


Chromatin and Cancer: Implications of Disrupted Chromatin Organization in Tumorigenesis and Its Diversification


Simple Summary
Abstract
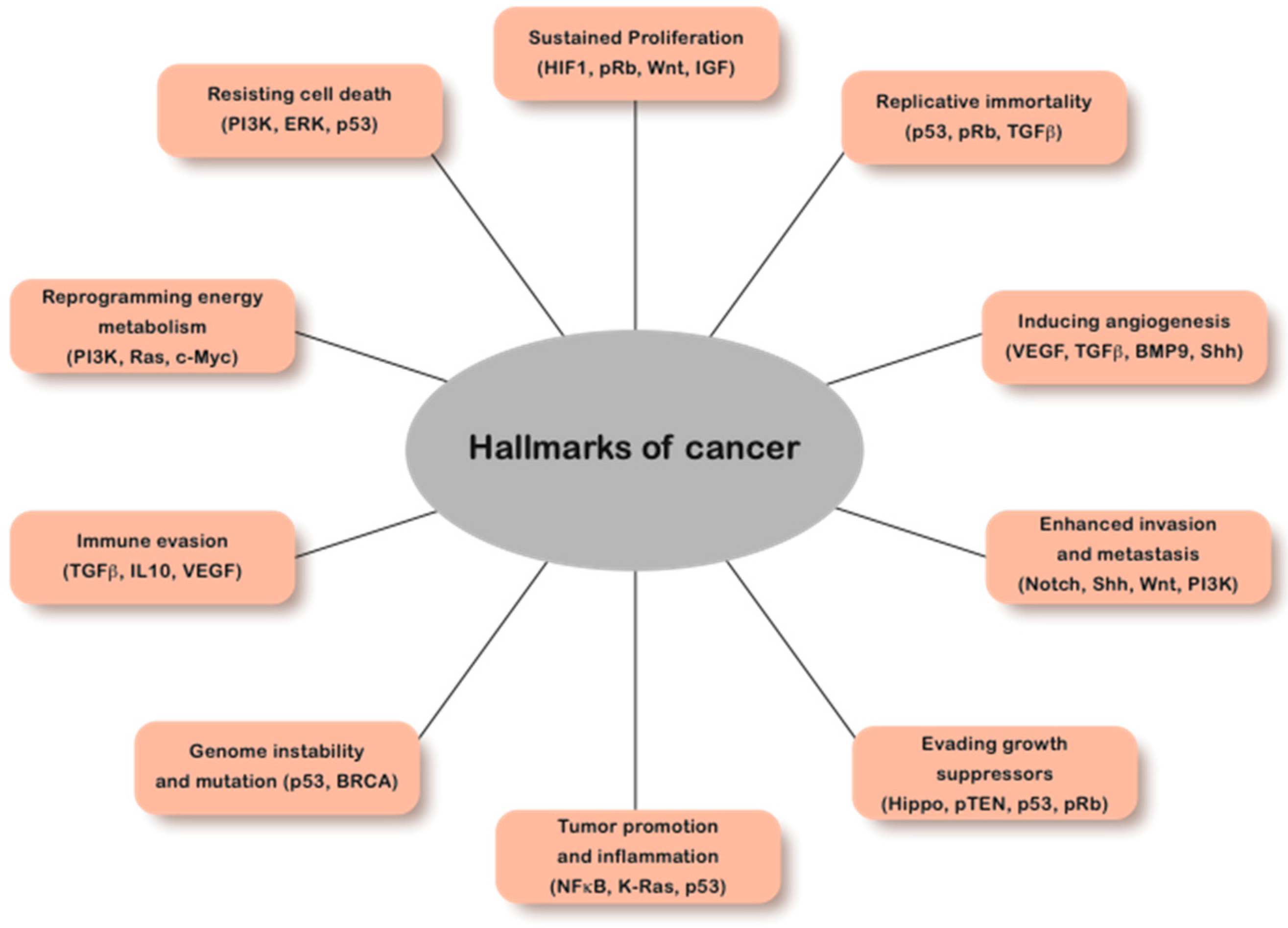
1. Introduction
2. Chromatin: The Fundamental Unit of Nuclear Functionality
3. Chromatin Organization and Function during Interphase
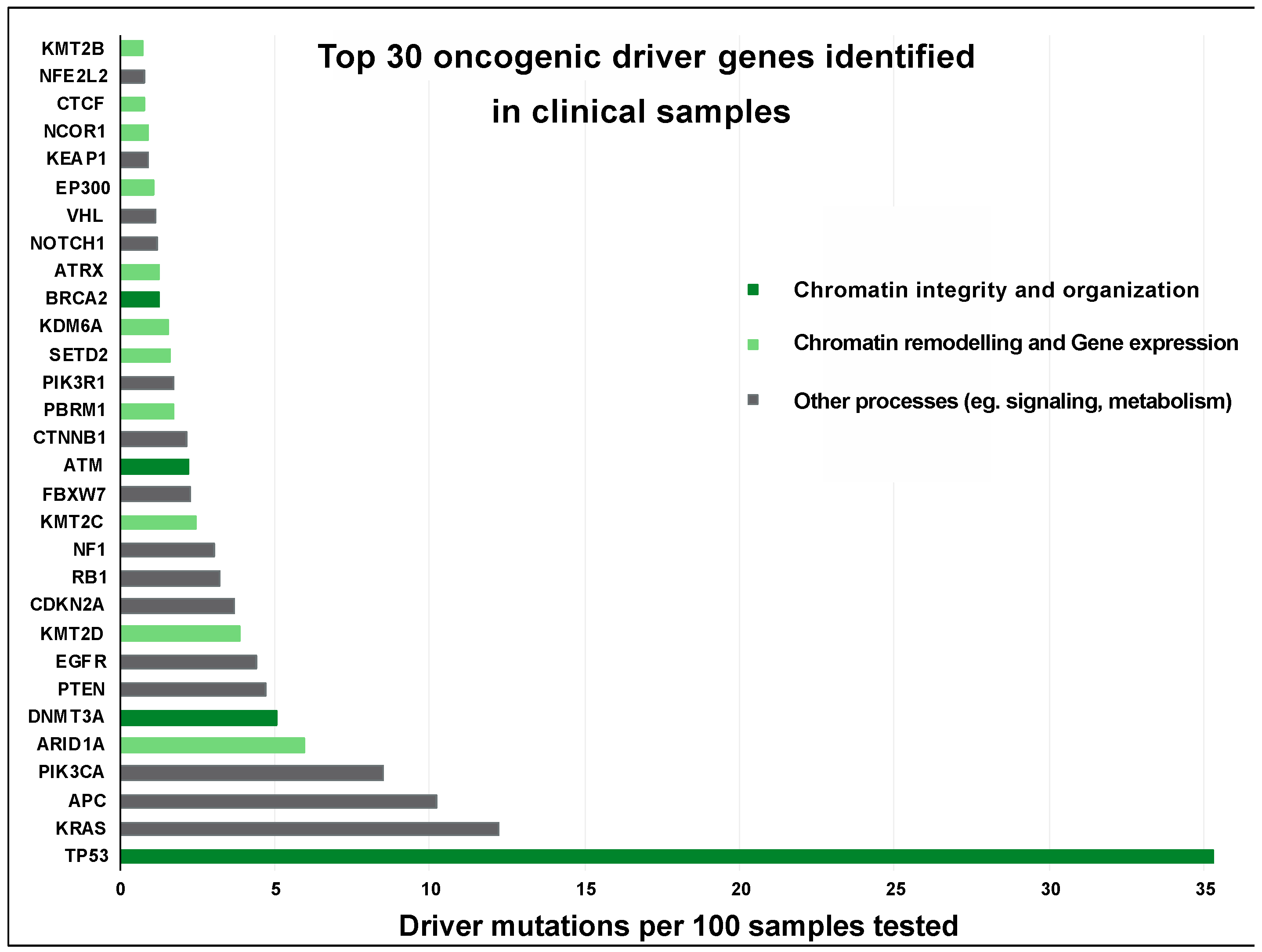
4. Pathological Consequences of Chromatin Abnormalities
4.1. Reactivation of X-Chromosome (XaXa) and Cancer
4.2. Chromosomal Rearrangements Induced by Repetitive Elements
4.3. Altered Long-Range Chromatin Contacts in Cancer
4.4. Altered DNA Methylation in Cancer
4.5. Chromatin and Alternative-Splicing of Pre-mRNA
5. Impact of Chromatin Disorganization on Cancer Progression
5.1. Diversification of Tumors by Mislocalized or Disorganized Chromatin
5.2. Aneuploidy and Evasion of Therapeutic Interventions
5.3. Increased Invasive Potential of Tumors
6. Targeted Cancer Therapeutics Aimed at Chromatin Modifiers
6.1. Direct Inhibition of Epigenetic Modifiers
6.2. Cancer-Associated Metabolic Enzymes and Metabolites
6.3. Other Chromatin Modulators in Cancer Therapeutics
6.4. Targeted Degradation of Proteins
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, N.; Erdal, E.; Mumcuoglu, M.; Akcali, K.C.; Yalcin, O.; Senturk, S.; Arslan-Ergul, A.; Gur, B.; Yulug, I.; Cetin-Atalay, R.; et al. Reprogramming of Replicative Senescence in Hepatocellular Carcinoma-Derived Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 2178–2183. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.Y.K.; Papa, A. Signaling Pathways in Cancer: Therapeutic Targets, Combinatorial Treatments, and New Developments. Cells 2021, 10, 659. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained Proliferation in Cancer: Mechanisms and Novel Therapeutic Targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef] [PubMed]
- Harry, J.A.; Ormiston, M.L. Novel Pathways for Targeting Tumor Angiogenesis in Metastatic Breast Cancer. Front. Oncol. 2021, 11, 772305. [Google Scholar] [CrossRef]
- Amin, A.R.M.R.; Karpowicz, P.A.; Carey, T.E.; Arbiser, J.; Nahta, R.; Chen, Z.G.; Dong, J.-T.T.; Kucuk, O.; Khan, G.N.; Huang, G.S.; et al. Evasion of Anti-Growth Signaling: A Key Step in Tumorigenesis and Potential Target for Treatment and Prophylaxis by Natural Compounds. Semin. Cancer Biol. 2015, 35, S55–S77. [Google Scholar] [CrossRef]
- Fernald, K.; Kurokawa, M. Evading Apoptosis in Cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic Instability-an Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Maeshima, K.; Iida, S.; Tamura, S. Physical Nature of Chromatin in the Nucleus. Cold Spring Harb. Perspect. Med. 2021, 13, a040675. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 340, 1546–1558. [Google Scholar] [CrossRef]
- Carone, D.M.; Lawrence, J.B. Heterochromatin Instability in Cancer: From the Barr Body to Satellites and the Nuclear Periphery. Semin. Cancer Biol. 2013, 23, 99–108. [Google Scholar] [CrossRef]
- Sengupta, S.; Rani, E. George Super-Enhancer-Driven Transcriptional Dependencies in Cancer. Trends Cancer 2017, 3, 269–281. [Google Scholar] [CrossRef]
- Reddy, K.L.; Feinberg, A.P. Higher Order Chromatin Organization in Cancer. Semin. Cancer Biol. 2013, 23, 109–115. [Google Scholar] [CrossRef]
- Luger, K.; Hansen, J.C. Nucleosome and Chromatin Fiber Dynamics. Curr. Opin. Struct. Biol. 2005, 15, 188–196. [Google Scholar] [CrossRef]
- Blank, T.A.; Becker, P.B. Electrostatic Mechanism of Nucleosome Spacing. J. Mol. Biol. 1995, 252, 305–313. [Google Scholar] [CrossRef]
- Dueva, R.; Akopyan, K.; Pederiva, C.; Trevisan, D.; Dhanjal, S.; Lindqvist, A.; Farnebo, M. Neutralization of the Positive Charges on Histone Tails by RNA Promotes an Open Chromatin Structure. Cell Chem. Biol. 2019, 26, 1436–1449.e5. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Berger, F. Emil Heitz, a True Epigenetics Pioneer. Nat. Rev. Mol. Cell Biol. 2019, 20, 572. [Google Scholar] [CrossRef]
- Allshire, R.C.; Madhani, H.D. Ten Principles of Heterochromatin Formation and Function. Nat. Rev. Mol. Cell Biol. 2018, 19, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten Things You Should Know about Transposable Elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Filion, G.J.; van Bemmel, J.G.; Braunschweig, U.; Talhout, W.; Kind, J.; Ward, L.D.; Brugman, W.; de Castro, I.J.; Kerkhoven, R.M.; Bussemaker, H.J.; et al. Systematic Protein Location Mapping Reveals Five Principal Chromatin Types in Drosophila Cells. Cell 2010, 143, 212–224. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.; Wang, Y.; Zhang, L.; Brinkman, E.K.; Adam, S.A.; Goldman, R.; Van Steensel, B.; Ma, J.; Belmont, A.S. Mapping 3D Genome Organization Relative to Nuclear Compartments Using TSA-Seq as a Cytological Ruler. J. Cell Biol. 2018, 217, 4025–4048. [Google Scholar] [CrossRef] [PubMed]
- Quinodoz, S.A.; Ollikainen, N.; Tabak, B.; Palla, A.; Schmidt, J.M.; Detmar, E.; Lai, M.M.; Shishkin, A.A.; Bhat, P.; Takei, Y.; et al. Higher-Order Inter-Chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 2018, 174, 744–757.e24. [Google Scholar] [CrossRef]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain Organization of Human Chromosomes Revealed by Mapping of Nuclear Lamina Interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef]
- Kind, J.; Pagie, L.; Ortabozkoyun, H.; Boyle, S.; De Vries, S.S.; Janssen, H.; Amendola, M.; Nolen, L.D.; Bickmore, W.A.; Van Steensel, B. Single-Cell Dynamics of Genome-Nuclear Lamina Interactions. Cell 2013, 153, 178–192. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Parnaik, V.K. Lamin A Rod Domain Mutants Target Heterochromatin Protein 1α and β for Proteasomal Degradation by Activation of F-Box Protein, FBXW10. PLoS ONE 2010, 5, e10620. [Google Scholar] [CrossRef]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and Lamin A/C Sequentially Tether Peripheral Heterochromatin and Inversely Regulate Differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef]
- Lemaître, C.; Bickmore, W.A. Chromatin at the Nuclear Periphery and the Regulation of Genome Functions. Histochem. Cell Biol. 2015, 144, 111–122. [Google Scholar] [CrossRef]
- Parnaik, V.K.; Chaturvedi, P.; Muralikrishna, B. Lamins, Laminopathies and Disease Mechanisms: Possible Role for Proteasomal Degradation of Key Regulatory Proteins. J. Biosci. 2011, 36, 471–479. [Google Scholar] [CrossRef]
- Parnaik, V.K.; Chaturvedi, P. Fluorescence Recovery after Photobleaching Studies Reveal Complexity of Nuclear Architecture. Int. J. Chem. 2015, 4, 297–302. [Google Scholar]
