
The Uncharted Landscape of Rare Endocrine Immune-Related Adverse Events

 ,
,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
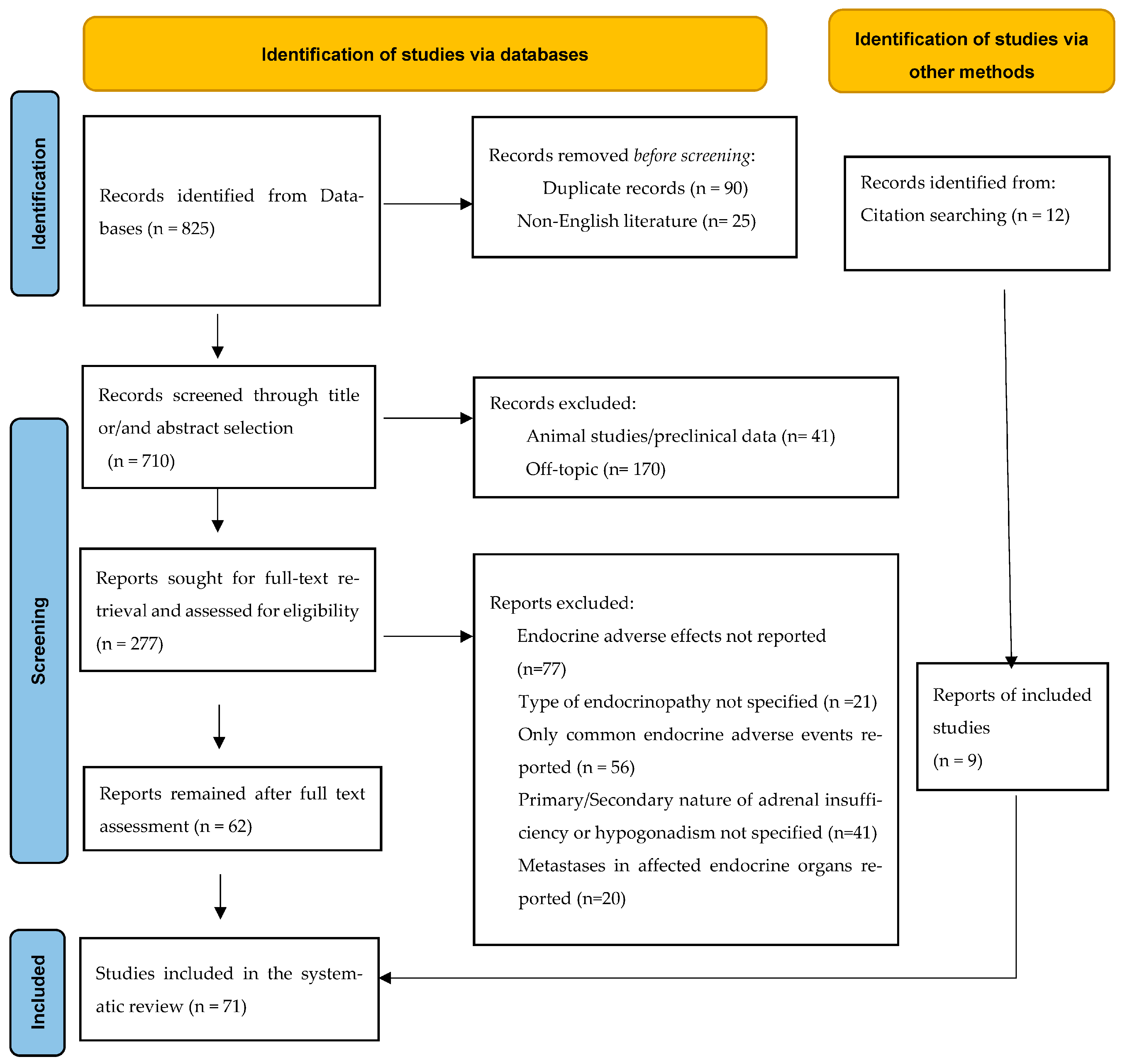
2. Materials and Methods
2.1. Data Sources and Search Strategy
2.2. Eligibility Criteria for Articles of Inclusion
3. Literature Review Results
3.1. Primary Adrenal Insufficiency (PAI)
3.1.1. Background
3.1.2. Case Studies
3.1.3. Pathophysiology
3.1.4. Clinical Presentation
3.1.5. Diagnosis
3.1.6. Management
3.2. Diabetes Insipidus (DI)
3.2.1. Background
3.2.2. Case Studies
3.2.3. Pathophysiology
3.2.4. Clinical Presentation
3.2.5. Diagnosis
3.2.6. Management
3.3. Hypoparathyroidism
3.3.1. Background
3.3.2. Case Studies
3.3.3. Pathophysiology
3.3.4. Clinical Presentation
3.3.5. Diagnosis
3.3.6. Management
3.4. Lipodystrophy
3.4.1. Background
3.4.2. Case Studies
3.4.3. Pathophysiology
3.4.4. Clinical Presentation
3.4.5. Diagnosis
3.4.6. Management
3.5. Osteoporosis
3.5.1. Background
3.5.2. Case studies
3.5.3. Pathophysiology
3.5.4. Clinical Presentation
3.5.5. Diagnosis
3.5.6. Management
3.6. Hypergonadotropic Hypogonadism
3.7. Cushing Disease (CD)
4. Future Aspects
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.; Obeid, M.; Spain, L.; Carbonnel, F.; Wang, Y.; Robert, C.; Lyon, A.R.; Wick, W.; Kostine, M.; Peters, S.; et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 33, 1217–1238. [Google Scholar] [CrossRef]
- Husebye, E.S.; Castinetti, F.; Criseno, S.; Curigliano, G.; Decallonne, B.; Fleseriu, M.; Higham, C.E.; Lupi, I.; Paschou, S.A.; Toth, M.; et al. Endocrine-related adverse conditions in patients receiving immune checkpoint inhibition: An ESE clinical practice guideline. Eur. J. Endocrinol. 2022, 187, G1–G21. [Google Scholar] [CrossRef] [PubMed]
- Barroso-Sousa, R.; Barry, W.T.; Garrido-Castro, A.C.; Hodi, F.S.; Min, L.; Krop, I.E.; Tolaney, S.M. Incidence of Endocrine Dysfunction Following the Use of Different Immune Checkpoint Inhibitor Regimens: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 173–182. [Google Scholar] [CrossRef]
- De Filette, J.; Andreescu, C.E.; Cools, F.; Bravenboer, B.; Velkeniers, B. A Systematic Review and Meta-Analysis of Endocrine-Related Adverse Events Associated with Immune Checkpoint Inhibitors. Horm. Metab. Res. 2019, 51, 145–156. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Bmj 2021, 372, n71. [Google Scholar] [CrossRef]
- Available online: https://health.ec.europa.eu/system/files/2016-11/smpc_guideline_rev2_en_0.pdf (accessed on 1 October 2022).
