04165772 (accessed on 13 January 2024).
- Keler, T.; Halk, E.; Vitale, L.; O’Neill, T.; Blanset, D.; Lee, S.; Srinivasan, M.; Graziano, R.F.; Davis, T.; Lonberg, N.; et al. Activity and safety of CTLA-4 blockade combined with vaccines in cynomolgus macaques. J. Immunol. 2003, 171, 6251–6259. [Google Scholar] [CrossRef]
- Buchbinder, E.; Stephen Hodi, F. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J. Clin. Investig. 2015, 125, 3377–3383. [Google Scholar] [CrossRef]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in tumor immunity. Int. J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef]
- Kumar, A.; Gautam, V.; Sandhu, A.; Rawat, K.; Sharma, A.; Saha, L. Current and emerging therapeutic approaches for colorectal cancer: A comprehensive review. World J. Gastrointest. Surg. 2023, 15, 495–519. [Google Scholar] [CrossRef]
- Leko, V.; Rosenberg, S.A. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell 2020, 38, 454–472. [Google Scholar] [CrossRef]
- Zimmermannova, O.; Caiado, I.; Ferreira, A.G.; Pereira, C.F. Cell Fate Reprogramming in the Era of Cancer Immunotherapy. Front. Immunol. 2021, 12, 714822. [Google Scholar] [CrossRef]
- Rouce, R.H.; Sharma, S.; Huynh, M.; Heslop, H.E. Recent advances in T-cell immunotherapy for haematological malignancies. Br. J. Haematol. 2017, 176, 688–704. [Google Scholar] [CrossRef]
- Gauthier, J.; Yakoub-Agha, I. Chimeric antigen-receptor T-cell therapy for hematological malignancies and solid tumors: Clinical data to date, current limitations and perspectives. Curr. Res. Transl. Med. 2017, 65, 93–102. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Garfall, A.L.; Maus, M.V.; Hwang, W.-T.; Lacey, S.F.; Mahnke, Y.D.; Melenhorst, J.J.; Zheng, Z.; Vogl, D.T.; Cohen, A.D.; Weiss, B.M.; et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; McGuirk, J.P. CAR T cells: Continuation in a revolution of immunotherapy. Lancet Oncol. 2020, 21, e168–e178. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, F.; Yang, J.; Zhaov, C.; Chu, Y. New chimeric antigen receptor design for solid tumors. Front. Immunol. 2017, 8, 1934. [Google Scholar] [CrossRef]
- Ye, B.; Stary, C.M.; Li, X.; Gao, Q.; Kang, C.; Xiong, X. Engineering chimeric antigen receptor-T cells for cancer treatment. Mol. Cancer 2018, 17, 32. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalían, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef]
- Turcotte, S.; Gros, A.; Hogan, K.; Tran, E.; Hinrichs, C.S.; Wunderlich, J.R.; Dudley, M.E.; Rosenberg, S.A. Phenotype and Function of T Cells Infiltrating Visceral Metastases from Gastrointestinal Cancers and Melanoma: Implications for Adoptive Cell Transfer Therapy. J. Immunol. 2013, 191, 2217–2225. [Google Scholar] [CrossRef]
- Immunotherapy Using Tumor Infiltrating Lymphocytes for Patients with Metastatic Human Papillomavirus-Associated Cancers. Available online: https://clinicaltrials.gov/study/NCT01174121 (accessed on 13 January 2024).