- Thanumalayan, S.; Sehgal, P.; Muralikrishna, B.; Ajay, G.; Rani, D.S.; Govindaraj, P.; Khullar, M.; Bahl, A.; Thangaraj, K.; Parnaik, V.K. A Rare Mutation in Lamin A Gene Is Associated with Dilated Cardiomyopathy in Indian Patients. Eur. J. Mol. Biol. Biochem. 2015, 2, 190–196. [Google Scholar]
- Sehgal, P.; Chaturvedi, P.; Kumaran, R.I.; Kumar, S.; Parnaik, V.K. Lamin A/C Haploinsufficiency Modulates the Differentiation Potential of Mouse Embryonic Stem Cells. PLoS ONE 2013, 8, e57891. [Google Scholar] [CrossRef]
- Sakthivel, K.M.; Sehgal, P. A Novel Role of Lamins from Genetic Disease to Cancer Biomarkers. Oncol. Rev. 2016, 10, 65–71. [Google Scholar] [CrossRef]
- Somsuan, K.; Peerapen, P.; Boonmark, W.; Plumworasawat, S.; Samol, R.; Sakulsak, N.; Thongboonkerd, V. ARID1A Knockdown Triggers Epithelial-Mesenchymal Transition and Carcinogenesis Features of Renal Cells: Role in Renal Cell Carcinoma. FASEB J. 2019, 33, 12226–12239. [Google Scholar] [CrossRef]
- Douet, J.; Corujo, D.; Malinverni, R.; Renauld, J.; Sansoni, V.; Posavec Marjanović, M.; Cantariño, N.; Valero, V.; Mongelard, F.; Bouvet, P.; et al. MacroH2A Histone Variants Maintain Nuclear Organization and Heterochromatin Architecture. J. Cell Sci. 2017, 130, 1570–1582. [Google Scholar] [CrossRef]
- Lee, S.; Ahn, Y.M.; Kim, J.Y.; Cho, Y.E.; Park, J.H. Downregulation of NOP53 Ribosome Biogenesis Factor Leads to Abnormal Nuclear Division and Chromosomal Instability in Human Cervical Cancer Cells. Pathol. Oncol. Res. 2020, 26, 453–459. [Google Scholar] [CrossRef]
- Rajshekar, S.; Yao, J.; Arnold, P.K.; Payne, S.G.; Zhang, Y.; Bowman, T.V.; Schmitz, R.J.; Edwards, J.R.; Goll, M. Pericentromeric Hypomethylation Elicits an Interferon Response in an Animal Model of ICF Syndrome. Elife 2018, 7, e39658. [Google Scholar] [CrossRef]
- Wazir, U.; Ahmed, M.H.; Bridger, J.M.; Harvey, A.; Jiang, W.G.; Sharma, A.K.; Mokbel, K. The Clinicopathological Significance of Lamin A/C, Lamin B1 and Lamin B Receptor MRNA Expression in Human Breast Cancer. Cell. Mol. Biol. Lett. 2013, 18, 595–611. [Google Scholar] [CrossRef]
- Jia, Y.; Vong, J.S.-L.; Asafova, A.; Garvalov, B.K.; Caputo, L.; Cordero, J.; Singh, A.; Boettger, T.; Günther, S.; Fink, L.; et al. Lamin B1 Loss Promotes Lung Cancer Development and Metastasis by Epigenetic Derepression of RET. J. Exp. Med. 2019, 216, 1377–1395. [Google Scholar] [CrossRef]
- Graziano, S.; Kreienkamp, R.; Coll-Bonfill, N.; Gonzalo, S. Causes and Consequences of Genomic Instability in Laminopathies: Replication Stress and Interferon Response. Nucleus 2018, 9, 258–275. [Google Scholar] [CrossRef]
- Bell, E.S.; Lammerding, J. Causes and Consequences of Nuclear Envelope Alterations in Tumour Progression. Eur. J. Cell Biol. 2016, 95, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.J.L.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.H.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.B.; et al. Heritable Somatic Methylation and Inactivation of MSH2 in Families with Lynch Syndrome Due to Deletion of the 3′ Exons of TACSTD1. Nat. Genet. 2009, 41, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Capo-chichi Callinice, D.; Cai Kathy, Q.; Testa Joseph, R.; Godwin Andrew, K.; Xiang-Xi, X. Loss of GATA6 Leads to Nuclear Deformation and Aneuploidy in Ovarian Cancer. Mol. Cell. Biol. 2009, 29, 4766–4777. [Google Scholar] [CrossRef] [PubMed]
- Muralikrishna, B.; Chaturvedi, P.; Sinha, K.; Parnaik, V.K. Lamin Misexpression Upregulates Three Distinct Ubiquitin Ligase Systems That Degrade ATR Kinase in HeLa Cells. Mol. Cell. Biochem. 2012, 365, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Liddane, A.G.; Holaska, J.M. The Role of Emerin in Cancer Progression and Metastasis. Int. J. Mol. Sci. 2021, 22, 11289. [Google Scholar] [CrossRef]
- Young, A.N.; Perlas, E.; Ruiz-Blanes, N.; Hierholzer, A.; Pomella, N.; Martin-Martin, B.; Liverziani, A.; Jachowicz, J.W.; Giannakouros, T.; Cerase, A. Deletion of LBR N-Terminal Domains Recapitulates Pelger-Huet Anomaly Phenotypes in Mouse without Disrupting X Chromosome Inactivation. Commun. Biol. 2021, 4, 478. [Google Scholar] [CrossRef]
- Hoffmann, K.; Dreger, C.K.; Olins, A.L.; Olins, D.E.; Shultz, L.D.; Lucke, B.; Karl, H.; Kaps, R.; Müller, D.; Vayá, A.; et al. Mutations in the Gene Encoding the Lamin B Receptor Produce an Altered Nuclear Morphology in Granulocytes (Pelger-Huët Anomaly). Nat. Genet. 2002, 31, 410–414. [Google Scholar] [CrossRef]
- Tufarelli, C.; Frischauf, A.M.; Hardison, R.; Flint, J.; Higgs, D.R. Characterization of a Widely Expressed Gene (LUC7-LIKE; LUC7L) Defining the Centromeric Boundary of the Human α-Globin Domain. Genomics 2001, 71, 307–314. [Google Scholar] [CrossRef][Green Version]
- Mancuso, A. Evidence-Based Medicine and Management of Hepatocellular Carcinoma in Thalassemia. BMC Gastroenterol. 2020, 20, 2–4. [Google Scholar] [CrossRef]
- Sugawara, S.; Okada, R.; Loo, T.M.; Tanaka, H.; Miyata, K.; Chiba, M.; Kawasaki, H.; Katoh, K.; Kaji, S.; Maezawa, Y.; et al. RNaseH2A Downregulation Drives Inflammatory Gene Expression via Genomic DNA Fragmentation in Senescent and Cancer Cells. Commun. Biol. 2022, 5, 1420. [Google Scholar] [CrossRef]
- Pageau, G.J.; Hall, L.L.; Ganesan, S.; Livingston, D.M.; Lawrence, J.B. The Disappearing Barr Body in Breast and Ovarian Cancers. Nat. Rev. Cancer 2007, 7, 628–633. [Google Scholar] [CrossRef]
- Ghosh, S.N.; Shah, P.N. Probable Mechanism for the Loss of Barr Body in Human Female Tumor with Special Reference to Breast Cancer. Med. Hypotheses 1981, 7, 1099–1104. [Google Scholar] [CrossRef]
- Hultborn, R.; Hanson, C.; Köpf, I.; Verbiené, I.; Warnhammar, E.; Weimarck, A. Prevalence of Klinefelter’s Syndrome in Male Breast Cancer Patients. Anticancer Res. 1997, 17, 4293–4297. [Google Scholar]
- Thakur, A.; Xu, H.; Wang, Y.; Bollig, A.; Biliran, H.; Liao, J.D. The Role of X-Linked Genes in Breast Cancer. Breast Cancer Res. Treat. 2005, 93, 135–143. [Google Scholar] [CrossRef]
- Tricarico, R.; Nicolas, E.; Hall, M.J.; Golemis, E.A. X- and Y-Linked Chromatin-Modifying Genes as Regulators of Sex-Specific Cancer Incidence and Prognosis. Clin. Cancer Res. 2020, 26, 5567–5578. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, O.S. The CBio Cancer Genomics. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal Complementary Data Sources and Analysis Options. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Xu, S.-J.; Wang, Y.-P.; Roe, B.; Pearson, W.R. Characterization of the Human Class Mu Glutathione S-Transferase Gene Cluster and the GSTM1 Deletion*. J. Biol. Chem. 1998, 273, 3517–3527. [Google Scholar] [CrossRef]