- Cui, K.; Wang, Z.; Zhang, Q.; Zhang, X. Immune checkpoint inhibitors and adrenal insufficiency: A large-sample case series study. Ann. Transl. Med. 2022, 10, 251. [Google Scholar] [CrossRef]
- Grouthier, V.; Lebrun-Vignes, B.; Moey, M.; Johnson, D.B.; Moslehi, J.J.; Salem, J.E.; Bachelot, A. Immune Checkpoint Inhibitor-Associated Primary Adrenal Insufficiency: WHO VigiBase Report Analysis. Oncologist 2020, 25, 696–701. [Google Scholar] [CrossRef]
- Lu, D.; Yao, J.; Yuan, G.; Gao, Y.; Zhang, J.; Guo, X. Immune checkpoint inhibitor-associated new-onset primary adrenal insufficiency: A retrospective analysis using the FAERS. J. Endocrinol. Investig. 2022, 45, 2131–2137. [Google Scholar] [CrossRef]
- Hasegawa, S.; Ikesue, H.; Nakao, S.; Shimada, K.; Mukai, R.; Tanaka, M.; Matsumoto, K.; Inoue, M.; Satake, R.; Yoshida, Y.; et al. Analysis of immune-related adverse events caused by immune checkpoint inhibitors using the Japanese Adverse Drug Event Report database. Pharmacoepidemiol. Drug Saf. 2020, 29, 1279–1294. [Google Scholar] [CrossRef] [PubMed]
- Paepegaey, A.C.; Lheure, C.; Ratour, C.; Lethielleux, G.; Clerc, J.; Bertherat, J.; Kramkimel, N.; Groussin, L. Polyendocrinopathy Resulting From Pembrolizumab in a Patient With a Malignant Melanoma. J. Endocr. Soc. 2017, 1, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Akarca, F.K.; Can, O.; Yalcinli, S.; Altunci, Y.A. Nivolumab, a new immunomodulatory drug, a new adverse effect; adrenal crisis. Turk. J. Emerg. Med. 2017, 17, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, K.; Agrawal, N. Lambert-Eaton Myasthenic Syndrome Secondary to Nivolumab and Ipilimumab in a Patient with Small-Cell Lung Cancer. Case Rep. Neurol. Med. 2019, 2019, 5353202. [Google Scholar] [CrossRef] [PubMed]
- Gunjur, A.; Klein, O.; Kee, D.; Cebon, J. Anti-programmed cell death protein 1 (anti-PD1) immunotherapy induced autoimmune polyendocrine syndrome type II (APS-2): A case report and review of the literature. J. Immunother. Cancer 2019, 7, 241. [Google Scholar] [CrossRef]
- Lanzolla, G.; Coppelli, A.; Cosottini, M.; Del Prato, S.; Marcocci, C.; Lupi, I. Immune Checkpoint Blockade Anti-PD-L1 as a Trigger for Autoimmune Polyendocrine Syndrome. J. Endocr. Soc. 2019, 3, 496–503. [Google Scholar] [CrossRef]
- Iqbal, I.; Khan, M.A.A.; Ullah, W.; Nabwani, D. Nivolumab-induced adrenalitis. BMJ Case Rep. 2019, 12, e231829. [Google Scholar] [CrossRef]
- Kagoshima, H.; Hori, R.; Kojima, T.; Okanoue, Y.; Fujimura, S.; Taguchi, A.; Shoji, K. Adrenal insufficiency following nivolumab therapy in patients with recurrent or metastatic head and neck cancer. Auris Nasus Larynx 2020, 47, 309–313. [Google Scholar] [CrossRef]
- Abdallah, D.; Johnson, J.; Goldner, W.; Addasi, N.; Desouza, C.; Kotwal, A. Adrenal Insufficiency From Immune Checkpoint Inhibitors Masquerading as Sepsis. JCO Oncol. Pract. 2021, 17, 212–214. [Google Scholar] [CrossRef]
- Harsch, I.A.; Gritsaenko, A.; Konturek, P.C. An analysis of early morning acth levels in the first case of pembrolizumab-induced adrenalitis as a delayed immune-related event (dire)—Case study. Wiad. Lek. 2020, 73, 396–400. [Google Scholar] [CrossRef]
- Özyurt, E.; Özçelik, S.; Sürmeli, H.; Çelik, M.; Ayhan, M.; Özçelik, M. Side effects of immune-checkpoint inhibitors: Can multiple side effects be seen in a patient? J. Oncol. Pharm. Pract. 2022, 28, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, J.; Fries, C.; Heer, A.; Hoffmann, F.; Meyer, C.; Landsberg, J.; Fenske, W.K. It’s Not Always SIAD: Immunotherapy-Triggered Endocrinopathies Enter the Field of Cancer-Related Hyponatremia. J. Endocr. Soc. 2022, 6, bvac036. [Google Scholar] [CrossRef] [PubMed]
- Trainer, H.; Hulse, P.; Higham, C.E.; Trainer, P.; Lorigan, P. Hyponatraemia secondary to nivolumab-induced primary adrenal failure. Endocrinol. Diabetes Metab. Case Rep. 2016, 2016, 16-0108. [Google Scholar] [CrossRef] [PubMed]
- Min, L.; Ibrahim, N. Ipilimumab-induced autoimmune adrenalitis. Lancet Diabetes Endocrinol. 2013, 1, e15. [Google Scholar] [CrossRef]
- Deligiorgi, M.V.; Trafalis, D.T. Reversible primary adrenal insufficiency related to anti-programmed cell-death 1 protein active immunotherapy: Insight into an unforeseen outcome of a rare immune-related adverse event. Int. Immunopharmacol. 2020, 89, 107050. [Google Scholar] [CrossRef]
- Salinas, C.; Renner, A.; Rojas, C.; Samtani, S.; Burotto, M. Primary Adrenal Insufficiency during Immune Checkpoint Inhibitor Treatment: Case Reports and Review of the Literature. Case Rep. Oncol. 2020, 13, 621–626. [Google Scholar] [CrossRef]
- Dasgupta, A.; Tsay, E.; Federman, N.; Lechner, M.G.; Su, M.A. Polyendocrine Autoimmunity and Diabetic Ketoacidosis Following Anti-PD-1 and Interferon α. Pediatrics 2022, 149, e2021053363. [Google Scholar] [CrossRef]
- Figueroa-Perez, N.; Kashyap, R.; Bal, D.; Anjum Khan, S.; Pattan, V. Autoimmune Myasthenia, Primary Adrenal Insufficiency, and Progressive Hypothyroidism Due to Pembrolizumab and Axitinib Combination Regimen. Cureus 2021, 13, e16933. [Google Scholar] [CrossRef]
- Bacanovic, S.; Burger, I.A.; Stolzmann, P.; Hafner, J.; Huellner, M.W. Ipilimumab-Induced Adrenalitis: A Possible Pitfall in 18F-FDG-PET/CT. Clin. Nucl. Med. 2015, 40, e518–e519. [Google Scholar] [CrossRef]
- Knight, T.; Cooksley, T. Emergency Presentations of Immune Checkpoint Inhibitor-Related Endocrinopathies. J. Emerg. Med. 2021, 61, 140–146. [Google Scholar] [CrossRef]
- Coskun, N.S.S.; Simsir, I.Y.; Göksel, T. A case with a primary adrenal insufficiency secondary to nivolumab. Eur. Respir. J. 2016, 48, PA4853. [Google Scholar] [CrossRef]
- Hobbs, K.B.; Yackzan, S. Adrenal Insufficiency: Immune Checkpoint Inhibitors and Immune-Related Adverse Event Management. Clin. J. Oncol. Nurs. 2020, 24, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Gaballa, S.; Hlaing, K.M.; Mahler, N.; Moursy, S.; Ahmed, A. A Rare Case of Immune-Mediated Primary Adrenal Insufficiency With Cytotoxic T-Lymphocyte Antigen-4 Inhibitor Ipilimumab in Metastatic Melanoma of Lung and Neck of Unknown Primary. Cureus 2020, 12, e8602. [Google Scholar] [CrossRef] [PubMed]
- Afreen Idris Shariff, D.A.D.A. Primary Adrenal Insufficiency from Immune Checkpoint Inhibitors. AACE Clin. Case Rep. 2018, 4, 232–234. [Google Scholar] [CrossRef]