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Fan, T.; Zhang, M.; Yang, J.; Zhu, Z.; Cao, W.; Dong, C. Therapeutic cancer vaccines: Advancements, challenges, and prospects. Signal Transduct. Target. Ther. 2023, 8, 450. [Google Scholar] [CrossRef]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef]
- Harris, J.E.; Ryan, L.; Hoover, H.C.; Stuart, R.K.; Oken, M.M.; Benson, A.B.; Mansour, E.; Haller, D.G.; Manola, J.; Hanna, M.G. Adjuvant active specific immunotherapy for stage II and III colon cancer with an autologous tumor cell vaccine: Eastern Cooperative Oncology Group study E5283. J. Clin. Oncol. 2000, 18, 148–157. [Google Scholar] [CrossRef]
- Jocham, D.; Richter, A.; Hoffmann, L.; Iwig, K.; Fahlenkamp, D.; Zakrzewski, G.; Schmitt, E.; Dannenberg, T.; Lehmacher, W.; Von Wietersheim, J.; et al. Adjuvant autologous renal tumour cell vaccine and risk of tumour progression in patients with renal-cell carcinoma after radical nephrectomy: Phase III, randomised controlled trial. Lancet 2004, 363, 594–599. [Google Scholar] [CrossRef]
- Berd, D.; Sato, T.; Maguire, H.C.; Kairys, J.; Mastrangelo, M.J. Immunopharmacologic analysis of an autologous, hapten-modified human melanoma vaccine. J. Clin. Oncol. 2004, 22, 403–415. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Acquavella, N.; Yu, Z.; Restifo, N.P. Therapeutic cancer vaccines: Are we there yet? Immunol. Rev. 2011, 239, 27–44. [Google Scholar] [CrossRef]
- Salman, B.; Zhou, D.; Jaffee, E.M.; Edil, B.H.; Zheng, L. Vaccine therapy for pancreatic cancer. Oncoimmunology 2013, 2, e26662. [Google Scholar] [CrossRef]
- Bartnik, A.; Nirmal, A.J.; Yang, S.-Y. Peptide Vaccine Therapy in Colorectal Cancer. Vaccines 2013, 1, 1–16. [Google Scholar] [CrossRef]
- Yi, H.; Rong, Y.; Yankai, Z.; Wentao, L.; Hongxia, Z.; Jie, W.; Rongyue, C.; Taiming, L.; Jingjing, L. Improved efficacy of DNA vaccination against breast cancer by boosting with the repeat beta-hCG C-terminal peptide carried by mycobacterial heat-shock protein HSP65. Vaccine 2006, 24, 2575–2584. [Google Scholar] [CrossRef]
- Bilusic, M.; Heery, C.R.; Arlen, P.M.; Rauckhorst, M.; Apelian, D.; Tsang, K.Y.; Tucker, J.A.; Jochems, C.; Schlom, J.; Gulley, J.L.; et al. Phase i trial of a recombinant yeast-CEA vaccine (GI-6207) in adults with metastatic CEA-expressing carcinoma. Cancer Immunol. Immunother. 2014, 63, 225–234. [Google Scholar] [CrossRef]
- Yamada, A.; Sasada, T.; Noguchi, M.; Itoh, K. Next-generation peptide vaccines for advanced cancer. Cancer Sci. 2013, 104, 15–21. [Google Scholar] [CrossRef]
- Speetjens, F.M.; Kuppen, P.J.K.; Welters, M.J.P.; Essahsah, F.; Van Den Brink, A.M.E.G.V.; Lantrua, M.G.K.; Valentijn, A.R.P.M.; Oostendorp, J.; Fathers, L.M.; Nijman, H.W.; et al. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin. Cancer Res. 2009, 15, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Larocca, C.; Schlom, J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011, 17, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, S.R.; Higgins, J.P.; Dreher, M.R.; Woods, D.L.; Reddy, G.; Wood, B.J.; Guha, C.; Hodge, J.W. Combination Therapy with Local Radiofrequency Ablation and Systemic Vaccine Enhances Antitumor Immunity and Mediates Local and Distal Tumor Regression. PLoS ONE 2013, 8, e70417. [Google Scholar] [CrossRef]