- Saitou, M.; Gokcumen, O. An Evolutionary Perspective on the Impact of Genomic Copy Number Variation on Human Health. J. Mol. Evol. 2020, 88, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Rothman, N.; Garcia-Closas, M.; Chatterjee, N.; Malats, N.; Wu, X.; Figueroa, J.D.; Real, F.X.; Van Den Berg, D.; Matullo, G.; Baris, D.; et al. A Multi-Stage Genome-Wide Association Study of Bladder Cancer Identifies Multiple Susceptibility Loci. Nat. Genet. 2010, 42, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Yunis, J.J.; Soreng, A.L. Constitutive Fragile Sites and Cancer. Science 1984, 226, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Penny, L.; Mattina, T.; Yu, S.; Baker, E.; Voullaire, L.; Langdon, W.Y.; Sutherland, G.R.; Richards, R.I.; Tunnacliffe, A. Association of a Chromosome Deletion Syndrome with a Fragile Site within the Proto-Oncogene CBL2. Nature 1995, 376, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Drier, Y.; Cotton, M.J.; Williamson, K.E.; Gillespie, S.M.; Ryan, R.J.H.; Kluk, M.J.; Carey, C.D.; Rodig, S.J.; Sholl, L.M.; Afrogheh, A.H.; et al. An Oncogenic MYB Feedback Loop Drives Alternate Cell Fates in Adenoid Cystic Carcinoma. Nat. Genet. 2016, 48, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Montefiori, L.E.; Bendig, S.; Gu, Z.; Chen, X.; Pölönen, P.; Ma, X.; Murison, A.; Zeng, A.; Garcia-Prat, L.; Dickerson, K.; et al. Enhancer Hijacking Drives Oncogenic BCL11B Expression in Lineage-Ambiguous Stem Cell Leukemia. Cancer Discov. 2021, 11, 2846–2867. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Imielinski, M.; Watanabe, H.; Cherniack, A.D.; Meyerson, M. Identification of Focally Amplified Lineage-Specific Super-Enhancers in Human Epithelial Cancers. Nat. Genet. 2016, 48, 176–182. [Google Scholar] [CrossRef]
- Deng, R.; Huang, J.H.; Wang, Y.; Zhou, L.H.; Wang, Z.F.; Hu, B.X.; Chen, Y.H.; Yang, D.; Mai, J.; Li, Z.L.; et al. Disruption of Super-Enhancer-Driven Tumor Suppressor Gene RCAN1.4 Expression Promotes the Malignancy of Breast Carcinoma. Mol. Cancer 2020, 19, 122. [Google Scholar] [CrossRef]
- Zhou, H.; Schmidt, S.C.S.; Jiang, S.; Willox, B.; Bernhardt, K.; Liang, J.; Johannsen, E.C.; Kharchenko, P.; Gewurz, B.E.; Kieff, E.; et al. Epstein-Barr Virus Oncoprotein Super-Enhancers Control B Cell Growth. Cell Host Microbe 2015, 17, 205–216. [Google Scholar] [CrossRef]
- Rhie, S.K.; Perez, A.A.; Lay, F.D.; Schreiner, S.; Shi, J.; Polin, J.; Farnham, P.J. A High-Resolution 3D Epigenomic Map Reveals Insights into the Creation of the Prostate Cancer Transcriptome. Nat. Commun. 2019, 10, 4154. [Google Scholar] [CrossRef]
- Vilarrasa-Blasi, R.; Soler-Vila, P.; Verdaguer-Dot, N.; Russiñol, N.; Di Stefano, M.; Chapaprieta, V.; Clot, G.; Farabella, I.; Cuscó, P.; Kulis, M.; et al. Dynamics of Genome Architecture and Chromatin Function during Human B Cell Differentiation and Neoplastic Transformation. Nat. Commun. 2021, 12, 651. [Google Scholar] [CrossRef]
- Adeel, M.M.; Jiang, H.; Arega, Y.; Cao, K.; Lin, D.; Cao, C.; Cao, G.; Wu, P.; Li, G. Structural Variations of the 3D Genome Architecture in Cervical Cancer Development. Front. Cell Dev. Biol. 2021, 9, 1–12. [Google Scholar] [CrossRef]
- Kim, T.; Han, S.; Chun, Y.; Yang, H.; Min, H.; Jeon, S.Y.; Kim, J.; Moon, H.-G.; Lee, D. Comparative Characterization of 3D Chromatin Organization in Triple-Negative Breast Cancers. Exp. Mol. Med. 2022, 54, 585–600. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA Methylation and Human Disease. Nat. Rev. Genet. 2005 68 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Narayan, A.; Ji, W.; Zhang, X.-Y.; Marrogi, A.; Graff, J.R.; Baylin, S.B.; Ehrlich, M. Hypomethylation of Pericentromeric DNA in Breast Adenocarcinomas. Int. J. Cancer 1998, 77, 833–838. [Google Scholar] [CrossRef]
- Qu, G.Z.; Dubeau, L.; Narayan, A.; Yu, M.C.; Ehrlich, M. Satellite DNA Hypomethylation vs. Overall Genomic Hypomethylation in Ovarian Epithelial Tumors of Different Malignant Potential. Mutat. Res. 1999, 423, 91–101. [Google Scholar] [CrossRef]
- Pappalardo, X.G.; Barra, V. Losing DNA Methylation at Repetitive Elements and Breaking Bad. Epigenet. Chromatin 2021, 14, 25. [Google Scholar] [CrossRef]
- Dumbović, G.; Biayna, J.; Banús, J.; Samuelsson, J.; Roth, A.; Diederichs, S.; Alonso, S.; Buschbeck, M.; Perucho, M.; Forcales, S.V. A Novel Long Non-Coding RNA from NBL2 Pericentromeric Macrosatellite Forms a Perinucleolar Aggregate Structure in Colon Cancer. Nucleic Acids Res. 2018, 46, 5504–5524. [Google Scholar] [CrossRef]
- Jintaridth, P.; Mutirangura, A. Distinctive Patterns of Age-Dependent Hypomethylation in Interspersed Repetitive Sequences. Physiol. Genom. 2010, 41, 194–200. [Google Scholar] [CrossRef]
- Costello, J.F.; Frühwald, M.C.; Smiraglia, D.J.; Rush, L.J.; Robertson, G.P.; Gao, X.; Wright, F.A.; Feramisco, J.D.; Peltomäki, P.; Lang, J.C.; et al. Aberrant CpG-Island Methylation Has Non-Random and Tumour-Type-Specific Patterns. Nat. Genet. 2000, 24, 132–138. [Google Scholar] [CrossRef]
- Bouras, E.; Karakioulaki, M.; Bougioukas, K.I.; Aivaliotis, M.; Tzimagiorgis, G.; Chourdakis, M. Gene Promoter Methylation and Cancer: An Umbrella Review. Gene 2019, 710, 333–340. [Google Scholar] [CrossRef] [PubMed]
- El Aliani, A.; El-Abid, H.; El Mallali, Y.; Attaleb, M.; Ennaji, M.M.; El Mzibri, M. Association between Gene Promoter Methylation and Cervical Cancer Development: Global Distribution and A Meta-Analysis. Cancer Epidemiol. Biomark. Prev. 2021, 30, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Nowacka-Zawisza, M.; Wiśnik, E. DNA Methylation and Histone Modifications as Epigenetic Regulation in Prostate Cancer (Review). Oncol. Rep. 2017, 38, 2587–2596. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA Methylomes at Base Resolution Show Widespread Epigenomic Differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Huang, Y.H.; Cui, X.; Wang, X.; Zhang, X.; Lei, Y.; Xu, J.; Lin, X.; Chen, K.; Lv, J.; et al. Homeobox Oncogene Activation by Pan-Cancer DNA Hypermethylation. Genome Biol. 2018, 19, 108. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative Splicing and Cancer: A Systematic Review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef]
- Alló, M.; Buggiano, V.; Fededa, J.P.; Petrillo, E.; Schor, I.; De La Mata, M.; Agirre, E.; Plass, M.; Eyras, E.; Elela, S.A.; et al. Control of Alternative Splicing through SiRNA-Mediated Transcriptional Gene Silencing. Nat. Struct. Mol. Biol. 2009, 16, 717–724. [Google Scholar] [CrossRef]
- Batsché, E.; Yaniv, M.; Muchardt, C. The Human SWI/SNF Subunit Brm Is a Regulator of Alternative Splicing. Nat. Struct. Mol. Biol. 2006, 13, 22–29. [Google Scholar] [CrossRef]
- Anczukow, O.; Krainer, A.R. Splicing-Factor Alterations in Cancers. RNA 2016, 22, 1285–1301. [Google Scholar] [CrossRef]
