
Identifying New Contributors to Brain Metastasis in Lung Adenocarcinoma: A Transcriptomic Meta-Analysis
 , ,
, ,  ,
,  ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA-Seq and Microarray Data Selection for Meta-Analysis
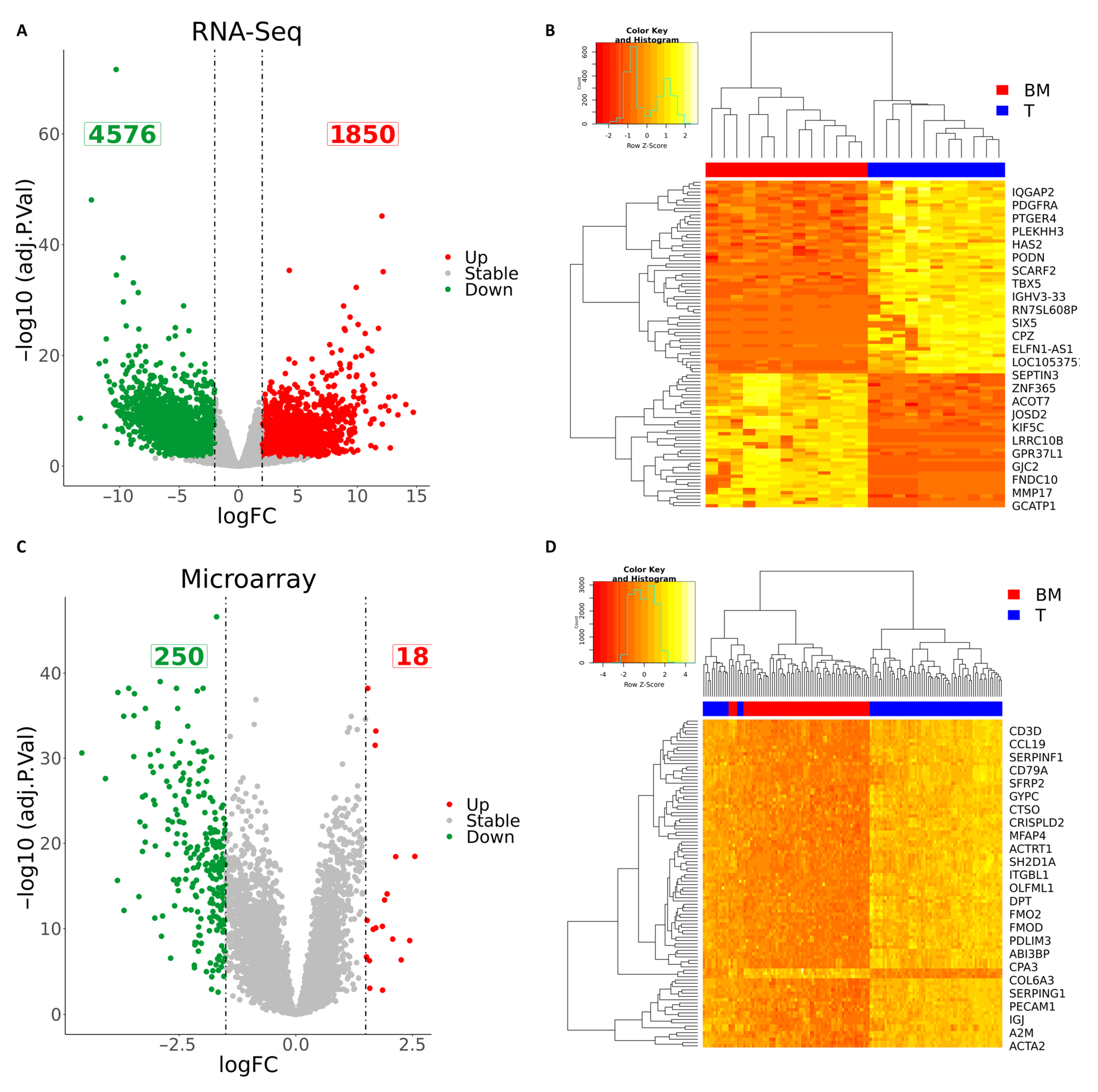