- Gulley, J.L.; Madan, R.A.; Tsang, K.Y.; Arlen, P.M.; Camphausen, K.; Mohebtash, M.; Kamrava, M.; Schlom, J.; Citrin, D. A pilot safety trial investigating a vector-based vaccine targeting carcinoembryonic antigen in combination with radiotherapy in patients with gastrointestinal malignancies metastatic to the liver. Expert Opin. Biol. Ther. 2011, 11, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Tuyaerts, S.; Aerts, J.L.; Corthals, J.; Neyns, B.; Heirman, C.; Breckpot, K.; Thielemans, K.; Bonehill, A. Current approaches in dendritic cell generation and future implications for cancer immunotherapy. Cancer Immunol. Immunother. 2007, 56, 1513–1537. [Google Scholar] [CrossRef] [PubMed]
- Wooster, A.L.; Girgis, L.H.; Brazeale, H.; Anderson, T.S.; Wood, L.M.; Lowe, D.B. Dendritic cell vaccine therapy for colorectal cancer. Pharmacol. Res. 2021, 164, 105374. [Google Scholar] [CrossRef] [PubMed]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynde, M.; Mlecnik, B.; Bindea, G.; Fredriksen, T.; Church, S.E.; Lafontaine, L.; Haicheur, N.; Marliot, F.; Angelova, M.; Vasaturo, A.; et al. The Link between the Multiverse of Immune Microenvironments in Metastases and the Survival of Colorectal Cancer Patients. Cancer Cell 2018, 34, 1012–1026.e3. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wang, C.; Qiu, X.; Pu, X.; Chang, P. Colorectal Cancer Immune Infiltrates: Significance in Patient Prognosis and Immunotherapeutic Efficacy. Front. Immunol. 2020, 11, 528129. [Google Scholar] [CrossRef]
- Fucà, G.; Cohen, R.; Lonardi, S.; Shitara, K.; Elez, M.E.; Fakih, M.; Chao, J.; Klempner, S.J.; Emmett, M.; Jayachandran, P.; et al. Ascites and resistance to immune checkpoint inhibition in dMMR/MSI-H metastatic colorectal and gastric cancers. J. Immunother. Cancer 2022, 10, e004001. [Google Scholar] [CrossRef]
- Castellucci, E.; He, T.; Goldstein, D.Y.; Halmos, B.; Chuy, J. DNA Polymerase ε Deficiency Leading to an Ultramutator Phenotype: A Novel Clinically Relevant Entity. Oncologist 2017, 22, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Freeman-Mills, L.; Rayner, E.; Glaire, M.; Briggs, S.; Vermeulen, L.; Fessler, E.; Medema, J.P.; Boot, A.; Morreau, H.; et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroenterol. Hepatol. 2016, 1, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.J.; Cech, T.R. Shaping human telomeres: From shelterin and CST complexes to telomeric chromatin organization. Nat. Rev. Mol. Cell Biol. 2021, 22, 283–298. [Google Scholar] [CrossRef]
- Vasilopoulos, E.; Fragkiadaki, P.; Kalliora, C.; Fragou, D.; Docea, A.O.; Vakonaki, E.; Tsoukalas, D.; Calina, D.; Buga, A.M.; Georgiadis, G.; et al. The association of female and male infertility with telomere length (Review). Int. J. Mol. Med. 2019, 44, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Razgonova, M.P.; Zakharenko, A.M.; Golokhvast, K.S.; Thanasoula, M.; Sarandi, E.; Nikolouzakis, K.; Fragkiadaki, P.; Tsoukalas, D.; Spandidos, D.A.; Tsatsakis, A. Telomerase and telomeres in aging theory and chronographic aging theory (Review). Mol. Med. Rep. 2020, 22, 1679–1694. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ju, Z.; Rudolph, K.L. Telomere shortening and ageing. Z. Gerontol. Geriatr. 2007, 40, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere dysfunction in ageing and age-related diseases. Nat. Cell Biol. 2022, 24, 135–147. [Google Scholar] [CrossRef]