- Saint-André, V.; Batsché, E.; Rachez, C.; Muchardt, C. Histone H3 Lysine 9 Trimethylation and HP1γ Favor Inclusion of Alternative Exons. Nat. Struct. Mol. Biol. 2011, 18, 337–344. [Google Scholar] [CrossRef]
- Huang, J.; Chang, S.; Lu, Y.; Wang, J.; Si, Y.; Zhang, L.; Cheng, S.; Jiang, W.G. Enhanced Osteopontin Splicing Regulated by RUNX2 Is HDAC-Dependent and Induces Invasive Phenotypes in NSCLC Cells. Cancer Cell Int. 2019, 19, 306. [Google Scholar] [CrossRef]
- Huang, J.; Hu, M.; Niu, H.; Wang, J.; Si, Y.; Cheng, S.; Ding, W. Osteopontin Isoform c Promotes the Survival of Cisplatin-Treated NSCLC Cells Involving NFATc2-Mediated Suppression on Calcium-Induced ROS Levels. BMC Cancer 2021, 21, 750. [Google Scholar] [CrossRef]
- Janiszewska, M. The Microcosmos of Intratumor Heterogeneity: The Space-Time of Cancer Evolution. Oncogene 2020, 39, 2031–2039. [Google Scholar] [CrossRef]
- Bure, I.; Geer, S.; Knopf, J.; Roas, M.; Henze, S.; Ströbel, P.; Agaimy, A.; Wiemann, S.; Hoheisel, J.D.; Hartmann, A.; et al. Long Noncoding RNA HOTAIR Is Upregulated in an Aggressive Subgroup of Gastrointestinal Stromal Tumors (GIST) and Mediates the Establishment of Gene-Specific DNA Methylation Patterns. Genes Chromosom. Cancer 2018, 57, 584–597. [Google Scholar] [CrossRef]
- Costa, A.L.; Moreira-Barbosa, C.; Lobo, J.; Vilela-Salgueiro, B.; Cantante, M.; Guimarães, R.; Lopes, P.; Braga, I.; Oliveira, J.; Antunes, L.; et al. DNA Methylation Profiling as a Tool for Testicular Germ Cell Tumors Subtyping. Epigenomics 2018, 10, 1511–1523. [Google Scholar] [CrossRef]
- Yadav, P.; Masroor, M.; Nandi, K.; Kaza, R.C.M.; Jain, S.K.; Khuarana, N.; Saxena, A. Promoter Methylation of BRCA1, DAPK1 and RASSF1A Is Associated with Increased Mortality among Indian Women with Breast Cancer. Asian Pacific J. Cancer Prev. 2018, 19, 443–448. [Google Scholar] [CrossRef]
- Dvojakovska, S.; Popovic-Monevska, D.; Grcev, A.; Pancevski, G.; Benedetti, A.; Popovski, V.; Dimovski, A.; Stamatoski, A. Promotor Hypermethylated Genes: Prospective Diagnostic Biomarkers in Oral Cancerogenesis. J. Cranio-Maxillofac. Surg. 2018, 46, 1737–1740. [Google Scholar] [CrossRef]
- Vastrad, B.; Vastrad, C.; Godavarthi, A.; Chandrashekar, R. Molecular Mechanisms Underlying Gliomas and Glioblastoma Pathogenesis Revealed by Bioinformatics Analysis of Microarray Data. Med. Oncol. 2017, 34, 182. [Google Scholar] [CrossRef]
- Ozyerli-Goknar, E.; Bagci-Onder, T. Epigenetic Deregulation of Apoptosis in Cancers. Cancers 2021, 13, 3210. [Google Scholar] [CrossRef]
- Kouidou, S.; Agidou, T.; Kyrkou, A.; Andreou, A.; Katopodi, T.; Georgiou, E.; Krikelis, D.; Dimitriadou, A.; Spanos, P.; Tsilikas, C.; et al. Non-CpG Cytosine Methylation of P53 Exon 5 in Non-Small Cell Lung Carcinoma. Lung Cancer 2005, 50, 299–307. [Google Scholar] [CrossRef]
- Li, C.; Xiong, W.; Liu, X.; Xiao, W.; Guo, Y.; Tan, J.; Li, Y. Hypomethylation at Non-CpG/CpG Sites in the Promoter of HIF-1α Gene Combined with Enhanced H3K9Ac Modification Contribute to Maintain Higher HIF-1α Expression in Breast Cancer. Oncogenesis 2019, 8, 26. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting Epigenetic Regulators for Cancer Therapy: Mechanisms and Advances in Clinical Trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Lin, M.; Li, C.; Liu, H.; Gong, C. A Comprehensive Review of the Roles of E2F1 in Colon Cancer. Am. J. Cancer Res. 2020, 10, 757–768. [Google Scholar] [PubMed]
- Sampath, D.; Liu, C.; Vasan, K.; Sulda, M.; Puduvalli, V.K.; Wierda, W.G.; Keating, M.J. Histone Deacetylases Mediate the Silencing of MiR-15a, MiR-16, and MiR-29b in Chronic Lymphocytic Leukemia. Blood 2012, 119, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Hada, M.; Opipari, A.W., Jr.; Castle, V.P.; Kwok, R.P.S. CREB-Binding Protein Regulates Ku70 Acetylation in Response to Ionization Radiation in Neuroblastoma. Mol. Cancer Res. 2013, 11, 173–181. [Google Scholar] [CrossRef]
- You, J.S.; Jones, P.A. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef]
- Ropero, S.; Ballestar, E.; Alaminos, M.; Arango, D.; Schwartz, S.; Esteller, M. Transforming Pathways Unleashed by a HDAC2 Mutation in Human Cancer. Oncogene 2008, 27, 4008–4012. [Google Scholar] [CrossRef]
- Cooke, M.; Magimaidas, A.; Casado-Medrano, V.; Kazanietz, M.G. Protein Kinase C in Cancer: The Top Five Unanswered Questions. Mol. Carcinog. 2017, 56, 1531–1542. [Google Scholar] [CrossRef]
- Bae, K.-M.; Wang, H.; Jiang, G.; Chen, M.G.; Lu, L.; Xiao, L. Protein Kinase Cε Is Overexpressed in Primary Human Non–Small Cell Lung Cancers and Functionally Required for Proliferation of Non–Small Cell Lung Cancer Cells in a P21/Cip1-Dependent Manner. Cancer Res. 2007, 67, 6053–6063. [Google Scholar] [CrossRef]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs Kinases—The Lessons from the Mouse Models: Inhibition ≠ Deletion. Cell Biosci. 2020, 10, 8. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Khanna, R.; Parnaik, V.K. Ubiquitin Ligase RNF123 Mediates Degradation of Heterochromatin Protein 1α and β in Lamin A/C Knock-Down Cells. PLoS ONE 2012, 7, e47558. [Google Scholar] [CrossRef]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 Phosphorylates Histone H3Y41 and Excludes HP1α from Chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Arfuso, F.; Arumugam, S.; Chinnathambi, A.; Jinsong, B.; Warrier, S.; Wang, L.Z.; Kumar, A.P.; Ahn, K.S.; Sethi, G.; et al. Role of Novel Histone Modifications in Cancer. Oncotarget 2017, 9, 11414–11426. [Google Scholar] [CrossRef]
- Mirabella, A.C.; Foster, B.M.; Bartke, T. Chromatin Deregulation in Disease. Chromosoma 2016, 125, 75–93. [Google Scholar] [CrossRef]
- Mullen, J.; Kato, S.; Sicklick, J.K.; Kurzrock, R. Targeting ARID1A Mutations in Cancer. Cancer Treat. Rev. 2021, 100, 1–9. [Google Scholar] [CrossRef]
- Zhu, Q.; Pao, G.M.; Huynh, A.M.; Suh, H.; Tonnu, N.; Nederlof, P.M.; Gage, F.H.; Verma, I.M. BRCA1 Tumour Suppression Occurs via Heterochromatin-Mediated Silencing. Nature 2011, 477, 179–184. [Google Scholar] [CrossRef]
- Gramling, S.; Reisman, D. Discovery of BRM Targeted Therapies: Novel Reactivation of an Anti-Cancer Gene. Lett. Drug Des. Discov. 2011, 8, 93–99. [Google Scholar] [CrossRef]
- Glaros, S.; Cirrincione, G.M.; Muchardt, C.; Kleer, C.G.; Michael, C.W.; Reisman, D. The Reversible Epigenetic Silencing of BRM: Implications for Clinical Targeted Therapy. Oncogene 2007, 26, 7058–7066. [Google Scholar] [CrossRef]
- Xu, N.; Liu, F.; Wu, S.; Ye, M.; Ge, H.; Zhang, M.; Song, Y.; Tong, L.; Zhou, J.; Bai, C. CHD4 Mediates Proliferation and Migration of Non-Small Cell Lung Cancer via the RhoA/ROCK Pathway by Regulating PHF5A. BMC Cancer 2020, 20, 262. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Zhao, B.; Zimmerman, D.L.; Belmont, A.S. Stable and Reproducible Transgene Expression Independent of Proliferative or Differentiated State Using BAC TG-EMBED. Gene Ther. 2018, 25, 376–391. [Google Scholar] [CrossRef]
