
Immunotherapy in Melanoma: Recent Advances and Future Directions



Abstract
Simple Summary
Abstract
1. Introduction
2. Anti-CTLA-4
3. Anti-PD-1
3.1. Pembrolizumab
3.2. Nivolumab
4. Ipilimumab and Nivolumab
5. Nivolumab and Relatimab
6. TLR-9 Agonists
7. T-VEC
8. Adoptive Cell Therapy
9. Fecal Microbiota Transplant
10. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Bedikian, A.Y.; Millward, M.; Pehamberger, H.; Conry, R.; Gore, M.; Trefzer, U.; Pavlick, A.C.; DeConti, R.; Hersh, E.M.; Hersey, P. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: The Oblimersen Melanoma Study Group. J. Clin. Oncol. 2006, 24, 4738–4745. [Google Scholar] [PubMed]
- Middleton, M.R.; Grob, J.; Aaronson, N.; Fierlbeck, G.; Tilgen, W.; Seiter, S.; Gore, M.; Aamdal, S.; Cebon, J.; Coates, A. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J. Clin. Oncol. 2000, 18, 158. [Google Scholar] [CrossRef] [PubMed]
- Korn, E.L.; Liu, P.-Y.; Lee, S.J.; Chapman, J.-A.W.; Niedzwiecki, D.; Suman, V.J.; Moon, J.; Sondak, V.K.; Atkins, M.B.; Eisenhauer, E.A. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. J. Clin. Oncol. 2008, 26, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R. CheckMate 067: 6.5-year outcomes in patients (pts) with advanced melanoma. J. Clin. Oncol. 2021, 39, 9506. [Google Scholar] [CrossRef]
- Davar, D.; Ding, F.; Saul, M.; Sander, C.; Tarhini, A.A.; Kirkwood, J.M.; Tawbi, H.A. High-dose interleukin-2 (HD IL-2) for advanced melanoma: A single center experience from the University of Pittsburgh Cancer Institute. J. Immunother. Cancer 2017, 5, 74. [Google Scholar]
- Chen, L.; Ashe, S.; Brady, W.A.; Hellström, I.; Hellström, K.E.; Ledbetter, J.A.; McGowan, P.; Linsley, P.S. Costimulation of antitumor immunity by the B7 counterreceptor for the T lymphocyte molecules CD28 and CTLA-4. Cell 1992, 71, 1093–1102. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti–CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.; Tan, P.; Bradshaw, J.; Ledbetter, J.A.; Anasetti, C.; Damle, N.K. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J. Exp. Med. 1992, 176, 1595–1604. [Google Scholar]
- 1Walunas, T.L.; Bakker, C.Y.; Bluestone, J.A. CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 1996, 183, 2541–2550. [Google Scholar]
- Hodi, F.S.; O’day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [PubMed]
- Ascierto, P.A.; Del Vecchio, M.; Robert, C.; Mackiewicz, A.; Chiarion-Sileni, V.; Arance, A.; Lebbé, C.; Bastholt, L.; Hamid, O.; Rutkowski, P. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2017, 18, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Chiarion-Sileni, V.; Grob, J.-J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N. Engl. J. Med. 2016, 375, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.A.; Lee, S.J.; Hodi, F.S.; Rao, U.N.; Cohen, G.I.; Hamid, O.; Hutchins, L.F.; Sosman, J.A.; Kluger, H.M.; Eroglu, Z. Phase III study of adjuvant ipilimumab (3 or 10 mg/kg) versus high-dose interferon alfa-2b for resected high-risk melanoma: North American Intergroup E1609. J. Clin. Oncol. 2020, 38, 567. [Google Scholar]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 616. [Google Scholar]
- Boussiotis, V.A. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.-J.; Weber, J.S. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [PubMed]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.G.; Dalle, S.; Haydon, A.M.; Meshcheryakov, A.; Khattak, A.; Carlino, M.S. Longer follow-up confirms recurrence-free survival benefit of adjuvant pembrolizumab in high-risk stage III melanoma: Updated results from the EORTC 1325-MG/KEYNOTE-054 trial. J. Clin. Oncol. 2020, 38, 3925. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Rutkowski, P.; Queirolo, P.; Del Vecchio, M.; Mackiewicz, J.; Chiarion-Sileni, V.; de la Cruz Merino, L.; Khattak, M.A.; Schadendorf, D.; Long, G.V. Pembrolizumab versus placebo as adjuvant therapy in completely resected stage IIB or IIC melanoma (KEYNOTE-716): A randomised, double-blind, phase 3 trial. Lancet 2022, 399, 1718–1729. [Google Scholar] [CrossRef]