- Vakonaki, E.; Tsiminikaki, K.; Plaitis, S.; Fragkiadaki, P.; Tsoukalas, D.; Katsikantami, I.; Vaki, G.; Tzatzarakis, M.N.; Spandidos, D.A.; Tsatsakis, A.M. Common mental disorders and association with telomere length (Review). Biomed. Rep. 2018, 8, 111–116. [Google Scholar] [CrossRef]
- Maciejowski, J.; De Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Han, W.; Xue, W.; Zou, Y.; Xie, C.; Du, J.; Jin, G. The association between telomere length and cancer risk in population studies. Sci. Rep. 2016, 6, 22243. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.L.; Coller, H.A.; Roberts, J.M. Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat. Cell Biol. 2003, 5, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.K.; et al. Telomerase directly regulates NF-B-dependent transcription. Nat. Cell Biol. 2012, 14, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Q.; Li, K.; Chen, L.; Li, W.; Hou, M.; Liu, T.; Yang, J.; Lindvall, C.; Björkholm, M.; et al. Telomerase reverse transcriptase promotes epithelial-mesenchymal transition and stem cell-like traits in cancer cells. Oncogene 2013, 32, 4203–4213. [Google Scholar] [CrossRef]
- Yochum, G.S.; Cleland, R.; Goodman, R.H. A Genome-Wide Screen for β-Catenin Binding Sites Identifies a Downstream Enhancer Element That Controls c -Myc Gene Expression. Mol. Cell. Biol. 2008, 28, 7368–7379. [Google Scholar] [CrossRef]
- Zhao, Z.; Pan, X.; Liu, L.; Liu, N. Telomere length maintenance, shortening, and lengthening. J. Cell. Physiol. 2014, 229, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; De Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [PubMed]
- Hug, N.; Lingner, J. Telomere length homeostasis. Chromosoma 2006, 115, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Flores, I.; Benetti, R.; Blasco, M.A. Telomerase regulation and stem cell behaviour. Curr. Opin. Cell Biol. 2006, 18, 254–260. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, L.; Lu, S. Role of TERRA in the regulation of telomere length. Int. J. Biol. Sci. 2015, 11, 316–323. [Google Scholar] [CrossRef]
- Azzalin, C.M.; Lingner, J. Telomeres: The silence is broken. Cell Cycle 2008, 7, 1161–1165. [Google Scholar] [CrossRef]
- Farnung, B.O.; Brun, C.M.; Arora, R.; Lorenzi, L.E.; Azzalin, C.M. Telomerase efficiently elongates highly transcribing telomeres in human cancer cells. PLoS ONE 2012, 7, e35714. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Kroupa, M.; Rachakonda, S.K.; Liska, V.; Srinivas, N.; Urbanova, M.; Jiraskova, K.; Schneiderova, M.; Vycital, O.; Vymetalkova, V.; Vodickova, L.; et al. Relationship of telomere length in colorectal cancer patients with cancer phenotype and patient prognosis. Br. J. Cancer 2019, 121, 344–350. [Google Scholar] [CrossRef]
- Lai, T.P.; Wright, W.E.; Shay, J.W. Comparison of telomere length measurement methods. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2018, 373, 20160451. [Google Scholar] [CrossRef]
- Vaiserman, A.; Krasnienkov, D. Telomere Length as a Marker of Biological Age: State-of-the-Art, Open Issues, and Future Perspectives. Front. Genet. 2021, 11, 630186. [Google Scholar] [CrossRef] [PubMed]
- Luu, H.N.; Qi, M.; Wang, R.; Adams-Haduch, J.; Miljkovic, I.; Opresko, P.L.; Jin, A.; Koh, W.P.; Yuan, J.M. Association between leukocyte telomere length and colorectal cancer risk in the Singapore Chinese health study. Clin. Transl. Gastroenterol. 2019, 10, e00043. [Google Scholar] [CrossRef] [PubMed]