- Kojadinovic, A.; Mundi, P.S. Primary Adrenal Insufficiency and Acute Cardiomyopathy in a Patient With Colorectal Cancer Treated With Dual Immune Checkpoint Inhibitors. Clin. Colorectal. Cancer 2021, 20, e249–e252. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.M.; Selamet, U.; Bui, P.; Sun, S.F.; Shenouda, O.; Nobakht, N.; Barsoum, M.; Arman, F.; Rastogi, A. Acute Kidney Injury after Pembrolizumab-Induced Adrenalitis and Adrenal Insufficiency. Case Rep. Nephrol. Dial. 2018, 8, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Galliazzo, S.; Morando, F.; Sartorato, P.; Bortolin, M.; De Menis, E. A Case of Cancer-Associated Hyponatraemia: Primary Adrenal Insufficiency Secondary to Nivolumab. Endocr. Metab. Immune Disord. Drug Targets 2022, 22, 363–366. [Google Scholar] [CrossRef]
- Hescot, S.; Haissaguerre, M.; Pautier, P.; Kuhn, E.; Schlumberger, M.; Berdelou, A. Immunotherapy-induced Addison’s disease: A rare, persistent and potentially lethal side-effect. Eur. J. Cancer 2018, 97, 57–58. [Google Scholar] [CrossRef]
- Colombo, C.; De Leo, S.; Di Stefano, M.; Vannucchi, G.; Persani, L.; Fugazzola, L. Primary Adrenal Insufficiency During Lenvatinib or Vandetanib and Improvement of Fatigue After Cortisone Acetate Therapy. J. Clin. Endocrinol. Metab. 2019, 104, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, S.J.; Tucker, M.A.; Engels, E.A.; Dores, G.M.; Sampson, J.N.; Shiels, M.S.; Chanock, S.J.; Morton, L.M. Immune-Related Adverse Events After Immune Checkpoint Inhibitors for Melanoma Among Older Adults. JAMA Netw. Open 2022, 5, e223461. [Google Scholar] [CrossRef] [PubMed]
- Hellesen, A.; Bratland, E.; Husebye, E.S. Autoimmune Addison’s disease—An update on pathogenesis. Ann. Endocrinol. 2018, 79, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.L.; Pearce, S.H. Autoimmune Addison disease: Pathophysiology and genetic complexity. Nat. Rev. Endocrinol. 2012, 8, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Husebye, E.S.; Anderson, M.S.; Kämpe, O. Autoimmune Polyendocrine Syndromes. N. Engl. J. Med. 2018, 378, 2542–2544. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.R.; Allolio, B.; Arlt, W.; Barthel, A.; Don-Wauchope, A.; Hammer, G.D.; Husebye, E.S.; Merke, D.P.; Murad, M.H.; Stratakis, C.A.; et al. Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 364–389. [Google Scholar] [CrossRef] [PubMed]
- Castinetti, F.; Borson-Chazot, F. Immunotherapy-induced endocrinopathies: Insights from the 2018 French Endocrine Society Guidelines. Bull. Cancer 2019, 106, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Johannsson, G.; Nilsson, A.G.; Bergthorsdottir, R.; Burman, P.; Dahlqvist, P.; Ekman, B.; Engström, B.E.; Olsson, T.; Ragnarsson, O.; Ryberg, M.; et al. Improved cortisol exposure-time profile and outcome in patients with adrenal insufficiency: A prospective randomized trial of a novel hydrocortisone dual-release formulation. J. Clin. Endocrinol. Metab. 2012, 97, 473–481. [Google Scholar] [CrossRef]
- Arima, H.; Cheetham, T.; Christ-Crain, M.; Cooper, D.; Gurnell, M.; Drummond, J.B.; Levy, M.; McCormack, A.I.; Verbalis, J.; Newell-Price, J.; et al. Changing the name of diabetes insipidus: A position statement of The Working Group for Renaming Diabetes Insipidus. Endocr. J. 2022, 69, 1281–1284. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Ippolito, S.; Lupi, I.; Caturegli, P. Hypophysitis induced by immune checkpoint inhibitors: A 10-year assessment. Expert Rev. Endocrinol. Metab. 2019, 14, 381–398. [Google Scholar] [CrossRef]
- Bai, X.; Lin, X.; Zheng, K.; Chen, X.; Wu, X.; Huang, Y.; Zhuang, Y. Mapping endocrine toxicity spectrum of immune checkpoint inhibitors: A disproportionality analysis using the WHO adverse drug reaction database, VigiBase. Endocrine 2020, 69, 670–681. [Google Scholar] [CrossRef]
- Zhai, Y.; Ye, X.; Hu, F.; Xu, J.; Guo, X.; Zhuang, Y.; He, J. Endocrine toxicity of immune checkpoint inhibitors: A real-world study leveraging US Food and Drug Administration adverse events reporting system. J. Immunother. Cancer 2019, 7, 286. [Google Scholar] [CrossRef]
- Brilli, L.; Calabrò, L.; Campanile, M.; Pilli, T.; Agostinis, C.; Cerase, A.; Maio, M.; Castagna, M.G. Permanent diabetes insipidus in a patient with mesothelioma treated with immunotherapy. Arch. Endocrinol. Metab. 2020, 64, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Terán Brage, E.; Heras Benito, M.; Navalón Jiménez, M.B.; Vidal Tocino, R.; Del Barco Morillo, E.; Fonseca Sánchez, E. Severe Hyponatremia Masking Central Diabetes Insipidus in a Patient with a Lung Adenocarcinoma. Case Rep Oncol. 2022, 15, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Grami, Z.; Manjappachar, N.; Reddy Dereddi, R. Diabetes Insipidus in Checkpoint Inhibitor Treatment and Acute Myeloid Leukemia. Crit. Care Med. 2020, 48, 144. [Google Scholar] [CrossRef]
- Dillard, T.; Yedinak, C.G.; Alumkal, J.; Fleseriu, M. Anti-CTLA-4 antibody therapy associated autoimmune hypophysitis: Serious immune related adverse events across a spectrum of cancer subtypes. Pituitary 2010, 13, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Nallapaneni, N.N.; Mourya, R.; Bhatt, V.R.; Malhotra, S.; Ganti, A.K.; Tendulkar, K.K. Ipilimumab-induced hypophysitis and uveitis in a patient with metastatic melanoma and a history of ipilimumab-induced skin rash. J. Natl. Compr. Canc. Netw. 2014, 12, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, F.; George, E.; Roberts, A. Combination immune checkpoint inhibitor therapy nivolumab and ipilimumab associated with multiple endocrinopathies. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018, 17-0146. [Google Scholar] [CrossRef] [PubMed]
- Tshuma, N.; Glynn, N.; Evanson, J.; Powles, T.; Drake, W.M. Hypothalamitis and severe hypothalamic dysfunction associated with anti-programmed cell death ligand 1 antibody treatment. Eur. J. Cancer 2018, 104, 247–249. [Google Scholar] [CrossRef]
- Barnabei, A.; Carpano, S.; Chiefari, A.; Bianchini, M.; Lauretta, R.; Mormando, M.; Puliani, G.; Paoletti, G.; Appetecchia, M.; Torino, F. Case Report: Ipilimumab-Induced Panhypophysitis: An Infrequent Occurrence and Literature Review. Front. Oncol. 2020, 10, 582394. [Google Scholar] [CrossRef]
- Deligiorgi, M.V.; Siasos, G.; Vergadis, C.; Trafalis, D.T. Central diabetes insipidus related to anti-programmed cell-death 1 protein active immunotherapy. Int. Immunopharmacol. 2020, 83, 106427. [Google Scholar] [CrossRef]
- Yu, M.; Liu, L.; Shi, P.; Zhou, H.; Qian, S.; Chen, K. Anti-PD-1 treatment-induced immediate central diabetes insipidus: A case report. Immunotherapy 2021, 13, 1255–1260. [Google Scholar] [CrossRef]