- Patel, S.; Othus, M.; Prieto, V.; Lowe, M.; Buchbinder, E.; Chen, Y.; Hyngstrom, J.; Lao, C.D.; Truong, T.G.; Chandra, S. LBA6 Neoadjvuant versus adjuvant pembrolizumab for resected stage III-IV melanoma (SWOG S1801). Ann. Oncol. 2022, 33, S1408. [Google Scholar]
- Long, G.V.; Spillane, A.J.; Pennington, T.E.; Shannon, K.F.; Stretch, J.; Gonzalez, M.; Saw, R.P.M.; Lo, S.N.; Scolyer, R.A.; Menzies, A.M. 793P NeoPeLe: A phase II trial of neoadjuvant (NAT) pembrolizumab (Pembro) combined with lenvatinib (Lenva) in resectable stage III melanoma. Ann. Oncol. 2022, 33, S906–S907. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L. Phase I study of single-agent anti–programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167. [Google Scholar] [CrossRef]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller Jr, W.H.; Lao, C.D. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Rutkowski, P.; Hassel, J.C.; McNeil, C.M.; Kalinka, E.A. Five-year outcomes with nivolumab in patients with wild-type BRAF advanced melanoma. J. Clin. Oncol. 2020, 38, 3937. [Google Scholar]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [PubMed]
- Eggermont, A.M.; Chiarion-Sileni, V.; Grob, J.-J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2015, 16, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Del Vecchio, M.; Mandalá, M.; Gogas, H.; Arance, A.M.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.-J.; Chiarion-Sileni, V. Adjuvant nivolumab versus ipilimumab in resected stage IIIB–C and stage IV melanoma (CheckMate 238): 4-year results from a multicentre, double-blind, randomised, controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1465–1477. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [PubMed]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Tarhini, A.; McDermott, D.; Ambavane, A.; Gupte-Singh, K.; Aponte-Ribero, V.; Ritchings, C.; Benedict, A.; Rao, S.; Regan, M.M.; Atkins, M. Clinical and economic outcomes associated with treatment sequences in patients with BRAF-mutant advanced melanoma. Immunotherapy 2019, 11, 283–295. [Google Scholar] [CrossRef]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Ribas, A.; Tarhini, A.A.; Truong, T.-G.; Davar, D.; O’Rourke, M.A.; Curti, B.D.; Brell, J.M. DREAMseq (Doublet, Randomized Evaluation in Advanced Melanoma Sequencing): A phase III trial—ECOG-ACRIN EA6134. J. Clin. Oncol. 2021, 39, 356154. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Mandalà, M.; Ferrucci, P.F.; Guidoboni, M.; Rutkowski, P.; Ferraresi, V.; Arance, A.; Guida, M.; Maiello, E.; Gogas, H. Sequencing of ipilimumab plus nivolumab and encorafenib plus binimetinib for untreated BRAF-mutated metastatic melanoma (SECOMBIT): A randomized, three-arm, open-label phase II trial. J. Clin. Oncol. 2022, 41, 212–221. [Google Scholar] [CrossRef]
- Hoejberg, L.; Bastholt, L.; Schmidt, H. Interleukin-6 and melanoma. Melanoma Res. 2012, 22, 327–333. [Google Scholar] [PubMed]
- Tartour, E.; Dorval, T.; Mosseri, V.; Deneux, L.; Mathiot, C.; Brailly, H.; Montero, F.; Joyeux, I.; Pouillart, P.; Fridman, W.H. Serum interleukin 6 and C-reactive protein levels correlate with resistance to IL-2 therapy and poor survival in melanoma patients. Br. J. Cancer 1994, 69, 911–913. [Google Scholar] [CrossRef]
- Valpione, S.; Pasquali, S.; Campana, L.G.; Piccin, L.; Mocellin, S.; Pigozzo, J.; Chiarion-Sileni, V. Sex and interleukin-6 are prognostic factors for autoimmune toxicity following treatment with anti-CTLA4 blockade. J. Transl. Med. 2018, 16, 94. [Google Scholar]