- Peacock, S.D.; Massey, T.E.; Vanner, S.J.; King, W.D. Telomere length in the colon is related to colorectal adenoma prevalence. PLoS ONE 2018, 13, e0205697. [Google Scholar] [CrossRef] [PubMed]
- Naing, C.; Aung, K.; Lai, P.K.; Mak, J.W. Association between telomere length and the risk of colorectal cancer: A meta-analysis of observational studies. BMC Cancer 2017, 17, 24. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, E.; Bertorelle, R.; Serra, L.; Terrin, L.; Candiotto, C.; Pucciarelli, S.; Del Bianco, P.; Nitti, D.; De Rossi, A. Relationship between telomere shortening, genetic instability, and site of tumour origin in colorectal cancers. Br. J. Cancer 2010, 102, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Aranda, C.; De Juan, C.; Diaz-Lopez, A.; Sanchez-Pernaute, A.; Torres, A.J.; Diaz-Rubio, E.; Balibrea, J.L.; Benito, M.; Iniesta, P. Correlations of telomere length, telomerase activity, and telomerlc-repeat binding factor 1 expression in colorectal carcinoma: Prognostic indications. Cancer 2006, 106, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Xin, X.; Pan, Y.; Zhang, H.; Tian, S.; Sun, C. Telomerase activity as a marker for differential diagnosis of pancreatic adenocarcinoma: A systematic review and meta-analysis. Int. J. Biol. Markers 2016, 31, e126–e137. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi-Tabata, R.; Lopez, F.; Fratantonio, S.; Kim, N.; Goldblum, J.; Tubbs, R.; Elson, P.; Lavery, I.; Bukowski, R.M.; Ganapathi, R.; et al. Telomerase activity in stage II colorectal carcinoma: Telomerase-negative tumors are correlated with poor prognosis. Cancer 2002, 95, 1834–1839. [Google Scholar] [CrossRef] [PubMed]
- Bertorelle, R.; Rampazzo, E.; Pucciarelli, S.; Nitti, D.; De Rossi, A. Telomeres, telomerase and colorectal cancer. World J. Gastroenterol. 2014, 20, 1940–1950. [Google Scholar] [CrossRef]
- Cong, Y.-S.; Wright, W.E.; Shay, J.W. Human Telomerase and Its Regulation. Microbiol. Mol. Biol. Rev. 2002, 66, 407–425. [Google Scholar] [CrossRef]
- Guilleret, I.; Yan, P.; Grange, F.; Braunschweig, R.; Bosman, F.T.; Benhattar, J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int. J. Cancer 2002, 101, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Bertorelle, R.; Briarava, M.; Rampazzo, E.; Biasini, L.; Agostini, M.; Maretto, I.; Lonardi, S.; Friso, M.L.; Mescoli, C.; Zagonel, V.; et al. Telomerase is an independent prognostic marker of overall survival in patients with colorectal cancer. Br. J. Cancer 2013, 108, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Terrin, L.; Rampazzo, E.; Pucciarelli, S.; Agostini, M.; Bertorelle, R.; Esposito, G.; DelBianco, P.; Nitti, D.; De Rossi, A. Relationship between tumor and plasma levels of hTERT mRNA in patients with colorectal cancer: Implications for monitoring of neoplastic disease. Clin. Cancer Res. 2008, 14, 7444–7451. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, E.; Del Bianco, P.; Bertorelle, R.; Boso, C.; Perin, A.; Spiro, G.; Bergamo, F.; Belluco, C.; Buonadonna, A.; Palazzari, E.; et al. The predictive and prognostic potential of plasma telomerase reverse transcriptase (TERT) RNA in rectal cancer patients. Br. J. Cancer 2018, 118, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Laish, I.; Levi, Z.; Mahajna, H.; Albshesh, A.; Horesh, N.; Katz, E.; Feldman, D.; Shinar, N.; Picard, O.; Yavzori, M.; et al. Characterization of blood-derived exosomal hTERT mRNA as a biomarker for colon cancer and Lynch syndrome. Front. Oncol. 2022, 12, 962473. [Google Scholar] [CrossRef] [PubMed]