2.2. Identification of Differentially Expressed Genes Using RNA-Seq Data
2.3. Identification of Differentially Expressed Genes Using Microarray Data
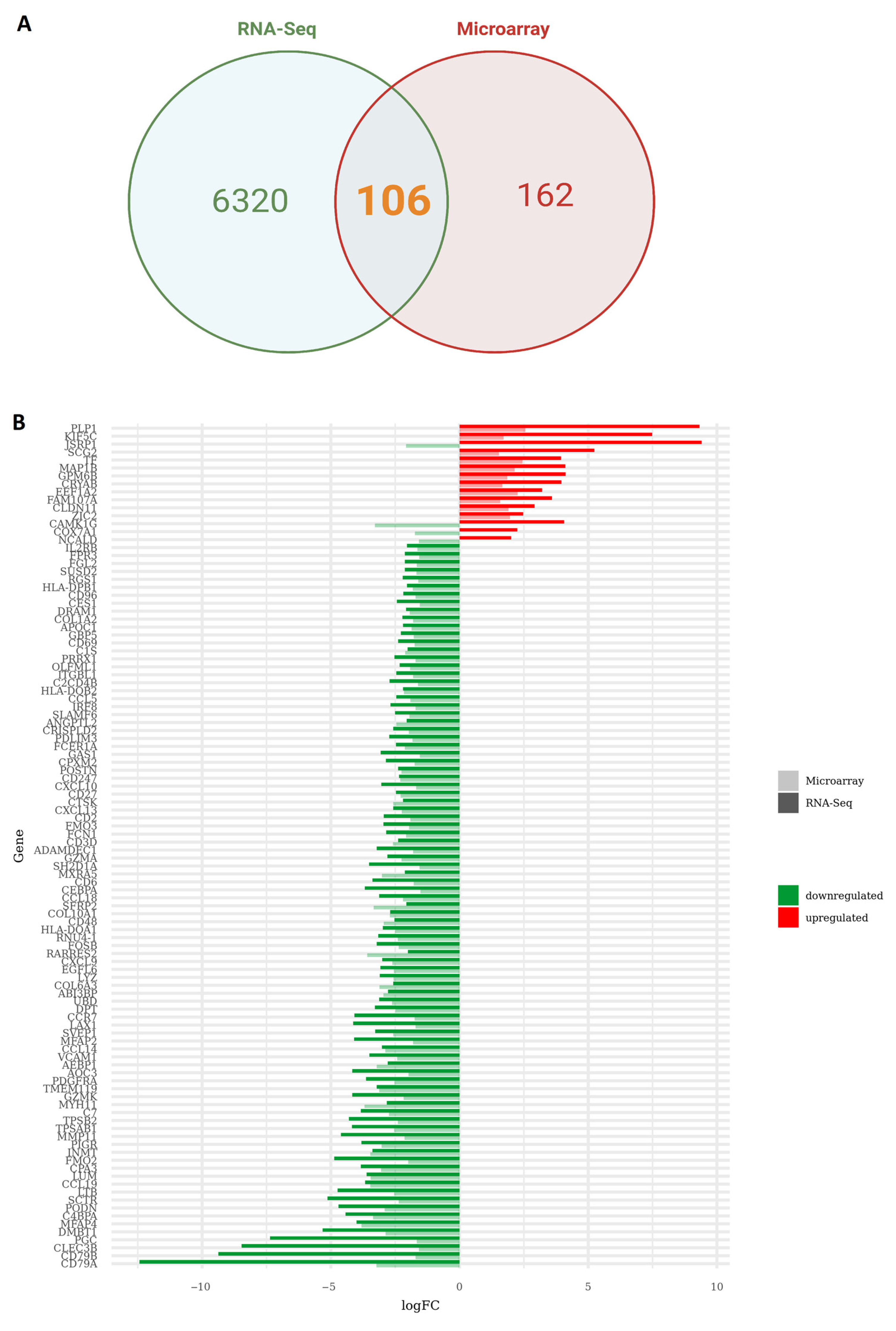
2.4. Identification of Differentially Expressed