- Weber, J.S.; Muramatsu, T.; Hamid, O.; Mehnert, J.; Hodi, F.S.; Krishnarajapet, S.; Malatyali, S.; Buchbinder, E.; Goldberg, J.; Sullivan, R. 1040O Phase II trial of ipilimumab, nivolumab and tocilizumab for unresectable metastatic melanoma. Ann. Oncol. 2021, 32, S869. [Google Scholar]
- Zimmer, L.; Livingstone, E.; Hassel, J.C.; Fluck, M.; Eigentler, T.; Loquai, C.; Haferkamp, S.; Gutzmer, R.; Meier, F.; Mohr, P. Adjuvant nivolumab plus ipilimumab or nivolumab monotherapy versus placebo in patients with resected stage IV melanoma with no evidence of disease (IMMUNED): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2020, 395, 1558–1568. [Google Scholar] [CrossRef]
- Long, G.V.; Schadendorf, D.; Vecchio, M.D.; Larkin, J.; Atkinson, V.; Schenker, M.; Pigozzo, J.; Gogas, H.J.; Dalle, S.; Meyer, N. Abstract CT004: Adjuvant therapy with nivolumab (NIVO) combined with ipilimumab (IPI) vs NIVO alone in patients (pts) with resected stage IIIB-D/IV melanoma (CheckMate 915). Cancer Res. 2021, 81, CT004. [Google Scholar] [CrossRef]
- Workman, C.J.; Cauley, L.S.; Kim, I.-J.; Blackman, M.A.; Woodland, D.L.; Vignali, D.A.A. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J. Immunol. 2004, 172, 5450–5455. [Google Scholar] [CrossRef]
- Baixeras, E.; Huard, B.; Miossec, C.a.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Piatier-Tonneau, D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 1992, 176, 327–337. [Google Scholar] [CrossRef]
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4-and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721. [Google Scholar] [CrossRef]
- Hemon, P.; Jean-Louis, F.; Ramgolam, K.; Brignone, C.; Viguier, M.; Bachelez, H.; Triebel, F.; Charron, D.; Aoudjit, F.; Al-Daccak, R. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J. Immunol. 2011, 186, 5173–5183. [Google Scholar] [CrossRef]
- Demeure, C.E.; Wolfers, J.; Martin-Garcia, N.; Gaulard, P.; Triebel, F. T Lymphocytes infiltrating various tumour types express the MHC class II ligand lymphocyte activation gene-3 (LAG-3): Role of LAG-3/MHC class II interactions in cell–cell contacts. Eur. J. Cancer 2001, 37, 1709–1718. [Google Scholar] [PubMed]
- Ascierto, P.A.; Bono, P.; Bhatia, S.; Melero, I.; Nyakas, M.S.; Svane, I.M.; Larkin, J.; Gomez-Roca, C.; Schadendorf, D.; Dummer, R. Efficacy of BMS-986016, a monoclonal antibody that targets lymphocyte activation gene-3 (LAG-3), in combination with nivolumab in pts with melanoma who progressed during prior anti–PD-1/PD-L1 therapy (mel prior IO) in all-comer and biomarker-enriched populations. Ann. Oncol. 2017, 28, v611–v612. [Google Scholar]
- Ascierto, P.A.; Melero, I.; Bhatia, S.; Bono, P.; Sanborn, R.E.; Lipson, E.J.; Callahan, M.K.; Gajewski, T.; Gomez-Roca, C.A.; Hodi, F.S. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. J. Clin. Oncol. 2017, 35, 9520. [Google Scholar]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Amaria, R.N.; Postow, M.A.; Tetzlaff, M.T.; Ross, M.I.; Glitza, I.C.; McQuade, J.L.; Wong, M.K.K.; Gershenwald, J.E.; Goepfert, R.; Keung, E.Z.-Y. Neoadjuvant and adjuvant nivolumab (nivo) with anti-LAG3 antibody relatlimab (rela) for patients (pts) with resectable clinical stage III melanoma. J. Clin. Oncol. 2021, 39, 9502. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Leifer, C.A.; Kennedy, M.N.; Mazzoni, A.; Lee, C.; Kruhlak, M.J.; Segal, D.M. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J. Immunol. 2004, 173, 1179–1183. [Google Scholar] [CrossRef]
- Agrawal, S.; Kandimalla, E.R. Synthetic agonists of Toll-like receptors 7, 8 and 9. Biochem. Soc. Trans. 2007, 35, 1461–1467. [Google Scholar] [CrossRef]