- Fosci, M.; Pigliaru, F.; Salcuni, A.S.; Ghiani, M.; Cherchi, M.V.; Calia, M.A.; Loviselli, A.; Velluzzi, F. Diabetes insipidus secondary to nivolumab-induced neurohypophysitis and pituitary metastasis. Endocrinol. Diabetes Metab. Case Rep. 2021, 2021, 20-0123. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Tella, S.H.; Del Rivero, J.; Kommalapati, A.; Ebenuwa, I.; Gulley, J.; Strauss, J.; Brownell, I. Anti-PD-L1 Treatment Induced Central Diabetes Insipidus. J. Clin. Endocrinol. Metab. 2018, 103, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Angelousi, A.; Papalexis, P.; Karampela, A.; Marra, M.; Misthos, D.; Ziogas, D.; Gogas, H. Diabetes insipidus: A rare endocrine complication of immune check point inhibitors: A case report and literature review. Exp. Ther. Med. 2023, 25, 10. [Google Scholar] [CrossRef] [PubMed]
- Amereller, F.; Deutschbein, T.; Joshi, M.; Schopohl, J.; Schilbach, K.; Detomas, M.; Duffy, L.; Carroll, P.; Papa, S.; Störmann, S. Differences between immunotherapy-induced and primary hypophysitis-a multicenter retrospective study. Pituitary 2022, 25, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Barnabei, A.; Corsello, A.; Paragliola, R.M.; Iannantuono, G.M.; Falzone, L.; Corsello, S.M.; Torino, F. Immune Checkpoint Inhibitors as a Threat to the Hypothalamus-Pituitary Axis: A Completed Puzzle. Cancers 2022, 14, 1057. [Google Scholar] [CrossRef]
- Caturegli, P.; Di Dalmazi, G.; Lombardi, M.; Grosso, F.; Larman, H.B.; Larman, T.; Taverna, G.; Cosottini, M.; Lupi, I. Hypophysitis Secondary to Cytotoxic T-Lymphocyte-Associated Protein 4 Blockade: Insights into Pathogenesis from an Autopsy Series. Am. J. Pathol. 2016, 186, 3225–3235. [Google Scholar] [CrossRef]
- Iervasi, E.; Strangio, A.; Saverino, D. Hypothalamic expression of PD-L1: Does it mediate hypothalamitis? Cell Mol. Immunol. China 2019, 16, 625–626. [Google Scholar] [CrossRef]
- Christ-Crain, M.; Bichet, D.G.; Fenske, W.K.; Goldman, M.B.; Rittig, S.; Verbalis, J.G.; Verkman, A.S. Diabetes insipidus. Nat. Rev. Dis. Prim. 2019, 5, 54. [Google Scholar] [CrossRef]
- Shoback, D. Clinical practice. Hypoparathyroidism. N. Engl. J. Med. 2008, 359, 391–403. [Google Scholar] [CrossRef]
- Dadu, R.; Rodgers, T.E.; Trinh, V.A.; Kemp, E.H.; Cubb, T.D.; Patel, S.; Simon, J.M.; Burton, E.M.; Tawbi, H. Calcium-sensing receptor autoantibody-mediated hypoparathyroidism associated with immune checkpoint inhibitor therapy: Diagnosis and long-term follow-up. J. Immunother. Cancer 2020, 8, e000687. [Google Scholar] [CrossRef]
- El Kawkgi, O.M.; Li, D.; Kotwal, A.; Wermers, R.A. Hypoparathyroidism: An Uncommon Complication Associated With Immune Checkpoint Inhibitor Therapy. In Mayo Clinic Proceedings: Innovations, Quality & Outcomes; Mayo Foundation for Medical Education and Research; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 4, pp. 821–825. [Google Scholar]
- Win, M.A.; Thein, K.Z.; Qdaisat, A.; Yeung, S.J. Acute symptomatic hypocalcemia from immune checkpoint therapy-induced hypoparathyroidism. Am. J. Emerg. Med. 2017, 35, e1035–e1039. [Google Scholar] [CrossRef] [PubMed]
- Piranavan, P.; Li, Y.; Brown, E.; Kemp, E.H.; Trivedi, N. Immune Checkpoint Inhibitor-Induced Hypoparathyroidism Associated With Calcium-Sensing Receptor-Activating Autoantibodies. J. Clin. Endocrinol. Metab. 2019, 104, 550–556. [Google Scholar] [CrossRef]
- Trinh, B.; Sanchez, G.O.; Herzig, P.; Läubli, H. Inflammation-induced hypoparathyroidism triggered by combination immune checkpoint blockade for melanoma. J. Immunother. Cancer 2019, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Umeguchi, H.; Takenoshita, H.; Inoue, H.; Kurihara, Y.; Sakaguchi, C.; Yano, S.; Hasuzawa, N.; Sakamoto, S.; Sakamoto, R.; Ashida, K. Autoimmune-Related Primary Hypoparathyroidism Possibly Induced by the Administration of Pembrolizumab: A Case Report. J. Oncol. Pract. 2018, 14, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Lupi, I.; Brancatella, A.; Cetani, F.; Latrofa, F.; Kemp, E.H.; Marcocci, C. Activating Antibodies to The Calcium-sensing Receptor in Immunotherapy-induced Hypoparathyroidism. J. Clin. Endocrinol. Metab. 2020, 105, 1581–1588. [Google Scholar] [CrossRef]
- Mahmood, I.; Kuhadiya, N.D.; Gonzalaes, M. Pembrolizumab-Associated Hypoparathyroidism: A Single Case Report. In AACE Clinical Case Reports; Elsevier Inc.: Amsterdam, The Netherlands, 2021; Volume 7, pp. 23–25. [Google Scholar]
- Horinouchi, H.; Yamamoto, N.; Fujiwara, Y.; Sekine, I.; Nokihara, H.; Kubota, K.; Kanda, S.; Yagishita, S.; Wakui, H.; Kitazono, S.; et al. Phase I study of ipilimumab in phased combination with paclitaxel and carboplatin in Japanese patients with non-small-cell lung cancer. Investig. New. Drugs 2015, 33, 881–889. [Google Scholar] [CrossRef]
- Pan, B.; Wang, A.; Pang, J.; Zhang, Y.; Cui, M.; Sun, J.; Liang, Z. Programmed death ligand 1 (PD-L1) expression in parathyroid tumors. Endocr. Connect. 2019, 8, 887–897. [Google Scholar] [CrossRef]
- Bollerslev, J.; Rejnmark, L.; Marcocci, C.; Shoback, D.M.; Sitges-Serra, A.; van Biesen, W.; Dekkers, O.M. European Society of Endocrinology Clinical Guideline: Treatment of chronic hypoparathyroidism in adults. Eur. J. Endocrinol. 2015, 173, G1–G20. [Google Scholar] [CrossRef]
- Husebye, E.S.; Perheentupa, J.; Rautemaa, R.; Kämpe, O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J. Intern. Med. 2009, 265, 514–529. [Google Scholar] [CrossRef]
- Husebye, E.S.; Anderson, M.S.; Kämpe, O. Autoimmune Polyendocrine Syndromes. N. Engl. J. Med. 2018, 378, 1132–1141. [Google Scholar] [CrossRef]
- Khan, A.A.; Koch, C.A.; Van Uum, S.; Baillargeon, J.P.; Bollerslev, J.; Brandi, M.L.; Marcocci, C.; Rejnmark, L.; Rizzoli, R.; Shrayyef, M.Z.; et al. Standards of care for hypoparathyroidism in adults: A Canadian and International Consensus. Eur. J. Endocrinol. 2019, 180, P1–P22. [Google Scholar] [CrossRef] [PubMed]
- Betterle, C.; Garelli, S.; Presotto, F. Diagnosis and classification of autoimmune parathyroid disease. Autoimmun. Rev. 2014, 13, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, C.G.; Chou, S.H.; Mantzoros, C.S. Lipodystrophy: Pathophysiology and advances in treatment. Nat. Rev. Endocrinol. 2011, 7, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Haddad, N.; Vidal-Trecan, T.; Baroudjian, B.; Zagdanski, A.M.; Arangalage, D.; Battistella, M.; Gautier, J.F.; Lebbe, C.; Delyon, J.; PATIO group. Acquired generalized lipodystrophy under immune checkpoint inhibition. Br. J. Dermatol. 2020, 182, 477–480. [Google Scholar] [CrossRef]
- Bedrose, S.; Turin, C.G.; Lavis, V.R.; Kim, S.T.; Thosani, S.N. A case of acquired generalized lipodystrophy associated with pembrolizumab in a patient with metastatic malignant melanoma. AACE Clin. Case Rep. 2020, 6, e40–e45. [Google Scholar] [CrossRef] [PubMed]
- Gnanendran, S.S.; Miller, J.A.; Archer, C.A.; Jain, S.V.; Hwang, S.J.E.; Peters, G.; Miller, A. Acquired lipodystrophy associated with immune checkpoint inhibitors. Melanoma Res. 2020, 30, 599–602. [Google Scholar] [CrossRef]