Genes (DEGs) Overlap between RNA-Seq Data and Microarray
2.5. Functional and Pathway Enrichment Analyses
2.6. Protein–Protein Interaction Network Construction for Selected Modules and Hub Genes Identification
2.7. Immunophenotype of Brain Metastasis from Lung Adenocarcinoma
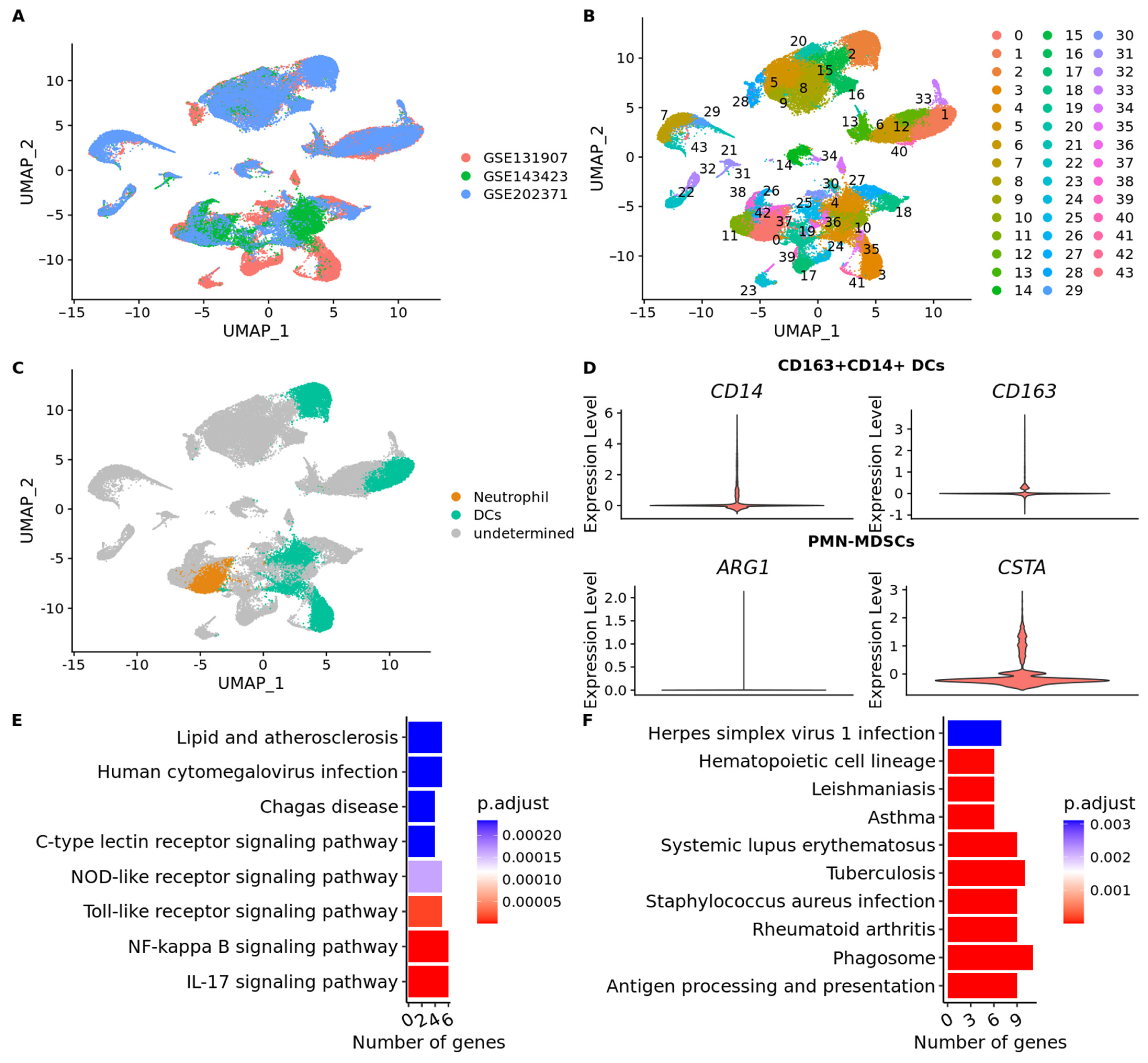
2.8. Single-Cell RNA-Sequencing Data Processing and Analysis
3. Results
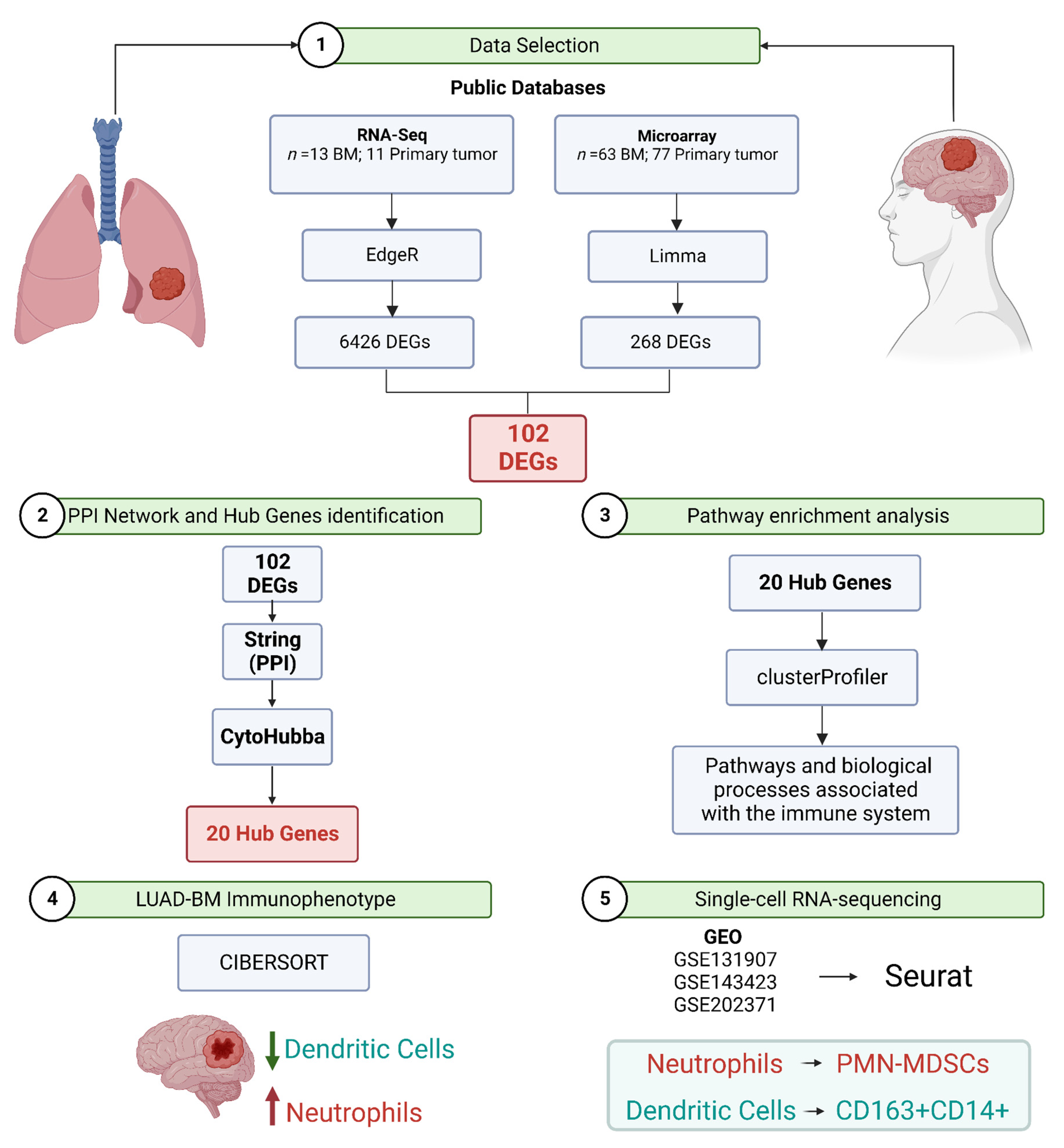
3.1. Datasets Selected for Meta-Analysis