- Kandimalla, E.R.; Bhagat, L.; Cong, Y.-P.; Pandey, R.K.; Yu, D.; Zhao, Q.; Agrawal, S. Secondary structures in CpG oligonucleotides affect immunostimulatory activity. Biochem. Biophys. Res. Commun. 2003, 306, 948–953. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar]
- Sato, S.; Sugiyama, M.; Yamamoto, M.; Watanabe, Y.; Kawai, T.; Takeda, K.; Akira, S. Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-κB and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 2003, 171, 4304–4310. [Google Scholar] [PubMed]
- Moynihan, K.D.; Irvine, D.J. Roles for innate immunity in combination immunotherapies. Cancer Res. 2017, 77, 5215–5221. [Google Scholar]
- Thompson, J.A.; Kuzel, T.; Drucker, B.J.; Urba, W.J.; Bukowski, R.M. Safety and efficacy of PF-3512676 for the treatment of stage IV renal cell carcinoma: An open-label, multicenter phase I/II study. Clin. Genitourin. Cancer 2009, 7, E58–E65. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Kors, C.; Audring, H.; Walden, P.; Sterry, W.; Trefzer, U. Phase 1 evaluation of intralesionally injected TLR9-agonist PF-3512676 in patients with basal cell carcinoma or metastatic melanoma. J. Immunother. 2008, 31, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Weihrauch, M.R.; Ansén, S.; Jurkiewicz, E.; Geisen, C.; Xia, Z.; Anderson, K.S.; Gracien, E.; Schmidt, M.; Wittig, B.; Diehl, V. Phase I/II Combined Chemoimmunotherapy with Carcinoembryonic Antigen–Derived HLA-A2–Restricted CAP-1 peptide and irinotecan, 5-fluorouracil, and leucovorin in patients with primary metastatic colorectal cancer. Clin. Cancer Res. 2005, 11, 5993–6001. [Google Scholar] [PubMed]
- Friedberg, J.W.; Kim, H.; McCauley, M.; Hessel, E.M.; Sims, P.; Fisher, D.C.; Nadler, L.M.; Coffman, R.L.; Freedman, A.S. Combination immunotherapy with a CpG oligonucleotide (1018 ISS) and rituximab in patients with non-Hodgkin lymphoma: Increased interferon-α/β–inducible gene expression, without significant toxicity. Blood 2005, 105, 489–495. [Google Scholar]
- Karapetyan, L.; Luke, J.J.; Davar, D. Toll-like receptor 9 agonists in cancer. OncoTargets Ther. 2020, 13, 10039. [Google Scholar] [CrossRef]
- Diem, S.; Hasan Ali, O.; Ackermann, C.J.; Bomze, D.; Koelzer, V.H.; Jochum, W.; Speiser, D.E.; Mertz, K.D.; Flatz, L. Tumor infiltrating lymphocytes in lymph node metastases of stage III melanoma correspond to response and survival in nine patients treated with ipilimumab at the time of stage IV disease. Cancer Immunol. Immunother. 2018, 67, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Madore, J.; Vilain, R.E.; Menzies, A.M.; Kakavand, H.; Wilmott, J.S.; Hyman, J.; Yearley, J.H.; Kefford, R.F.; Thompson, J.F.; Long, G.V. PD-L1 expression in melanoma shows marked heterogeneity within and between patients: Implications for anti-PD-1/PD-L 1 clinical trials. Pigment. Cell Melanoma Res. 2015, 28, 245–253. [Google Scholar]
- Diab, A.; Haymaker, C.; Uemura, M.; Murthy, R.; James, M.; Geib, J.; Cornfeld, M.; Swann, S.; Yee, C.; Wargo, J. A Phase 1/2 trial of intratumoral (it) IMO-2125 (IMO) in combination with checkpoint inhibitors (CPI) in PD-(L) 1-refractory melanoma. Ann. Oncol. 2017, 28, v421. [Google Scholar] [CrossRef]
- Milhem, M.M.; Long, G.V.; Hoimes, C.J.; Amin, A.; Lao, C.D.; Conry, R.M.; Hunt, J.; Daniels, G.A.; Almubarak, M.; Shaheen, M.F. Phase 1b/2, open label, multicenter, study of the combination of SD-101 and pembrolizumab in patients with advanced melanoma who are naïve to anti-PD-1 therapy. J. Clin. Oncol. 2019, 37, 9534. [Google Scholar] [CrossRef]
- Davar, D.; Karunamurthy, A.; Hartman, D.; DeBlasio, R.; Chauvin, J.-M.; Ding, Q.; Pagliano, O.; Rose, A.; Kirkwood, J.; Zarour, H. 303 Phase II trial of neoadjuvant nivolumab (Nivo) and intra-tumoral (IT) CMP-001 in high-risk resectable melanoma (Neo-C-Nivo): Final results. J. Immunother. Cancer 2020, 8, A185–A186. [Google Scholar]