- García, J.M.; García, V.; Peña, C.; Domínguez, G.; Silva, J.; Diaz, R.; Espinosa, P.; Citores, M.J.; Collado, M.; Bonilla, F. Extracellular plasma RNA from colon cancer patients is confined in a vesicle-like structure and is mRNA-enriched. RNA 2008, 14, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Dang, S.; Gabrani, R. Recent Patents on Anti-Telomerase Cancer Therapy. Recent Pat. Anticancer. Drug Discov. 2011, 7, 102–117. [Google Scholar] [CrossRef]
- Xu, L.; Li, S.; Stohr, B.A. The role of telomere biology in cancer. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 49–78. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Lee, Y.; Choi, B.K.; Park, B.M.; Kim, Y.H.; Yun, T.; Lee, W.J.; Yoo, H.; Baek, J.Y.; Woo, S.M.; et al. Phase 1 trial of 4-1BB-based adoptive T-cell therapy targeting human telomerase reverse transcriptase in patients with advanced refractory solid tumors. Cytotherapy 2023, 25, 1236–1241. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.J.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Tian, X.X.; Pang, J.C.S.; Zheng, J.; Chen, J.; To, S.S.T.; Ng, H.K. Antisense epidermal growth factor receptor RNA transfection in human glioblastoma cells down-regulates telomerase activity and telomere length. Br. J. Cancer 2002, 86, 1328–1332. [Google Scholar] [CrossRef] [PubMed]
- Kunimura, C.; Kikuchi, K.; Ahmed, N.; Shimizi, A.; Yasumoto, S. Telomerase activity in a specific cell subset co-expressing integrin β1/EGFR but not p75(NGFR)/bcl2/integrin β4 in normal human epithelial cells. Oncogene 1998, 17, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Heeg, S.; Hirt, N.; Queisser, A.; Schmieg, H.; Thaler, M.; Kunert, H.; Quante, M.; Goessel, G.; Von Werder, A.; Harder, J.; et al. EGFR overexpression induces activation of telomerase via PI3K/AKT-mediated phosphorylation and transcriptional regulation through Hif1-alpha in a cellular model of oral-esophageal carcinogenesis. Cancer Sci. 2011, 102, 351–360. [Google Scholar] [CrossRef]
- Douillard, J.-Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab–FOLFOX4 Treatment and RAS Mutations in Colorectal Cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Augustine, T.A.; Baig, M.; Sood, A.; Budagov, T.; Atzmon, G.; Mariadason, J.M.; Aparo, S.; Maitra, R.; Goel, S. Telomere length is a novel predictive biomarker of sensitivity to anti-EGFR therapy in metastatic colorectal cancer. Br. J. Cancer 2015, 112, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 2, 4172. [Google Scholar] [CrossRef]
- Puterman, E.; Lin, J.; Blackburn, E.; O’Donovan, A.; Adler, N.; Epel, E. The power of exercise: Buffering the effect of chronic stress on telomere length. PLoS ONE 2010, 5, e10837. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: A key player in cancer development. J. Hematol. Oncol. 2020, 13, 151. [Google Scholar] [CrossRef]
- Rufer, N.; Brümmendorf, T.H.; Kolvraa, S.; Bischoff, C.; Christensen, K.; Wadsworth, L.; Schulzer, M.; Lansdorp, P.M. Telomere fluorescence measurements in granulocytes and T lymphocyte subsets point to a high turnover of hematopoietic stem cells and memory T cells in early childhood. J. Exp. Med. 1999, 190, 157–167. [Google Scholar] [CrossRef]