- Drexler, K.; Zenderowski, V.; Berneburg, M.; Haferkamp, S. Facial lipodystrophy after immunotherapy with Nivolumab. J. Dtsch. Dermatol. Ges. 2021, 19, 1513–1515. [Google Scholar] [CrossRef]
- Jehl, A.; Cugnet-Anceau, C.; Vigouroux, C.; Legeay, A.L.; Dalle, S.; Harou, O.; Marchand, L.; Lascols, O.; Caussy, C.; Thivolet, C.; et al. Acquired Generalized Lipodystrophy: A New Cause of Anti-PD-1 Immune-Related Diabetes. Diabetes Care 2019, 42, 2008–2010. [Google Scholar] [CrossRef]
- Falcao, C.K.; Cabral, M.C.S.; Mota, J.M.; Arbache, S.T.; Costa-Riquetto, A.D.; Muniz, D.Q.B.; Cury-Martins, J.; Almeida, M.Q.; Kaczemorska, P.C.; Nery, M.; et al. Acquired Lipodystrophy Associated With Nivolumab in a Patient With Advanced Renal Cell Carcinoma. J. Clin. Endocrinol. Metab. 2019, 104, 3245–3248. [Google Scholar] [CrossRef]
- Eigentler, T.; Lomberg, D.; Machann, J.; Stefan, N. Lipodystrophic Nonalcoholic Fatty Liver Disease Induced by Immune Checkpoint Blockade. Ann. Intern. Med. USA 2020, 172, 836–837. [Google Scholar] [CrossRef]
- Brown, R.J.; Araujo-Vilar, D.; Cheung, P.T.; Dunger, D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.A.; Patni, N.; Rother, K.I.; et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 4500–4511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, R.; Rong, X.; Zhu, S.; Cui, Y.; Liu, H.; Li, M. Immunoporosis: Role of immune system in the pathophysiology of different types of osteoporosis. Front. Endocrinol. 2022, 13, 965258. [Google Scholar] [CrossRef] [PubMed]
- Moseley, K.F.; Naidoo, J.; Bingham, C.O.; Carducci, M.A.; Forde, P.M.; Gibney, G.T.; Lipson, E.J.; Shah, A.A.; Sharfman, W.H.; Cappelli, L.C. Immune-related adverse events with immune checkpoint inhibitors affecting the skeleton: A seminal case series. J. Immunother. Cancer 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- Filippini, D.M.; Gatti, M.; Di Martino, V.; Cavalieri, S.; Fusaroli, M.; Ardizzoni, A.; Raschi, E.; Licitra, L. Bone fracture as a novel immune-related adverse event with immune checkpoint inhibitors: Case series and large-scale pharmacovigilance analysis. Int. J. Cancer 2021, 149, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Pantano, F.; Tramontana, F.; Iuliani, M.; Leanza, G.; Simonetti, S.; Piccoli, A.; Paviglianiti, A.; Cortellini, A.; Spinelli, G.P.; Longo, U.G.; et al. Changes in bone turnover markers in patients without bone metastases receiving immune checkpoint inhibitors: An exploratory analysis. J. Bone Oncol. 2022, 37, 100459. [Google Scholar] [CrossRef]
- Yamazaki, N.; Kiyohara, Y.; Uhara, H.; Iizuka, H.; Uehara, J.; Otsuka, F.; Fujisawa, Y.; Takenouchi, T.; Isei, T.; Iwatsuki, K.; et al. Cytokine biomarkers to predict antitumor responses to nivolumab suggested in a phase 2 study for advanced melanoma. Cancer Sci. 2017, 108, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Weitzmann, M.N. The Role of Inflammatory Cytokines, the RANKL/OPG Axis, and the Immunoskeletal Interface in Physiological Bone Turnover and Osteoporosis. Scientifica 2013, 2013, 125705. [Google Scholar] [CrossRef]
- Kanis, J.A.; Cooper, C.; Rizzoli, R.; Reginster, J.Y. European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int. 2019, 30, 3–44. [Google Scholar] [CrossRef]
- Garutti, M.; Lambertini, M.; Puglisi, F. Checkpoint inhibitors, fertility, pregnancy, and sexual life: A systematic review. ESMO Open 2021, 6, 100276. [Google Scholar] [CrossRef]
- Brunet-Possenti, F.; Opsomer, M.A.; Gomez, L.; Ouzaid, I.; Descamps, V. Immune checkpoint inhibitors-related orchitis. Ann. Oncol. Engl. 2017, 28, 906–907. [Google Scholar] [CrossRef]
- Quach, H.T.; Robbins, C.J.; Balko, J.M.; Chiu, C.Y.; Miller, S.; Wilson, M.R.; Nelson, G.E.; Johnson, D.B. Severe Epididymo-Orchitis and Encephalitis Complicating Anti-PD-1 Therapy. Oncologist 2019, 24, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, M.J.; Kohn, T.P.; Peña, V.N.; Samarska, I.V.; Matoso, A.; Herati, A.S. Onset of azoospermia in man treated with ipilimumab/nivolumab for BRAF negative metastatic melanoma. In Urology Case Reports; Elsevier Inc.: Amsterdam, The Netherlands, 2021; Volume 34, p. 101488. [Google Scholar]
- Salzmann, M.; Tosev, G.; Heck, M.; Schadendorf, D.; Maatouk, I.; Enk, A.H.; Hartmann, M.; Hassel, J.C. Male fertility during and after immune checkpoint inhibitor therapy: A cross-sectional pilot study. Eur. J. Cancer 2021, 152, 41–48. [Google Scholar] [CrossRef]
- Scovell, J.M.; Benz, K.; Samarska, I.; Kohn, T.P.; Hooper, J.E.; Matoso, A.; Herati, A.S. Association of Impaired Spermatogenesis With the Use of Immune Checkpoint Inhibitors in Patients With Metastatic Melanoma. JAMA Oncol. 2020, 6, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.E.; Nelson, A.; Stimpert, K.; Flyckt, R.L.; Thirumavalavan, N.; Baker, K.C.; Weinmann, S.C.; Hoimes, C.J. Minding the Bathwater: Fertility and Reproductive Toxicity in the Age of Immuno-Oncology. JCO Oncol. Pract. 2022, 18, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Lupu, J.; Pages, C.; Laly, P.; Delyon, J.; Laloi, M.; Petit, A.; Basset-Seguin, N.; Oueslati, I.; Zagdanski, A.M.; Young, J.; et al. Transient pituitary ACTH-dependent Cushing syndrome caused by an immune checkpoint inhibitor combination. Melanoma Res. 2017, 27, 649–652. [Google Scholar] [CrossRef]
- Shalit, A.; Sarantis, P.; Koustas, E.; Trifylli, E.M.; Matthaios, D.; Karamouzis, M.V. Predictive Biomarkers for Immune-Related Endocrinopathies following Immune Checkpoint Inhibitors Treatment. Cancers 2023, 15, 375. [Google Scholar] [CrossRef]


{kind=link}
| Reference | Type of Study, (n) | Age (y) | Sex (M, Male and F, Female) | Malignancy | Drug | ICI Category | Previous Therapies | Laboratory Evaluation | Adrenal Imaging Findings after ICI Initiation (Method) | Grade of AE | Onset after ICI Initiation (Days) | Outcome of AE | Follow-Up (Days) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abdallah et al., 2020 [20] | Case report (n = 1) | 70 | F | Pancreatic adenocarcinoma | Nivolumab | PD-1 Ab | ND | Normal sodium and potassium levels, low F and increased ACTH levels, 21-OH Abs (-) | Normal (ND) | IV | After 3rd dose | ND | 90 |
| Agrawal et al., 2019 [15] | Case report (n = 1) | 59 | M | SCLC | Ipilimumab + Nivolumab | CTLA-4+PD-1 Ab | Lung radiotherapy and chemotherapy | Low F and increased ACTH levels | ND | II | 120 | ND | ND |
| Akarca et al., 2017 [14] | Case report (n = 1) | 52 | M | NSCLC | Nivolumab | PD-1 Ab | ND | Hyponatremia and hyperkalemia, low F and increased ACTH levels | Normal (ND) | IV | 14 | ND | ND |
| Bacanovic et al., 2015 [30] | Case report (n = 1) | 79 | ND | ND | Ipilimumab | CTLA-4 Ab | ND | ND | Symmetrically and smoothly enlarged, hypermetabolic (FDG-PET/CT) | ND | ND | ND | ND |