- Milhem, M.; Zakharia, Y.; Davar, D.; Buchbinder, E.; Medina, T.; Daud, A.; Ribas, A.; Niu, J.; Gibney, G.; Margolin, K. 304 Intratumoral injection of CMP-001, a toll-like receptor 9 (TLR9) agonist, in combination with pembrolizumab reversed programmed death receptor 1 (PD-1) blockade resistance in advanced melanoma. J. Immunother. Cancer 2020, 8, A186–A187. [Google Scholar]
- Russell, S.J.; Peng, K.-W. Viruses as anticancer drugs. Trends Pharmacol. Sci. 2007, 28, 326–333. [Google Scholar]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P. ICP34. 5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Hawkins, L.K.; Lemoine, N.R.; Kirn, D. Oncolytic biotherapy: A novel therapeutic platform. Lancet Oncol. 2002, 3, 17–26. [Google Scholar]
- Kaufman, H.L.; Amatruda, T.; Reid, T.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; Nemunaitis, J.; Zloza, A.; Wolf, M. Systemic versus local responses in melanoma patients treated with talimogene laherparepvec from a multi-institutional phase II study. J. Immunother. Cancer 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III–IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Ribas, A.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.S. Efficacy analysis of MASTERKEY-265 phase 1b study of talimogene laherparepvec (T-VEC) and pembrolizumab (pembro) for unresectable stage IIIB-IV melanoma. J. Clin. Oncol. 2016, 34, 9568. [Google Scholar]
- Sun, L.; Funchain, P.; Song, J.M.; Rayman, P.; Tannenbaum, C.; Ko, J.; McNamara, M.; Marcela Diaz-Montero, C.; Gastman, B. Talimogene Laherparepvec combined with anti-PD-1 based immunotherapy for unresectable stage III-IV melanoma: A case series. J. Immunother. Cancer 2018, 6, 36. [Google Scholar] [PubMed]
- Lee, S.; Margolin, K. Tumor-infiltrating lymphocytes in melanoma. Curr. Oncol. Rep. 2012, 14, 468–474. [Google Scholar] [PubMed]
- Wu, R.; Forget, M.-A.; Chacon, J.; Bernatchez, C.; Haymaker, C.; Chen, J.Q.; Hwu, P.; Radvanyi, L. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: Current status and future outlook. Cancer J. 2012, 18, 160. [Google Scholar] [CrossRef]
- Rohaan, M.W.; van den Berg, J.H.; Kvistborg, P.; Haanen, J.B.A.G. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: A viable treatment option. J. Immunother. Cancer 2018, 6, 102. [Google Scholar] [PubMed]
- Dudley, M.E.; Gross, C.A.; Langhan, M.M.; Garcia, M.R.; Sherry, R.M.; Yang, J.C.; Phan, G.Q.; Kammula, U.S.; Hughes, M.S.; Citrin, D.E. CD8+ Enriched “Young” Tumor Infiltrating Lymphocytes Can Mediate Regression of Metastatic MelanomaCD8+ Enriched Young TIL. Clin. Cancer Res. 2010, 16, 6122–6131. [Google Scholar] [CrossRef]
- Itzhaki, O.; Hovav, E.; Ziporen, Y.; Levy, D.; Kubi, A.; Zikich, D.; Hershkovitz, L.; Treves, A.J.; Shalmon, B.; Zippel, D. Establishment and large-scale expansion of minimally cultured “young” tumor infiltrating lymphocytes for adoptive transfer therapy. J. Immunother. 2011, 34, 212–220. [Google Scholar] [CrossRef]
- Besser, M.J.; Shapira-Frommer, R.; Treves, A.J.; Zippel, D.; Itzhaki, O.; Hershkovitz, L.; Levy, D.; Kubi, A.; Hovav, E.; Chermoshniuk, N. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2010, 16, 2646–2655. [Google Scholar]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.; Citrin, D.E.; Leitman, S.F. Adoptive cell therapy for patients with metastatic melanoma: Evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008, 26, 5233. [Google Scholar]