- Effros, R.B. Replicative senescence of CD8 T cells: Effect on human ageing. Exp. Gerontol. 2004, 39, 517–524. [Google Scholar] [CrossRef]
- Honda, M.; Mengesha, E.; Albano, S.; Stephen Nichols, W.; Wallace, D.J.; Metzger, A.; Klinenberg, J.R.; Linker-Israeli, M. Telomere shortening and decreased replicative potential, contrasted by continued proliferation of telomerase-positive CD8+CD2810 T cells in patients with systemic lupus erythematosus. Clin. Immunol. Immunopathol. 2001, 99, 211–221. [Google Scholar]
- Son, N.H.; Murray, S.; Yanovski, J.; Hodes, R.J.; Weng, N. Lineage-Specific Telomere Shortening and Unaltered Capacity for Telomerase Expression in Human T and B Lymphocytes with Age. J. Immunol. 2000, 165, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- van de Berg, P.J.E.J.; Griffiths, S.J.; Yong, S.-L.; Macaulay, R.; Bemelman, F.J.; Jackson, S.; Henson, S.M.; ten Berge, I.J.M.; Akbar, A.N.; van Lier, R.A.W. Cytomegalovirus Infection Reduces Telomere Length of the Circulating T Cell Pool. J. Immunol. 2010, 184, 3417–3423. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Colorectal Cancer Immunotherapy | ||||
|---|---|---|---|---|
| Category | ICIs | ACT | Anti-Tumor Vaccines | |
| Type | Anti PDL1Abs | Atezolimumab | CAR-T | Whole tumor |
| Avelumab | ||||
| Durvalumab | ||||
| Anti PD-1 Abs | Pembrolizumab | TILs | Peptide antigen | |
| Nivolumab | ||||
| Cemiplimab | ||||
| Anti-CTLA-4 Abs | Ipilimumab | Viral vector | ||
| Dendritic cells | ||||
| Anti-Tumor Vaccines | ||||
|---|---|---|---|---|
| Type | Sample | Advantages | Disadvantages | Conclusion |
| Whole tumor | Cancer tissue (Autologous) | Patient-specific | Cross reaction with normal cells | No significant benefits in OS/DFS |
| Peptide antigen | Tumor-specific peptides (Heterologous) | Less cross reactions with normal cells |
| Positive results |
| Viral vector | Recombined Tumor-specific peptides (Heterologous) with viral/bacterial vectors |
| Positive results | |
| Dendritic cells | Dendritic cells (Autologous) | High specificity |
| Mixed results |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolouzakis, T.K.; Chrysos, E.; Docea, A.O.; Fragkiadaki, P.; Souglakos, J.; Tsiaoussis, J.; Tsatsakis, A. Current and Future Trends of Colorectal Cancer Treatment: Exploring Advances in Immunotherapy. Cancers 2024, 16, 1995. https://doi.org/10.3390/cancers16111995
Nikolouzakis TK, Chrysos E, Docea AO, Fragkiadaki P, Souglakos J, Tsiaoussis J, Tsatsakis A. Current and Future Trends of Colorectal Cancer Treatment: Exploring Advances in Immunotherapy. Cancers. 2024; 16(11):1995. https://doi.org/10.3390/cancers16111995
Chicago/Turabian StyleNikolouzakis, Taxiarchis Konstantinos, Emmanuel Chrysos, Anca Oana Docea, Persefoni Fragkiadaki, John Souglakos, John Tsiaoussis, and Aristidis Tsatsakis. 2024. "Current and Future Trends of Colorectal Cancer Treatment: Exploring Advances in Immunotherapy" Cancers 16, no. 11: 1995. https://doi.org/10.3390/cancers16111995
APA StyleNikolouzakis, T. K., Chrysos, E., Docea, A. O., Fragkiadaki, P., Souglakos, J., Tsiaoussis, J., & Tsatsakis, A. (2024). Current and Future Trends of Colorectal Cancer Treatment: Exploring Advances in Immunotherapy. Cancers, 16(11), 1995. https://doi.org/10.3390/cancers16111995