| Bischoff et al., 2022 [23] | Case report (n = 1) | 53 | F | Melanoma | Pembrolizuma | PD-1 Ab | Surgery | Hyponatremia, hyperkalemia, low F and increased ACTH levels, ACA (+), 21-OH Abs (+) | Normal (FDG-PET/CT) | III-IV | 168 | ND | ND |
| Coskun et al., 2016 [32] | Case report (n = 1) | 50 | M | Lung adenocarcinoma | Nivolumab | PD-1 Ab | ND | Hyponatremia, hyperkalemia, low F and increased ACTH levels | Normal (Ultrasound) | III | 10 | ND | ND |
| Dasgupta et al., 2022 [28] | Case report-APS-2 (n = 1) | 14 | F | Hepatocellular carcinoma | Nivolumab | PD-1 Ab | Chemotherapy | Normal morning F and ACTH levels, 21-OH Abs (+) a | ND | I | ND | ND | ND |
| Deligiorgi et al., 2020 [26] | Case report (n = 1) | 42 | M | Rectal adenocarcinoma | Nivolumab | PD-1 Ab | Surgery and chemotherapy | Hyponatremia, low F and increased ACTH levels, low PAC levels, 21-OH Abs (+) | Normal (CT) | III–IV | 112 | Recovery after 12 weeks | 630 |
| Figuerora-Perez et al., 2021 [29] | Case report (n = 1) | 73 | M | Renal cell carcinoma | Pembrolizumab | PD-1 Ab | Surgery and axitinib | Low F and increased ACTH levels, low PAC levels, high PRA, 21-OH Abs (-) | ND | II | ND | ND | ND |
| Gaballa et al., 2020 [34] | Case report (n = 1) | 76 | M | Melanoma | Ipilimumab | CTLA-4 Ab | None | Hyponatremia with normal potassium, low F and elevated ACTH levels, PAC levels undetectable, increased PRA levels | Normal (CT) | III | After 4 cycles | Recovery | 16 cycles of nivolumab |
| Galliazzo et al., 2022 [38] | Case report (n = 1) | 74 | M | NSCLC | Nivolumab | PD-1 Ab | ND | Hyponatremia, low F and increased ACTH levels, low PAC levels, 21-OHAbs (-) | Normal (CT) | ND | ND | ND | ND |
| Gunjur et al., 2019 [16] | Case report- APS-2 (n = 1) | 78 | F | Melanoma | Pembrolizumab | PD-1 Ab | None | Hyponatremia with normal potassium, Pathological cosyntropin stimulation test (Synacthen), HLA-DRB1*04 genotype (DR4 serotype) | Normal (FDG-PET/CT) | III–IV | 63 | Persistence | 365 |
| Hanna et al., 2018 [37] | Case report (n = 1) | 70 | M | Lung adenocarcinoma | Pembrolizumab | PD-1 Ab | None | Pathological cosyntropin stimulation test (Synacthen) | ND | III–IV | ND | ND | ND |
| Harsch et al., 2020 [21] | Case report (n = 1) | 62 | F | Melanoma | Pembrolizumab | PD-1 Ab | None | Hyponatremia with normal potassium, low F and increased ACTH levels | Inconspicuous adrenal glands (CT) | III–IV | 365 | ND | ND |
| Hescot et al., 2018 [39] | Case report (n = 1) | 33 | F | Cervical squamous cell cancer | Pembrolizumab | PD-1 Ab | ND | Hyponatremia with normal potassium, low F and increased ACTH levels, 21-OH Abs (+) | Adrenal hypoplasia (CT) | III–IV | 147 | Recurrence | 365 |
| Hobbs et al., 2020 [33] | Case report (n = 1) | 58 | M | ND | Ipilimumab + Nivolumab | CTLA-4 Ab+ PD-1 Ab | ND | Hyponatremia with hyperkalemia, low F and increased ACTH levels | ND | III | After 4 cycles | ND | ND |
| Iqbal et al., 2019 [18] | Case report (n = 1) | 65 | F | NCSLC | Nivolumab | PD-1 Ab | ND | Hyponatremia with hyperkalemia, low F and increased ACTH levels, low PAC levels with increased PRA | Normal (CT) | III | ND | Persistence | ND |
| Kagoshima et al., 2019 [19] | Case report (n = 1) | 57 | F | Tongue squamous cell carcinoma | Nivolumab | PD-1 Ab | Surgery, radiotherapy and chemotherapy | Low F and normal ACTH levels, CRH test in favor of PAI | ND | II | ND | ND | ND |
| Knight et al., 2021 [31] | Prospective study (n = 1) | 59 | M | Renal cell carcinoma | Ipilimumab + Nivolumab | CTLA-4 Ab+ PD-1 Ab | ND | Hyponatremia, low F and increased ACTH levels | ND | III–IV | ND | ND | ND |
| Kojadinovic et al., 2021 [36] | Case report (n = 1) | 64 | Μ | Colorectal cancer | Pembrolizumab (9 cycles), pembrolizumab+ Ipilimumab | PD-1 Ab +CTLA-4 Ab | Surgery and multiple cycles of chemotherapy | Hyponatremia and low F levels, pathological cosyntropin stimulation test (Synacthen) | ND | II | 14 after initiation of dual therapy | Persistence | 567 |
| Lanzolla et al., 2019 [17] | Case report- APS-2 (n = 1) | 50 | M | Lung adenocarcinoma | Atezolizumab | PD-L1 Ab | Chemotherapy | Hyponatremia with normal potassium, low F and increased ACTH levels, low PAC levels with increased PRA, 21-OH Abs (+), HLA typing: DRB1*04 and DQB1*03 haplotypes | Normal (CT) | III | 84 | ND | ND |
| Min et al., 2013 [25] | Case report- mixed AI (PAI + SAI) (n = 1) | 56 | F | Melanoma | Ipilimumab | CTLA-4 Ab | ND | Low F and increased ACTH levels | Reversible bilateral enlargement (CT) | II | After 4 doses | ND | ND |
| Ozyurt et al., 2021 [22] | Case report (n = 1) | 66 | M | Renal cell carcinoma | Nivolumab | PD-1 Ab | Sunitinib | Hyponatremia with hyperkalemia, low F and increased ACTH levels, (under steroids) | ND | II | 21 | Persistence | 60 |
| Paepegaey et al., 2017 [13] | Case report-APS-2 (n = 1) | 55 | F | Melanoma | Pembrolizumab | PD-1 Ab | Surgery, chemotherapy and sorafenib | Hyponatremia with hyperkalemia, low F and increased ACTH levels, PAC undetectable with increased PRA levels, ACA (+), 21-OH Abs (+) | Atrophied adrenal glands (CT) | IV | 258 | ND | ND |
| Salinas et al., 2020 [27] | Case series (n = 3) | 60, 65, 76 | M(all) | Renal cell carcinoma(all) | Ipilimumab + nivolumab (n = 1) Nivolumab (n = 2) | -CTLA-4 Ab+ PD-1 Ab -PD-1 Ab (n = 2) | Cabozantinib (n = 1), none (n = 2) | Hyponatremia and pathological cosyntropin stimulation test (Synacthen) (n = 2) Pathological cosyntropin stimulation test (Synacthen) (n = 1) | Normal (n = 3) (CT) | IV IV III | 140 183 150 | ND | ND |
| Shariff et al., 2018 [35] | Case report (n = 1) | 49 | M | Melanoma | Ipilimumab + nivolumab | CTLA-4 Ab+ PD-1 Ab | Chemotherapy | Hyponatremia with hypokalemia, low F levels and increased ACTH levels, low PAC with increased PRA levels, 21-OH Abs (-) | Normal (ND) | III | 74 | Persistence | 420 |
| Trainer et al., 2016 [24] | Case study (n = 1) | 43 | F | Melanoma | Nivolumab | PD-1 Ab | Surgery | Hyponatremia, low F and increased ACTH levels, low PAC with increased PRA levels | Symmetrically and smoothly enlarged, increased FDG activity in both adrenal glands (FDG-PET/CT) | III | 56 | Persistence | 365 |
| Reference | Type of Study, (n) | Age (y) | Sex (M, Male and F, Female) | Malignancy | Drug | ICI Category | Previous Therapies | Dysfunction of Pituitary | Dysfunction of Hypothalamus | Onset after Initiation of ICI (Days) | Outcome of AE | Laboratory Evaluation | MRI Findings | Grade of AE | Follow-Up (Days) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amereller et al., 2021 [65] | Retrospective study (n = 2) | ND | M (n = 1), F (n = 1) | ND | Ipilimumab | CTLA-4 Ab | ND | ND | ND | ND | ND | Serum and urine osmolarity, serum Na, water deprivation test (+) | ND | ND | ND |