- Mullinax, J.E.; Hall, M.; Prabhakaran, S.; Weber, J.; Khushalani, N.; Eroglu, Z.; Brohl, A.S.; Markowitz, J.; Royster, E.; Richards, A. Combination of ipilimumab and adoptive cell therapy with tumor-infiltrating lymphocytes for patients with metastatic melanoma. Front. Oncol. 2018, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Geukes Foppen, M.H.; Donia, M.; Borch, T.H.; Met, Ö.; Blank, C.U.; Pronk, L.; Van Thienen, J.V.; Svane, I.M.; Haanen, J.B.A.G. Randomized phase III study comparing non-myeloablative lymphocyte depleting regimen of chemotherapy followed by infusion of tumor-infiltrating lymphocytes and interleukin-2 to standard ipilimumab treatment in metastatic melanoma. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Haanen, J.; Rohaan, M.; Borch, T.H.; van den Berg, J.H.; Met, Ö.; Foppen, M.G.; Granhøj, J.S.; Nuijen, B.; Nijenhuis, C.; Beijnen, J.H. LBA3 Treatment with tumor-infiltrating lymphocytes (TIL) versus ipilimumab for advanced melanoma: Results from a multicenter, randomized phase III trial. Ann. Oncol. 2022, 33, S1406. [Google Scholar] [CrossRef]
- Sarnaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Thomas, S.S.; Domingo-Musibay, E.; Pavlick, A.C.; Whitman, E.D. Lifileucel, a tumor-infiltrating lymphocyte therapy, in metastatic melanoma. J. Clin. Oncol. 2021, 39, 2656–2666. [Google Scholar] [PubMed]
- Russo, E.; Taddei, A.; Ringressi, M.N.; Ricci, F.; Amedei, A. The interplay between the microbiome and the adaptive immune response in cancer development. Ther. Adv. Gastroenterol. 2016, 9, 594–605. [Google Scholar] [CrossRef]
- Holmgren, J.; Czerkinsky, C.; Lycke, N.; Svennerholm, A.-M. Mucosal immunity: Implications for vaccine development. Immunobiology 1992, 184, 157–179. [Google Scholar] [PubMed]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef]
- Kim, C.H. Immune regulation by microbiome metabolites. Immunology 2018, 154, 220–229. [Google Scholar]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar]
- Shaw, M.H.; Kamada, N.; Kim, Y.-G.; Núñez, G. Microbiota-induced IL-1β, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J. Exp. Med. 2012, 209, 251–258. [Google Scholar]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.-M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O. Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: Mechanisms of action, efficacy, and limitations. Front. Oncol. 2018, 8, 86. [Google Scholar]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; Wang, Y.; DePeaux, K. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar]
- Luo, M.; Wang, H.; Wang, Z.; Cai, H.; Lu, Z.; Li, Y.; Du, M.; Huang, G.; Wang, C.; Chen, X. A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol. 2017, 12, 648–654. [Google Scholar]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING agonists as cancer therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef]
- Nakamura, T.; Sato, T.; Endo, R.; Sasaki, S.; Takahashi, N.; Sato, Y.; Hyodo, M.; Hayakawa, Y.; Harashima, H. STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. J. Immunother. Cancer 2021, 9, e002852. [Google Scholar] [CrossRef]
- Marcus, A.; Mao, A.J.; Lensink-Vasan, M.; Wang, L.; Vance, R.E.; Raulet, D.H. Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity 2018, 49, 754–763. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [PubMed]
- Kakavand, H.; Jackett, L.A.; Menzies, A.M.; Gide, T.N.; Carlino, M.S.; Saw, R.P.M.; Thompson, J.F.; Wilmott, J.S.; Long, G.V.; Scolyer, R.A. Negative immune checkpoint regulation by VISTA: A mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod. Pathol. 2017, 30, 1666–1676. [Google Scholar] [PubMed]
- Rotte, A.; Jin, J.Y.; Lemaire, V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann. Oncol. 2018, 29, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S. CAR T Cell Receptor Immunotherapy Targeting VEGFR2 for Patients with Metastatic Cancer. ClinicalTrials.gov Identifier: NCT01218867. Updated 10 December 2019. Available online: https://clinicaltrials.gov/ct2/show/results/NCT01218867 (accessed on 2 February 2023).