- Dutta, P.; Zhang, L.; Zhang, H.; Peng, Q.; Montgrain, P.R.; Wang, Y.; Song, Y.; Li, J.; Li, W.X. Unphosphorylated STAT3 in Heterochromatin Formation and Tumor Suppression in Lung Cancer. BMC Cancer 2020, 20, 145. [Google Scholar] [CrossRef] [PubMed]
- Gurrion, C.; Uriostegui, M.; Zurita, M. Heterochromatin Reduction Correlates with the Increase of the KDM4B and KDM6A Demethylases and the Expression of Pericentromeric DNA during the Acquisition of a Transformed Phenotype. J. Cancer 2017, 8, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Jones, D.T.W.; Zapatka, M.; Stütz, A.M.; Zichner, T.; Weischenfeldt, J.; Jäger, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome Sequencing of Pediatric Medulloblastoma Links Catastrophic DNA Rearrangements with TP53 Mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Rode, A.; Maass, K.K.; Willmund, K.V.; Lichter, P.; Ernst, A. Chromothripsis in Cancer Cells: An Update. Int. J. Cancer 2016, 138, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Remvikos, Y.; Vogt, N.; Muleris, M.; Salmon, R.J.; Malfoy, B.; Dutrillaux, B. DNA-Repeat Instability Is Associated with Colorectal Cancers Presenting Minimal Chromosome Rearrangements. Genes Chromosom. Cancer 1995, 12, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Zucca, E.; Bekradda, M.; Gomez-Roca, C.; Delord, J.-P.; de La Motte Rouge, T.; Uro-Coste, E.; de Braud, F.; Pelosi, G.; French, C.A. Clinical Response of Carcinomas Harboring the BRD4–NUT Oncoprotein to the Targeted Bromodomain Inhibitor OTX015/MK-8628. Cancer Discov. 2016, 6, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Chen, Z.; Lin, X.; Tian, L.; Su, Q.; An, P.; Li, W.; Wu, Y.; Du, J.; Shan, H.; et al. Inhibition of BRD4 Suppresses the Malignancy of Breast Cancer Cells via Regulation of Snail. Cell Death Differ. 2020, 27, 255–268. [Google Scholar] [CrossRef]
- Vad-Nielsen, J.; Nielsen, A.L. Beyond the Histone Tale: HP1α Deregulation in Breast Cancer Epigenetics. Cancer Biol. Ther. 2015, 16, 189–200. [Google Scholar] [CrossRef]
- Pradhan, S.; Solomon, R.; Gangotra, A.; Yakubov, G.E.; Willmott, G.R.; Whitby, C.P.; Hale, T.K.; Williams, M.A.K. Depletion of HP1α Alters the Mechanical Properties of MCF7 Nuclei. Biophys. J. 2021, 120, 2631–2643. [Google Scholar] [CrossRef]
- Norwood, L.E.; Moss, T.J.; Margaryan, N.V.; Cook, S.L.; Wright, L.; Seftor, E.A.; Hendrix, M.J.C.; Kirschmann, D.A.; Wallrath, L.L. A Requirement for Dimerization of HP1Hsα in Suppression of Breast Cancer Invasion. J. Biol. Chem. 2006, 281, 18668–18676. [Google Scholar] [CrossRef]
- Kirschmann, D.A.; Lininger, R.A.; Gardner, L.M.G.; Seftor, E.A.; Odero, V.A.; Ainsztein, A.M.; Earnshaw, W.C.; Wallrath, L.L.; Hendrix, M.J.C. Down-Regulation of HP1(Hsα) Expression Is Associated with the Metastatic Phenotype in Breast Cancer. Cancer Res. 2000, 60, 3359–3363. [Google Scholar]
- Yang, H.; Zhang, H.; Luan, Y.; Liu, T.; Yang, W.; Roberts, K.G.; Qian, M.; Zhang, B.; Yang, W.; Perez-Andreu, V.; et al. Noncoding Genetic Variation in GATA3 Increases Acute Lymphoblastic Leukemia Risk through Local and Global Changes in Chromatin Conformation. Nat. Genet. 2022, 54, 170–179. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Wilmink, J.W. IDH1/2 Mutations in Cancer Stem Cells and Their Implications for Differentiation Therapy. J. Histochem. Cytochem. 2022, 70, 83–97. [Google Scholar] [CrossRef]
- Kon, A.; Shih, L.-Y.; Minamino, M.; Sanada, M.; Shiraishi, Y.; Nagata, Y.; Yoshida, K.; Okuno, Y.; Bando, M.; Nakato, R.; et al. Recurrent Mutations in Multiple Components of the Cohesin Complex in Myeloid Neoplasms. Nat. Genet. 2013, 45, 1232–1237. [Google Scholar] [CrossRef]
- Lau, C.-H.; Suh, Y. CRISPR-Based Strategies for Studying Regulatory Elements and Chromatin Structure in Mammalian Gene Control. Mamm. Genome 2018, 29, 205–228. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator Dysfunction and Oncogene Activation in IDH Mutant Gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of Proto-Oncogenes by Disruption of Chromosome Neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef]
- Cremer, M.; Küpper, K.; Wagler, B.; Wizelman, L.; Hase, J.V.; Weiland, Y.; Kreja, L.; Diebold, J.; Speicher, M.R.; Cremer, T. Inheritance of Gene Density-Related Higher Order Chromatin Arrangements in Normal and Tumor Cell Nuclei. J. Cell Biol. 2003, 162, 809–820. [Google Scholar] [CrossRef]
- Sathitruangsak, C.; Righolt, C.H.; Klewes, L.; Tung Chang, D.; Kotb, R.; Mai, S. Distinct and Shared Three-Dimensional Chromosome Organization Patterns in Lymphocytes, Monoclonal Gammopathy of Undetermined Significance and Multiple Myeloma. Int. J. Cancer 2017, 140, 400–410. [Google Scholar] [CrossRef]
- Fritz, A.J.; Stojkovic, B.; Ding, H.; Xu, J.; Bhattacharya, S.; Gaile, D.; Berezney, R. Wide-Scale Alterations in Interchromosomal Organization in Breast Cancer Cells: Defining a Network of Interacting Chromosomes. Hum. Mol. Genet. 2014, 23, 5133–5146. [Google Scholar] [CrossRef]
- Meister, P.; Taddei, A. Building Silent Compartments at the Nuclear Periphery: A Recurrent Theme. Curr. Opin. Genet. Dev. 2013, 23, 96–103. [Google Scholar] [CrossRef] [PubMed]
- García-Nieto, P.E.; Schwartz, E.K.; King, D.A.; Paulsen, J.; Collas, P.; Herrera, R.E.; Morrison, A.J. Carcinogen Susceptibility Is Regulated by Genome Architecture and Predicts Cancer Mutagenesis. EMBO J. 2017, 36, 2829–2843. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Turner, K.M.; Nguyen, N.; Raviram, R.; Erb, M.; Santini, J.; Luebeck, J.; Rajkumar, U.; Diao, Y.; Li, B.; et al. Circular EcDNA Promotes Accessible Chromatin and High Oncogene Expression. Nature 2019, 575, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Schep, R.; Brinkman, E.K.; Leemans, C.; Vergara, X.; van der Weide, R.H.; Morris, B.; van Schaik, T.; Manzo, S.G.; Peric-Hupkes, D.; van den Berg, J.; et al. Impact of Chromatin Context on Cas9-Induced DNA Double-Strand Break Repair Pathway Balance. Mol. Cell 2021, 81, 2216–2230.e10. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Nguyen, N.P.; Turner, K.; Wu, S.; Gujar, A.D.; Luebeck, J.; Liu, J.; Deshpande, V.; Rajkumar, U.; Namburi, S.; et al. Extrachromosomal DNA Is Associated with Oncogene Amplification and Poor Outcome across Multiple Cancers. Nat. Genet. 2020, 52, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Selmecki, A.M.; Maruvka, Y.E.; Richmond, P.A.; Guillet, M.; Shoresh, N.; Sorenson, A.L.; De, S.; Kishony, R.; Michor, F.; Dowell, R.; et al. Polyploidy Can Drive Rapid Adaptation in Yeast. Nature 2015, 519, 349–351. [Google Scholar] [CrossRef]