| Angelousi et al., 2022 [64] | Case report (n = 1) | 53 | F | Melanoma | Nivolumab | PD-1 Ab | Multiple surgeries | Panhypopituitarism | ND | 240 | Persisted | Low urine osmolality, increased plasma osmolality, water deprivation test (+), low baseline copeptin levels | Absent bright spot | II | 180 |
| Barnabei et al., 2020 [59] | Case report (n = 1) | 64 | M | Melanoma | Ipilimumab | CTLA-4 Ab | Ocular proton beam radiotherapy | Panhypopituitarism | No | 60 | Transient (5 days) | Low urine osmolality, increased plasma osmolality, normal serum Na | Absent bright spot | I | 1230 |
| Brage et al., 2022 [53] | Case report (n = 1) | 46 | M | Adenocarcinoma of the lung | Nivolumab | PD-1 Ab | Whole brain radiotherapy, erlotinib osimertinib and chemotherapy | Panhypopituitarism | No | 62 | ND | Low urine osmolarity, water deprivation test (+) | ND | I | 0 |
| Brilli et al., 2020 [52] | Case report (n = 1) | 68 | M | Mesothelioma | Tremelimumab and durvalumab | CTLA-4 Ab + PD-L1 Ab | None | Isolated posterior pituitary | No | 60 | Persisted | Normal levels of serum sodium, plasma osmolality and urinary specific gravity test, water deprivation test (+) | Normal | ND | 570 |
| Deligiorgi et al., 2020 [60] | Case report (n = 1) | 71 | M | Adenocarcinoma of the lung | Nivolumab | PD-1 Ab | Surgery and chemotherapy | Isolated posterior pituitary | No | 90 | ND | Hypernatremia, high plasma osmolarity and hyposthenuria, undetectable serum AVP | Normal | IV | ND a |
| Dillard et al., 2009 [55] | Case report (n = 1) | 50 | M | Adenocarcinoma of prostate | Ipilimumab | CTLA-4 Ab | ND | Panhypopituitarism | No | 84 | Transient (3 weeks) | ND | Normal | III | ND |
| Fosci et al., 2021 [62] | Case report (n = 1) | 62 | M | Hypopharynx cancer | Nivolumab | PD-1 Ab | Surgery and chemotherapy | Panhypopituitarism | No | 35 | 50 days b | Low urine osmolarity, high plasma osmolality, response to desmopressin | Enlarged stalk | I | 50 b |
| Grami et al., 2019 [54] | Case report (n = 1) | 30 | M | Acute myeloid leukemia | Ipilimumab + nivolumab | CTLA-4 Ab + PD-1 Ab | Chemotherapy and allogenic stem cell transplant | Panhypopituitarism | No | ND | ND | Low urine osmolarity, high serum Na, response to desmopressin | ND | III | ND |
| Gunawan et al., 2018 [57] | Case report (n = 1) | 52 | M | Melanoma | Ipilimumab+ nivolumab | CTLA-4Ab + PD-1 Ab | Small bowel resection | Isolated posterior pituitary | No | 28 | ND | High serum Na, response to desmopressin | ND | I | ND |
| Nallapanemi et al., 2014 [56] | Case report (n = 1) | 62 | M | Melanoma | Ipilimumab | CTLA-4 Ab | Vemurafenib+IL-2 | Panhypopituitarism | No | 121 | 5mo | Water deprivation test (+) | ND | II | 180 |
| Tshuma et al., 2018 [58] | Case report (n = 1) | 74 | F | Bladder cancer | Atezolizumab | PD-L1 Ab | Surgery + neoadjuvant chemotherapy | Panhypopituitarism | Yes | 270 | ΝD | High serum Na, low urinary Na | Hypothalamic mass | I | 365 |
| Yu et al., 2021 [61] | Case report (n = 1) | 60 | M | Hodgkin lymphoma | Sintilimab | PD-1Ab | Chemotherapy | Isolated posterior pituitary | No | Immediate | Transient (3 months) | High serum osmolality, high serum Na, low urine-specific gravity, response to desmopressin | Nodular signal | II | 90 |
| Zhao et al., 2017 [63] | Case report (n = 1) | 73 | M | Merkel cell carcinoma | Avelumab | PD-L1 Ab | None | Isolated posterior pituitary | No | 112 | Transients (6 weeks) | High serum os- molarity, low urine osmolarity, low urine specific gravity, high serum Na, response to desmopressin | Normal | I | 240 |
| Reference | Type of Study | Age (y) | Sex (M, Male and F, Female) | Malignancy | Drug | ICI Category | Previous Therapies | Laboratory Evaluation | Grade of AE | Onset after Initiation of ICI (Days) | Outcome of AE | Follow-Up (Days) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dadu et al., 2020 [71] | Case report (n = 1) | 73 | M | Melanoma | Ipilimumab + nivolumab | CTLA-4Ab + PD-1 Ab | None | Low Ca, P and Mg levels, undetectable PTH levels, low 25-OHD3 CaSR-Abs (+), NALP5 Abs (-), Cytokine Abs (-) | IV | 28 | Persisted | 1185 |
| Horinouchi et al., 2015 [79] | Phase I study (n = 1) | ND | ND | NSCLC | Ipilimumab | CTLA-4 Ab | Chemotherapy | ND | I/II | ND | ND | ND |
| Kawkgi et al., 2020 [72] | Case report (n = 1) | 76 | M | Melanoma | Ipilimumab + nivolumab | CTLA-4Ab + PD-1 Ab | ND | Low Ca, P and Mg levels, undetectable PTH levels, normal 25-OHD3 levels, anti-PTH Abs (-) | III | 220(combination therapy), 160 (nivolumab monotherapy) | Persisted | 77 |
| Lupi et al., 2020 [77] | Case report (n = 1) | 53 | M | Lung adenocarcinoma | Pembrolizumab | PD-1 Ab | ND | Low Ca, normal P and Mg levels, inappropriate normal PTH levels, low25-OHD3 CaSR Abs (+) | IV | 510 | Persisted | 270 |
| Mahmood et al., 2020 [78] | Case report (n = 1) | 71 | M | Lung adenocarcinoma | Pembrolizumab | PD-1 Ab | Surgery and lung radiotherapy | Low Ca and PTH levels | II | 45 | Persisted | 210 |
| Piranavan et al., 2018 [74] | Case report (n = 1) | 61 | F | SCLC | Nivolumab | PD-1 Ab | Chemotherapy and radiotherapy | Low Ca, P and Mg levels, low PTH levels, normal 25-OHD3 levels, CaSR Abs (+), NALP5 Abs (-), Cytokine Abs (-) | IV | 120 | ND | ND |
| Trinh et al., 2019 [75] | Case report (n = 1) | 53 | ND | Melanoma | Ipilimumab + nivolumab | CTLA-4Ab + PD-1 Ab | ND | Low Ca and Mg levels, normal P, normal 25-OHD3 levels, low PTH levels, CaSR Abs insignificant titers | IV | 28 | Persisted | 14 |
| Umeguchi et al., 2018 [76] | Case report (n = 1) | 64 | M | NSCLC | Pembrolizumab | PD-1 Ab | Chemotherapy and lung radiotherapy | Low Ca levels, increased P levels, normal 1,25-(OH)2 D3, low PTH levels, CaSR Abs (+) | III | 42 | Persisted | ND |
| Win et al., 2017 [73] | Case report (n = 1) | 73 | M | Melanoma | Ipilimumab + nivolumab | CTLA-4Ab +PD-1 Ab | Local excision | Low Ca and Mg levels, low 25-OHD3 levels, low PTH levels | IV | 45 | Persisted | 120 |
| Reference | Type of Study, (n) | Age(y) | Sex (M, Male and F, Female) | Malignancy | Drug | ICI Category | Previous Therapies | Type of Lipodystrophy | Laboratory Evaluation | Onset after Initiation of ICI (Days) | Grade of AE | Treatment of AE | Outcome of AE |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bedrose et al., 2020 [88] | Case report (n = 1) | 67 | M | Melanoma | Pembrolizumab | PD-1 Ab | None | Generalized | Hyperglycemia and hypertriglyceridemia, normal values of liver enzymes, low leptin and adiponectin levels | 42 | II | Insulin + pioglitazone, Statin + fibrate+ omega-3 fatty acids | ND |