- Yvon, E.; Del Vecchio, M.; Savoldo, B.; Hoyos, V.; Dutour, A.; Anichini, A.; Dotti, G.; Brenner, M.K. Immunotherapy of metastatic melanoma using genetically engineered GD2-specific T cells. Clin. Cancer Res. 2009, 15, 5852–5860. [Google Scholar] [PubMed]
- Simon, B.; Harrer, D.C.; Schuler-Thurner, B.; Schuler, G.; Uslu, U. Arming T Cells with a gp100-Specific TCR and a CSPG4-Specific CAR Using Combined DNA- and RNA-Based Receptor Transfer. Cancers 2019, 11, 696. [Google Scholar]
- Yang, M.; Tang, X.; Zhang, Z.; Gu, L.; Wei, H.; Zhao, S.; Zhong, K.; Mu, M.; Huang, C.; Jiang, C. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics 2020, 10, 7622. [Google Scholar] [PubMed]



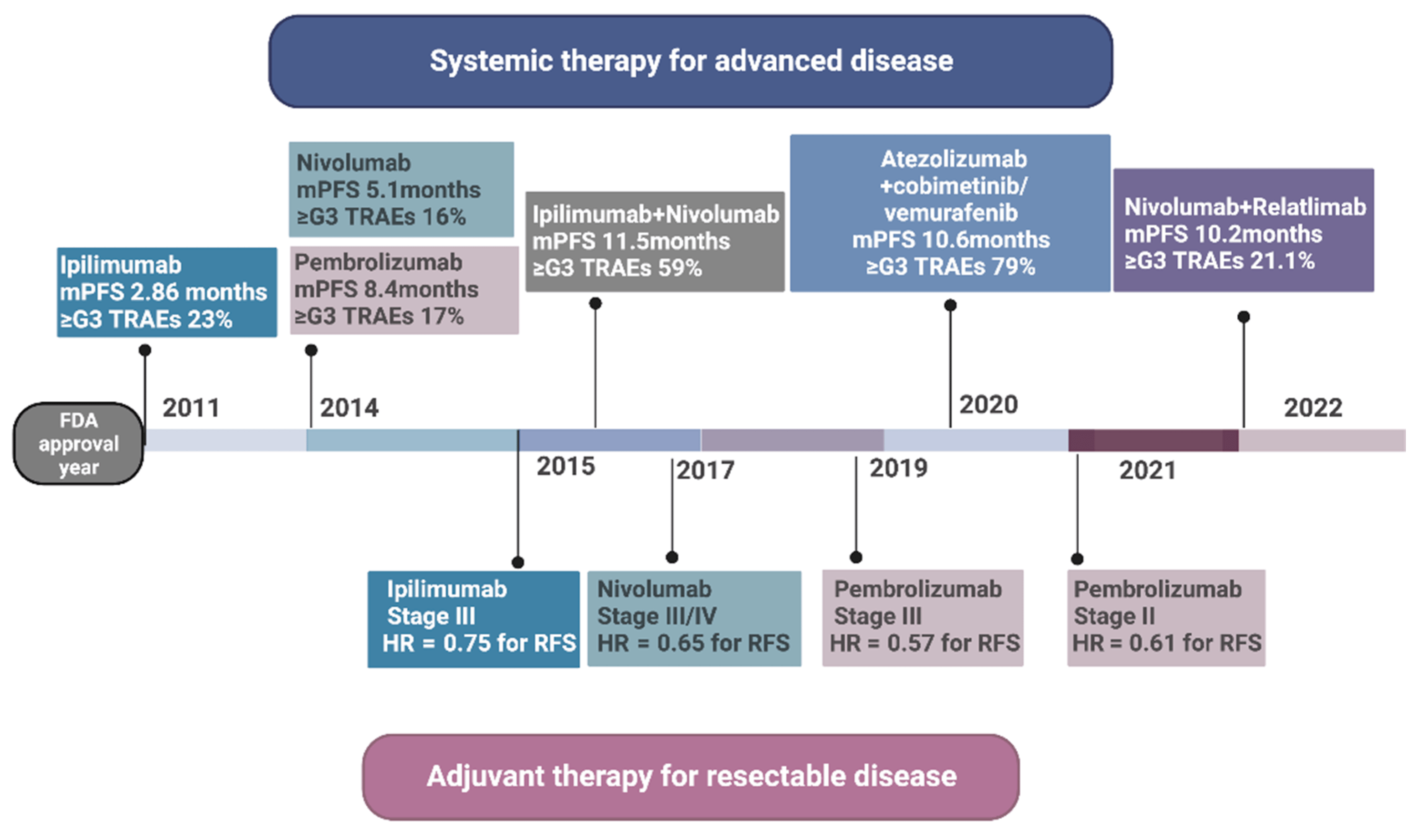{kind=link}
{kind=link}
| Medication | Study Phase | Clinical Setting | Efficacy |
|---|---|---|---|
| Ipilimumab vs. Placebo | Phase III | Adjuvant | 5-year RFS: 40.8% Ipilimumab vs. 30.3% placebo HR for recurrence/death: 0.75 |
| Nivolumab vs. Ipilimumab | Phase III | Adjuvant | 1-year RFS 70.5% Nivolumab vs. 60.8% Ipilimumab HR for recurrence/death: 0.66 |
| Pembrolizumab vs. Placebo | Phase III | Adjuvant | 1-year RFS 75.4% Pembrolizumab vs. 61.0% placebo |
| Relatimab + Nivolumab | Phase II | Adjuvant | 1-year RFS: 100%, pCR 59% |
| Ipilimumab + gp100 vs. ipilimumab vs. gp100 | Phase III | Unresectable or Metastatic | Median OS: 10.0 mo Ipilimumab + gp100 vs. 10.1 mo Ipilimumab vs. 6.4 mo gp100 |
| Ipilimumab + Nivolumab vs. Ipilimumab | Phase III | Unresectable or Metastatic | Median OS: 72.1 mo Ipilimumab + Nivolumab vs. 36.9 mo Nivolumab vs. 19.9 mo Ipilimumab |