- Rutledge, S.D.; Douglas, T.A.; Nicholson, J.M.; Vila-Casadesús, M.; Kantzler, C.L.; Wangsa, D.; Barroso-Vilares, M.; Kale, S.D.; Logarinho, E.; Cimini, D. Selective Advantage of Trisomic Human Cells Cultured in Non-Standard Conditions. Sci. Rep. 2016, 6, 22828. [Google Scholar] [CrossRef]
- Voronina, N.; Wong, J.K.L.; Hübschmann, D.; Hlevnjak, M.; Uhrig, S.; Heilig, C.E.; Horak, P.; Kreutzfeldt, S.; Mock, A.; Stenzinger, A.; et al. The Landscape of Chromothripsis across Adult Cancer Types. Nat. Commun. 2020, 11, 2320. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Gerlitz, G.; Bustin, M. Efficient Cell Migration Requires Global Chromatin Condensation. J. Cell Sci. 2010, 123, 2207–2217. [Google Scholar] [CrossRef]
- Gerlitz, G.; Livnat, I.; Ziv, C.; Yarden, O.; Bustin, M.; Reiner, O. Migration Cues Induce Chromatin Alterations. Traffic 2007, 8, 1521–1529. [Google Scholar] [CrossRef]
- Brandt, A.; Papagiannouli, F.; Wagner, N.; Wilsch-Bräuninger, M.; Braun, M.; Furlong, E.E.; Loserth, S.; Wenzl, C.; Pilot, F.; Vogt, N.; et al. Developmental Control of Nuclear Size and Shape by Kugelkern and Kurzkern. Curr. Biol. 2006, 16, 543–552. [Google Scholar] [CrossRef]
- Strom, A.R.; Biggs, R.J.; Banigan, E.J.; Wang, X.; Chiu, K.; Herman, C.; Collado, J.; Yue, F.; Politz, J.C.R.; Tait, L.J.; et al. Hp1α Is a Chromatin Crosslinker That Controls Nuclear and Mitotic Chromosome Mechanics. Elife 2021, 10, 1–30. [Google Scholar] [CrossRef]
- Sharma, G.G.; Hwang, K.; Pandita, R.K.; Gupta, A.; Dhar, S.; Parenteau, J.; Agarwal, M.; Worman, H.J.; Wellinger, R.J.; Pandita, T.K. Human Heterochromatin Protein 1 Isoforms HP1(Hsalpha) and HP1(Hsbeta) Interfere with HTERT-Telomere Interactions and Correlate with Changes in Cell Growth and Response to Ionizing Radiation. Mol. Cell. Biol. 2003, 23, 8363–8376. [Google Scholar] [CrossRef]
- Inoue, A.; Hyle, J.; Lechner, M.S.; Lahti, J.M. Perturbation of HP1 Localization and Chromatin Binding Ability Causes Defects in Sister-Chromatid Cohesion. Mutat. Res.-Genet. Toxicol. Environ. Mutagen. 2008, 657, 48–55. [Google Scholar] [CrossRef]
- Liu, L.; Luo, Q.; Sun, J.; Ju, Y.; Morita, Y.; Song, G. Chromatin Organization Regulated by EZH2-Mediated H3K27me3 Is Required for OPN-Induced Migration of Bone Marrow-Derived Mesenchymal Stem Cells. Int. J. Biochem. Cell Biol. 2018, 96, 29–39. [Google Scholar] [CrossRef]
- Gerlitz, G.; Bustin, M. The Role of Chromatin Structure in Cell Migration. Trends Cell Biol. 2011, 21, 6–11. [Google Scholar] [CrossRef]
- Dahl, K.N.; Engler, A.J.; Pajerowski, J.D.; Discher, D.E. Power-Law Rheology of Isolated Nuclei with Deformation Mapping of Nuclear Substructures. Biophys. J. 2005, 89, 2855–2864. [Google Scholar] [CrossRef]
- Gurova, K.V. Chromatin Stability as a Target for Cancer Treatment. BioEssays 2019, 41, 1–23. [Google Scholar] [CrossRef]
- Debes, J.D.; Sebo, T.J.; Heemers, H.V.; Kipp, B.R.; Haugen, D.A.L.; Lohse, C.M.; Tindall, D.J. P300 Modulates Nuclear Morphology in Prostate Cancer. Cancer Res. 2005, 65, 708–712. [Google Scholar] [CrossRef]
- Fischer, A.H.; Bond, J.A.; Taysavang, P.; Battles, O.E.; Wynford-Thomas, D. Papillary Thyroid Carcinoma Oncogene (RET/PTC) Alters the Nuclear Envelope and Chromatin Structure. Am. J. Pathol. 1998, 153, 1443–1450. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roberti, A.; Valdes, A.F.; Torrecillas, R.; Fraga, M.F.; Fernandez, A.F. Epigenetics in Cancer Therapy and Nanomedicine. Clin. Epigenetics 2019, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Liou, J.P. Recent Developments in Epigenetic Cancer Therapeutics: Clinical Advancement and Emerging Trends. J. Biomed. Sci. 2021, 28, 27. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.G.; Mandloi, T.; Kunte, P.; Natu, A.; Rashid, M.; Reddy, D.; Gadewal, N.; Gupta, S. HISTome2: A Database of Histone Proteins, Modifiers for Multiple Organisms and Epidrugs. Epigenet. Chromatin 2020, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective Inhibition of BET Bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Eder, J.P.; LoRusso, P.M. BET Inhibitors: A Novel Epigenetic Approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef]
- Hoy, S.M. Tazemetostat: First Approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Li, X.; Egervari, G.; Wang, Y.; Berger, S.L.; Lu, Z. Regulation of Chromatin and Gene Expression by Metabolic Enzymes and Metabolites. Nat. Rev. Mol. Cell Biol. 2018, 19, 563–578. [Google Scholar] [CrossRef]
- Janke, R.; Dodson, A.E.; Rine, J. Metabolism and Epigenetics. Annu. Rev. Cell Dev. Biol. 2015, 31, 473–496. [Google Scholar] [CrossRef]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An Inhibitor of Mutant IDH1 Delays Growth and Promotes Differentiation of Glioma Cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT Promotes Epigenetic Remodeling in Cancer by Creating a Metabolic Methylation Sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef]
- Roberti, A.; Fernández, A.F.; Fraga, M.F. Nicotinamide N-Methyltransferase: At the Crossroads between Cellular Metabolism and Epigenetic Regulation. Mol. Metab. 2021, 45, 101165. [Google Scholar] [CrossRef]
- Neelakantan, H.; Vance, V.; Wetzel, M.D.; Wang, H.Y.L.; McHardy, S.F.; Finnerty, C.C.; Hommel, J.D.; Watowich, S.J. Selective and Membrane-Permeable Small Molecule Inhibitors of Nicotinamide N-Methyltransferase Reverse High Fat Diet-Induced Obesity in Mice. Biochem. Pharmacol. 2018, 147, 141–152. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA Damage Response Signaling Pathways and Targets for Radiotherapy Sensitization in Cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Siklos, M.; Kubicek, S. Therapeutic Targeting of Chromatin: Status and Opportunities. FEBS J. 2022, 289, 1276–1301. [Google Scholar] [CrossRef]
- Suzuki, H.; Gabrielson, E.; Chen, W.; Anbazhagan, R.; Van Engeland, M.; Weijenberg, M.P.; Herman, J.G.; Baylin, S.B. A Genomic Screen for Genes Upregulated by Demethylation and Histone Deacetylase Inhibition in Human Colorectal Cancer. Nat. Genet. 2002, 31, 141–149. [Google Scholar] [CrossRef]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of Demethylation and Histone Deacetylase Inhibition in the Re-Expression of Genes Silenced in Cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Gore, S.D.; Baylin, S.; Sugar, E.; Carraway, H.; Miller, C.B.; Carducci, M.; Grever, M.; Galm, O.; Dauses, T.; Karp, J.E.; et al. Combined DNA Methyltransferase and Histone Deacetylase Inhibition in the Treatment of Myeloid Neoplasms. Cancer Res. 2006, 66, 6361–6369. [Google Scholar] [CrossRef]