| Drexler et al., 2021 [90] | Case report (n = 1) | 41 | F | Melanoma | Nivolumab | PD-1 Ab | Inguinal lymph node dissection and local excision | Facial | Normal values of cholesterol, triglycerides, HbA1C | 474 | II | Steroids | Persistence |
| Eigentler et al., 2019 [93] | Case report (n = 1) | 45 | F | Melanoma | Nivolumab | PD-1 Ab | Local excision and IFN-a | Generalized | Hyperglycemia, hypertriglyceridemia, increased liver enzymes | 360 | II | Steroids, insulin and then, overlapping courses of empagliflozin, liraglutide and pioglitazone | Improvement in metabolic abnormalities |
| Gnanendran et al., 2020 [89] | Case report (n = 1) | 34 | F | Melanoma | Nivolumab | PD-1 Ab | Local excision | Generalized | Normal values of glucose, HbA1C, LDL high leptin levels | 270 | II | Steroids | Persistence |
| Haddad et al., 2019 [87] | Case report (n = 1) | 47 | F | Melanoma | Pembrolizumab | PD-1 Ab | None | Generalized | Prediabetes, low leptin and adiponectin levels, hypertriglyceridemia | 60 | III | Treatment for metabolic abnormalities | Persistence |
| Jehl et al., 2019 [91] | Case report (n = 1) | 62 | F | Melanoma | Nivolumab | PD-1 Ab | Local excision | Generalized | DM, hypertriglyceridemia, increased liver enzymes, low leptin and adiponectin levels | 540 | III | Insulin + metfromin | Improvement in metabolic abnormalities |
| Kruschewsky Falcao et al., 2019 [92] | Case report (n = 1) | 57 | F | Renal cell carcinoma | Nivolumab | PD-1 Ab | Local excision and sunitinib, pazobanib | Generalized | DM, hypertriglyceridemia, high LDL, low leptin levels | 60 | II | Steroids | Improvement in metabolic abnormalities |
| Reference | Type of Study | Age (y) | Sex (M, Male and F, Female) | Malignancy | Drug/ICI Category | Previous Therapies | Skeletal AE | Laboratory Evaluation | Grade of AE | Onset after ICI Initiation |
|---|---|---|---|---|---|---|---|---|---|---|
| Filippini et al., 2021 [97] | Case series (n = 4) | 67.8 (mean age) | M (n = 1), F (n = 3) | Squamous cell carcinoma (n = 4) | Anti-PD-1 Ab (n = 2) Anti-PD-L1 Ab (n = 2) | ND | Dorsal vertebral (D12) fracture Calcaneal fracture Lumbar vertebral (L1) fracture Multiple vertebral (D7-L5) fractures | ND | II | From 2.5 to 15.5 months |
| Moseley et al., 2018 [96] | Case series (n = 6) | 59.3 (mean age) | M (n = 5), F (n = 1) | Melanoma (n = 4), RCC (n = 1), lung adenocarcinoma (n = 1) | Pembrolizumab/PD-1 Ab (n = 2), Nivolumab/PD-1 Ab (n = 2), Nivolumab + ipilimumab/PD-1 Ab + CTLA-4 Ab (n = 2) | Wide local excision + axillary lymph node dissection + GM-CSF secreting allogeneic melanoma cell vaccine (n = 1) wide local excision + IFN-a+IL-2 (n = 1), none (n = 4) | 1st patient: compression vertebral fractures (T6, T7, T10, T11, and T12); rib and pelvic fractures 2nd patient: compression vertebral fractures (T6–12, L1) 3rd patient: compression vertebral fracture (T11); lumbar osteomalacia 4th patient: Resorptive bone lesion of left shoulder 5th patient: Resorptive bone lesion of right wrist 6th patient: Resorptive bone lesion of right clavicle | Elevated or high normal CTX and/or bsALP levels (n = 5), Elevated CRP and/or ESR (n = 6) | II | 1st patient: After 20 doses of pembrolizumab therapy 2nd patient: 8 cycles of nivolumab and IL-21 3rd patient: 10 months 4th patient: 8 months 5th patient: 18 months 6th patient: ND |
| Pantano et al., 2022 [98] | Case series (n = 4) | ND | ND | ND | ND | ND | Lumbar fractures | Relatively increased CTX-1 levels 1 Relatively decreased PINP levels 1 | II | ND |
| Reference | Type of Study, (n) | Age (y) | Sex (M, Male and F, Female) | Malignancy | Drug | ICI Category | Previous Therapies | Clinical Presentation | Laboratory Evaluation | Testicular Biopsy | Onset after Initiation of ICI (Days) | Duration of AE (Days) | Follow up (Days) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brunet-Possenti et al., 2016 [103] | Case report (n = 1) | 54 | M | Melanoma | Ipilimumab + nivolumab | CTLA-4 Ab+ PD-1 Ab | ND | Bilateral orchitis | Low testosterone with high LH levels | ND | 14 | 7 | 28 |
| Quach et al., 2019 [104] | Case report (n = 1) | 69 | M | Melanoma | Pembrolizumab | PD-1 Ab | Partial hepatectomy and RFA of liver lesions. | Bilateral epididymo-orchitis | ND | ND | 60 | 35 | 80 |
| Rabinowitz et al., 2021 [105] | Case report (n = 1) | 30 | M | Melanoma | Ipilimumab + nivolumab | CTLA-4 Ab+ PD-1 Ab | None | Infertility | Spermogram: Azoospermia, Hormone profile: Low testosterone levels with high FSH and normal LH levels | Sertoli-only pathology | 730 (time of evaluation) | Persisted | 180 |
| Salzamann et al., 2021 [106] | Cross-sectional pilot study (n = 4) | 44, 51, 30, 36 | M | ND | Ipilimumab + nivolumab (n = 2), Pembrolizumab(n = 1), PD-L1 Ab (n = 1) | CTLA-4 Ab+ PD-1 Ab (n = 2), PD-1 Ab (n = 1), PD-L1 Ab (n = 1) | RT to inguinal lymph nodes (n = 1), Chemotherapy (4 years before) (n = 1), ND (n = 2) | None | Spermogram: Azoospermia (n = 3), Oligoasthenoteratozoospermia (n = 1) Hormone profile: normal (n = 2), high FSH levels (n = 2) | No signs of inflammation (n = 2), Inflammation infiltrate (n = 2) | >120 | ND | ND |
| Scovell et al., 2020 [107] | Cohort study (n = 6) | ND | M | Melanoma | Ipilimumab/nivolumab/pembrolizumab | CTLA-4 Ab/ PD-1 Ab | None | ND | ND | Sertoli-only syndrome (n = 3), focal active spermatogenesis (n = 1), hypospermatogenesis (n = 2) | ND | ND | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mytareli, C.; Ziogas, D.C.; Karampela, A.; Papalexis, P.; Siampanopoulou, V.; Lafioniatis, A.; Benopoulou, O.; Gogas, H.; Angelousi, A. The Uncharted Landscape of Rare Endocrine Immune-Related Adverse Events. Cancers 2023, 15, 2016. https://doi.org/10.3390/cancers15072016
Mytareli C, Ziogas DC, Karampela A, Papalexis P, Siampanopoulou V, Lafioniatis A, Benopoulou O, Gogas H, Angelousi A. The Uncharted Landscape of Rare Endocrine Immune-Related Adverse Events. Cancers. 2023; 15(7):2016. https://doi.org/10.3390/cancers15072016
Chicago/Turabian StyleMytareli, Chrysoula, Dimitrios C. Ziogas, Athina Karampela, Petros Papalexis, Vasiliki Siampanopoulou, Alexandros Lafioniatis, Olga Benopoulou, Helen Gogas, and Anna Angelousi. 2023. "The Uncharted Landscape of Rare Endocrine Immune-Related Adverse Events" Cancers 15, no. 7: 2016. https://doi.org/10.3390/cancers15072016
APA StyleMytareli, C., Ziogas, D. C., Karampela, A., Papalexis, P., Siampanopoulou, V., Lafioniatis, A., Benopoulou, O., Gogas, H., & Angelousi, A. (2023). The Uncharted Landscape of Rare Endocrine Immune-Related Adverse Events. Cancers, 15(7), 2016. https://doi.org/10.3390/cancers15072016