| Relatimab + Nivolumab vs. Nivolumab | Phase II/III | Unresectable or Metastatic | Median PFS: 10.1 mo Relatimab + Nivolumab vs. 4.6 mo Nivolumab |
| Lifileucel (Adoptive Cell Therapy) | Phase II | Unresectable or Metastatic | Objective response rate 36% (CI 25 to 49%) |
| Adoptive Cell Therapy vs. Ipilimumab | Phase III | Unresectable or Metastatic | Median PFS: 7.2 mo ACT vs. 3.1 mo Ipilimumab |
| Pembrolizumab | Phase II | Neoadjuvant vs. Adjuvant | EFS (recurrence or progression, HR 0.59, CI 0.40–0.86) |
| Vidutolimod | Phase II | Neoadjuvant | Pathologic response rate: 70% (21/30 patients) |
| Fecal Microbiota Transplantation + Pembrolizumab | Phase II | Unresectable or Metastatic | Objective response rate: 20% (3/15 patients) |
| Pembrolizumab + T-VEC vs. Pembrolizumab + Placebo | Phase III | Unresectable or Metastatic | HR for progression or death 0.86, CI 0.71–1.04 HR for death 0.96, CI 0.76–1.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knight, A.; Karapetyan, L.; Kirkwood, J.M. Immunotherapy in Melanoma: Recent Advances and Future Directions. Cancers 2023, 15, 1106. https://doi.org/10.3390/cancers15041106
Knight A, Karapetyan L, Kirkwood JM. Immunotherapy in Melanoma: Recent Advances and Future Directions. Cancers. 2023; 15(4):1106. https://doi.org/10.3390/cancers15041106
Chicago/Turabian StyleKnight, Andrew, Lilit Karapetyan, and John M. Kirkwood. 2023. "Immunotherapy in Melanoma: Recent Advances and Future Directions" Cancers 15, no. 4: 1106. https://doi.org/10.3390/cancers15041106
APA StyleKnight, A., Karapetyan, L., & Kirkwood, J. M. (2023). Immunotherapy in Melanoma: Recent Advances and Future Directions. Cancers, 15(4), 1106. https://doi.org/10.3390/cancers15041106