- Pruitt, K.; Zinn, R.L.; Ohm, J.E.; McGarvey, K.M.; Kang, S.H.L.; Watkins, D.N.; Herman, J.G.; Baylin, S.B. Inhibition of SIRT1 Reactivates Silenced Cancer Genes without Loss of Promoter DNA Hypermethylation. PLoS Genet. 2006, 2, 0344–0352. [Google Scholar] [CrossRef]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted Protein Degradation: Expanding the Toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Chaturvedi, P.; Zimmerman, D.L.; Belmont, A.S. Efficient and Reproducible Multigene Expression after Single-Step Transfection Using Improved Bac Transgenesis and Engineering Toolkit. ACS Synth. Biol. 2020, 9, 1100–1116. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inherited Pathology | Involved Tissue | Mutant Protein/RNA | Mechanism | References |
|---|---|---|---|---|
| Abnormal nuclear morphology | Renal cell carcinoma (RCC), gastrointestinal cancers, breast cancer, and cervical cancer | Depletion of AT-rich interactive domain 1A (ARID1A) | Increase in nuclear volume, cell proliferation, migration, and chemoresistance | [36] |
| Abnormal nuclear morphology | Melanoma, and bladder cancer | Loss of macroH2A1 and macroH2A2 histone variants | Defects in nuclear organization, including disruption of nucleoli and a global loss of dense heterochromatin | [37] |
| Abnormal nuclear morphology | Human cervical cancer | Reduction of NOP53 ribosome biogenesis factor (NOP53) | Increased chromosomal instability, multinucleated cells, nuclear budding | [38] |
| ICF syndrome | Immunodeficiency due to reduced or absent serum immunoglobulins, facial abnormalities, and developmental delay | DNA Methyltransferase 3B (DNMT3B), Zinc-finger & BTB domain containing 24 (ZBTB24), Cell division cycle associated 7 (CDCA7) or Helicase, lymphoid-specific (HELLS) | Hypomethylation of satellite repeats at the pericentromeric heterochromatin activating interferon-mediated innate immune response | [39] |
| Laminopathy | Small-cell lung cancer, prostate cancer, pancreatic cancer, and melanoma | Altered LMNB1/2 expression | Epigenetic derepression of the RET proto-oncogene by loss of PRC2 recruitment | [31,40,41] |
| Laminopathy | Breast Cancer, colorectal cancer, melanoma, gastric cancer, leukemia, and lymphoma | Mutations or reduced expression of LMNA | Destabilization of retinoblastoma (pRb) or hyperactivation of MAPK, PI3K/AKT pathways | [35,42,43] |
| Lynch syndrome hereditary non-polyposis colorectal cancer (HNPCC) | Colorectal, ovarian, and endometrial cancers | Epimutation (Deletion in TACSTD1) or mutation in associated genes (MLH1, MSH2, MSH6, PMS2 and EPCAM) | Mosaic and allele-specific hypermethylation of the downstream MSH2 promoter | [44] |
| Nuclear envelopathies | Ovarian cancer, prostate cancer, lung cancer, breast cancer, colorectal cancer | Emerin, Nesprin-1, and Nesprin-2 | Aneuploidy & chromosomal numerical instability. Altered chromatin conformation reduces GATA6 expression | [45,46,47] |
| Pelger-Huët anomaly and Greenberg Dysplasia | Multilobed, hypo-segmented nuclei form in white blood cells. Increased LBR expression is seen in aggressive breast cancers | Lamin B Receptor (LBR) | Mislocalization of inactive X (Xi) to the nuclear interior causes its genes to express | [40,48,49] |
| Thalassemia | Hepatocellular carcinoma | Epimutation (Deletion in LUC7L gene) | HBA2 gene silencing induced by promoter hypermethylation | [50,51] |
| Werner’s Syndrome, Aicardi-Goutières syndrome | Colorectal adenocarcinoma, metastatic prostate cancer, leukemia, cervical cancer, and ovarian cancer | Reduced expression of RNaseH2A | Genomic instability, increased metastasis, cellular senescence, ageing symptoms | [52] |
| Chromatin Modification | Gene/Region Involved | Known Cancer Association | References |
|---|---|---|---|
| DNA Methylation | |||
| Promoter Hypermethylation | RASSF1 | Hepatocellular carcinoma, oral squamous cell carcinoma, lung, breast, colorectal, bladder, cervical, and prostate cancers | [81,83,94,95,96] |
| CDH1 | Prostate cancer, hepatocellular carcinoma, Non-small cell lung carcinoma (NSCLC), esophageal, gastric, breast and bladder cancers | [81,83,97] | |
| DAPK1 | Breast, cervical, and bladder cancers | [96,97,98] | |
| CDKN2A | Melanoma, glioblastoma, bladder cancer | [81] | |
| Hypomethylation | HOX11 | Leukemia | [99] |
| pS2 | Breast cancer | [99] | |
| c-n-RAS | Most adult cancers | [99] | |
| C-MYC | Colorectal cancer | [99] | |
| LINE-1 repeats | Prostate cancer | [83] | |
| Genebody Methylation | p53-exon5 | Non-small cell lung carcinoma (NSCLC) | [100] |
| HIF-1α | Breast cancer | [101] | |
| Histone modifications | |||
| Methylation | EZH2 | Most adult cancers | [99,102] |
| KMT2D | Breast cancer | [102] | |
| SETD2 | Renal cell carcinoma, Lung cancer | [99,102] | |
| Acetylation | E2F1 | Colon cancer | [103] |
| Mcl-1 | Chronic myeloid leukemia (CML) | [104] | |
| Ku70 | Neuroblastoma, hepatocellular carcinoma | [105] | |
| EP300 | Breast, colorectal, pancreatic cancer | [106] | |
| HDAC2 | All major cancers | [102,107] | |
| Phosphorylation | PKC | Chronic Lymphocytic Leukemia (CLL), colorectal carcinoma, melanoma, invasive ductal breast cancer, NSCLC | [108,109] |
| ATM/ATR | Epithelial, breast, and pancreatic cancers, leukemias, lymphomas | [110,111] | |
| H3tyr41 | Leukemia | [112] | |
| Aurora B | Breast and colorectal cancers | [113] | |
| Chromatin Remodeling/pre-mRNA Splicing | ARID1A | Colon cancer, ovarian clear cell cancers, uterine endometrial cancers, renal cell carcinoma | [36,114,115] |
| BRCA1 | Breast and ovarian cancer | [116] | |
| BRM | Prostate cancer, basal cell carcinoma, Lung cancer | [117,118] | |
| CHD4/5 | NSCLC, Colorectal, gastric, ovarian, and Prostate cancers | [119,120] | |
| ASXL | Myelodysplastic syndromes, acute myeloid leukemia (AML) | [114] | |
| Structural changes | |||
| Loss of heterochromatin | Barr body | Breast cancer, Ovarian cancer | [13,53,54] |
| Pericentromeric and telomeric heterochromatin | Most adult cancers, Lung cancer | [121,122] | |
| Rearrangements | Genomewide local clustered rearrangements/Chromothripsis | Sonic-Hedgehog medulloblastoma, AML, aggressive tumors | [123,124] |
| Satellite repeats | Colorectal cancers | [125] | |
| TET1 | Osteosarcoma, AML | [99] | |
| BRD4 | Midline carcinoma, breast and colon cancer, AML | [126,127] | |
| Chromatin conformation and stiffness | HP1α | Breast cancer | [128,129,130,131] |
| GATA3 | Acute lymphoblastic leukemia | [132] | |
| IDH1/2 | Glioma, Chondrosarcoma, Cholangiocarcinoma, Myelodysplastic syndrome (MDS), AML | [133] | |
| STAG2, RAD21, SMC1A and SMC3 (Cohesin complex) | Myeloid leukemia, Breast cancer, Lung adenocarcinoma | [134] | |
| Long-Range interactions | |||
| Enhancer hijacking | MYB | Adenoid cystic carcinoma | [65] |
| BCL11B | Lineage-ambiguous leukemia | [66] | |
| KLF5 | Head and neck squamous cell carcinoma, esophagial carcinoma | [14,67,135] | |
| Super-Enhancer deletion | RCAN1.4 | Breast cancer | [68] |
| Enhancer Focal amplification | MYC | Lung adenocarcinoma, endometrial carcinoma | [67] |
| PARD6B | Liver hepatocellular carcinoma | [67] | |
| USP12 | Colorectal cancer | [67] | |
| TAD disruption | AR, FOXA1 | Prostrate cancer | [70] |
| PDGFRA | Glioma | [136] | |
| TAL1 | T cell acute lymphoblastic leukemia | [14,137] | |
| LMO2 | T cell acute lymphoblastic leukemia | [14,137] | |
| Inhibitor Category | Prominent Examples | Generic Name of FDA Approved Drug | Brand Name and Manufacturer | Therapeutic Use |
|---|---|---|---|---|
| Acetylated Histone binding protein inhibitor (PAHi) | CPI203, RVX-208, I-BET-726 | - | - | - |
| Bromodomain (BRD) and extra-terminal domain (BET) protein inhibitor (BETi) | OTX15, I-BET762, I-BET151, JQ1, Pelabresib (CPI-0610), Molibresib (GSK525762), INCB054329, INCB057643, ODM-207, Ten-010 (RO-6870810), BAY 1238097, SF-1126, Trotabresib (CC-90010), AZD-5153, PLX-51107 | Nivolumab (BMS-986158) | OPDIVO® by Bristol-Myers Squibb Pharma, NY, USA | Advanced NSCLC, melanoma, renal cell carcinoma, squamous cell carcinoma, hepatocellular carcinoma, urothelial carcinoma, colorectal cancer, classical Hodgkin’s lymphoma, malignant pleural mesothelioma |
| DNA Methyl Transferase inhibitor (DNMTi) | Epigallocatechin-3-gallate, Zebularine, Equol, Genistein, Guadecitabine (SGI-110), Procaine, Nanaomycin A, Disulfiram, Lomeguatrib, RG108, SGI-1027, MG98, CP-4200, Hinokitiol, DC_517, DC-05, Isothiocyanate, Fazarabine (Arabinosyl-5-azacytidine), DHAC (5,6-dihydro-5-azacytidine) | Decitabine (5-aza-2′deoxycytidine) 5-Azacytidine Procainamide | Dacogen® by MGI Pharma, Inc., NJ, USA Vidaza®, Onureg®. Both by Bristol-Myers Squibb Pharma, NY, USA Pronestyl® by Nicholas Piramal India Ltd., Mumbai, India and Bristol-Myers Squibb Pharma, NY, USA | Myelodysplastic syndrome (MDS) Myelomonocytic leukemia (CMML) Cardiac arrythmia |
| Histone Acetyl Transferase inhibitor (HATi) | Gallic acid, Garcinol, Anacardic acid, Procyanidin, MB-3, CTK7A, Plumbagin, Embelin, Curcumin, A-485, C646, DS17701585, Remodelin hydrobromide, Butyrolactone 3, CPTH2 | - | - | - |
| Histone Deacetylase inhibitor (HDACi) | Givinostat, AR-42, Entinostat, Apicidin, Pracinostat, Abexinostat, Resminostat, CUDC-101, Toxoflavin, Inauhzin, Cambinol, Salermide, Trichostatin A, CG-1521, OSU-HDAC-42, HC-toxin, Plitidepsin, Tasquinimod, Sodium butyrate, Mocetinostat, Tefinostat, CHR-3996, QUISINOSTAT, Sodium phenylbutyrate, Pivanex, Butyroyloxymethyl-diethyl phosphate, Resveratrol, Dacinostat, Droxinostat, Psammaplin A, ITF-A, ITF-B, OSU-HDAC-44, Ricolinostat, Tubastatin A, RGFP966, TMP195, Fimepinostat, LMK-235, ACY-738, PCI-34051, Nexturastat A, CAY10603, ACY-775, WT-161, MC1568, RGFP109, Citarinostat, Scriptaid, Tucidinostat, Santacruzamate A, EDO-S101, Oxamflatin, HPOB, BML-210, Pomiferin, Domatinostat, BG45, Bufexamac, Sinapinic acid, FT895, CHDI-390576 | Vorinostat, Panobinostat (LBH589), Belinostat (PXD101), Romidepsin (FK228, Depsipeptide), Valproic acid, Valproic acid and divalproex sodium Carbamazepine | Zolinza® by Merck & Co., Inc., NJ, USA Farydak® by Novartis, Basel, Switzerland Beleodaq® by Acrotech Biopharma Inc., NJ, USA Istodax® by Bristol-Myers Squibb Pharma, NY, USA Stavzor® by Noven Pharmaceuticals, FL, USA Depakene, Depakote by Abbott Laboratories, IL, USA Tegretol® by Novartis, Basel, Switzerland | Cutaneous T-cell lymphoma (CTCL) Multiple myeloma Multiple myeloma (discontinued) Elapsed or refractory peripheral T-cell lymphoma (PTCL) Cutaneous T-cell lymphoma (CTCL) Anticonvulsant Anticonvulsant (advanced-stage trials for breast cancer) Anticonvulsant |
| Histone Demethylase inhibitor (HDMi) | Pargyline, Clorgyline, Bizine, GSK2879552, KDM5-C70, JIB-04, ORY-1001, SID 85736331, Namoline, CBB1007, Methylstat, GSKJ4, GSKJ1, QC6352, SP2509, KDOAM-25, T-448, Daminozide, CPI-455, NCGC00244536, NCGC00247743, GSK-J2, Corin, GSK690, PBIT, S 2101, T-3775440 hydrochloride, INCB059872, CC-90011 | Tranylcypromine, Phenelzine | Parnate® by GlaxoSmithKline, Brentford, UK Nardil® by Pfizer, NY, USA | Antidepressant (being investigated for anticancer properties) Antidepressant (Phase 2 trials for prostate cancer) |
| Histone Kinase inhibitor (HKi) | Ruxolitinib, KU-55933, VE-821 | - | - | - |
| Histone Methyl Transferase inhibitor (HMTi) | UNC0321, UNC0224, EPZ-6438, DZNep, GSK343, Chaetocin, BIX-01338, BIX-01294, UNC0638, EPZ005687, GSK126, EPZ-5676, EPZ004777, SGC0946, E72, A-366, UNC1999, CPI360, UNC0965, BIX-01337, EI1, GSK503, BCI-121, LLY-507, EPZ015666, UNC0642, AZ505 ditrifluoroacetate, GSK3326595, MS023, JNJ-64619178, CM-579, EED226, MI-503, EPZ015866, MI-463, MI-538, MS049, CPI-169, BRD9539, LLY-283, EML741, OTS186935, SGC3027, Pinometostat | Tazemetostat (E7438/EPZ6438) | Tazverik® by Epizyme, MA, USA | Advanced epithelioid sarcoma, follicular lymphoma |
| Methylated Histone binding protein inhibitor (PMHi) | UNC669, UNC1215 | - | - | - |
| Poly (ADP-Ribose) Polymerase inhibitor (PARPi) | AMF-26, Talazoparib, Ilimaquinone, Veliparib, Niraparib, Rucaparib | Olaparib (AZD-2281) | Lynparza® by AstraZeneca, Cambridge, UK and Merck & Co., Inc., NJ, USA | BRCA-mutated advanced ovarian cancer |
| Protein Arginine Demethylase inhibitor (PADi) | YW3-56, YW4-03, YW4-15, D-o-F-amidine, Cl-amidine, GSK484 | - | - | - |
| Protein arginine methyltransferases (PRMTs) inhibitor (PRMTi) | AMI-1, Sinefungin | - | - | - |
| Ubiquitin Signaling Inhibitor (USi) | PTC209, GW7647, PRT4165, ML323 | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sehgal, P.; Chaturvedi, P. Chromatin and Cancer: Implications of Disrupted Chromatin Organization in Tumorigenesis and Its Diversification. Cancers 2023, 15, 466. https://doi.org/10.3390/cancers15020466
Sehgal P, Chaturvedi P. Chromatin and Cancer: Implications of Disrupted Chromatin Organization in Tumorigenesis and Its Diversification. Cancers. 2023; 15(2):466. https://doi.org/10.3390/cancers15020466
Chicago/Turabian StyleSehgal, Poonam, and Pankaj Chaturvedi. 2023. "Chromatin and Cancer: Implications of Disrupted Chromatin Organization in Tumorigenesis and Its Diversification" Cancers 15, no. 2: 466. https://doi.org/10.3390/cancers15020466
APA StyleSehgal, P., & Chaturvedi, P. (2023). Chromatin and Cancer: Implications of Disrupted Chromatin Organization in Tumorigenesis and Its Diversification. Cancers, 15(2), 466. https://doi.org/10.3390/cancers15020466