3.2. Integration of RNA-Seq and Microarray Datasets Identified 102 Differentially Expressed Genes in Brain Metastasis from Lung Adenocarcinoma
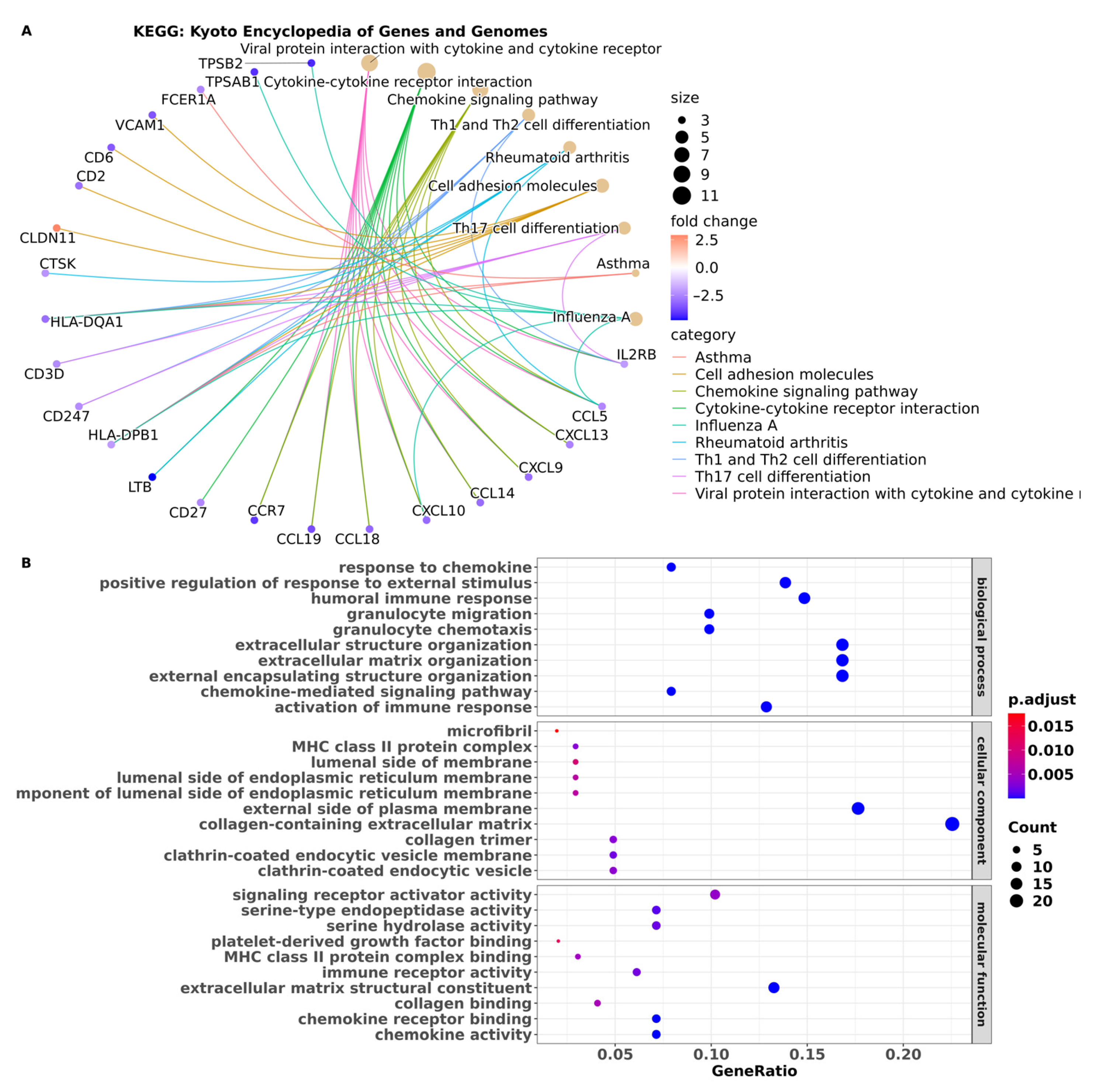
3.3. Pathway Enrichment Analysis Showed Pathways in BM Are Associated with the Immune System
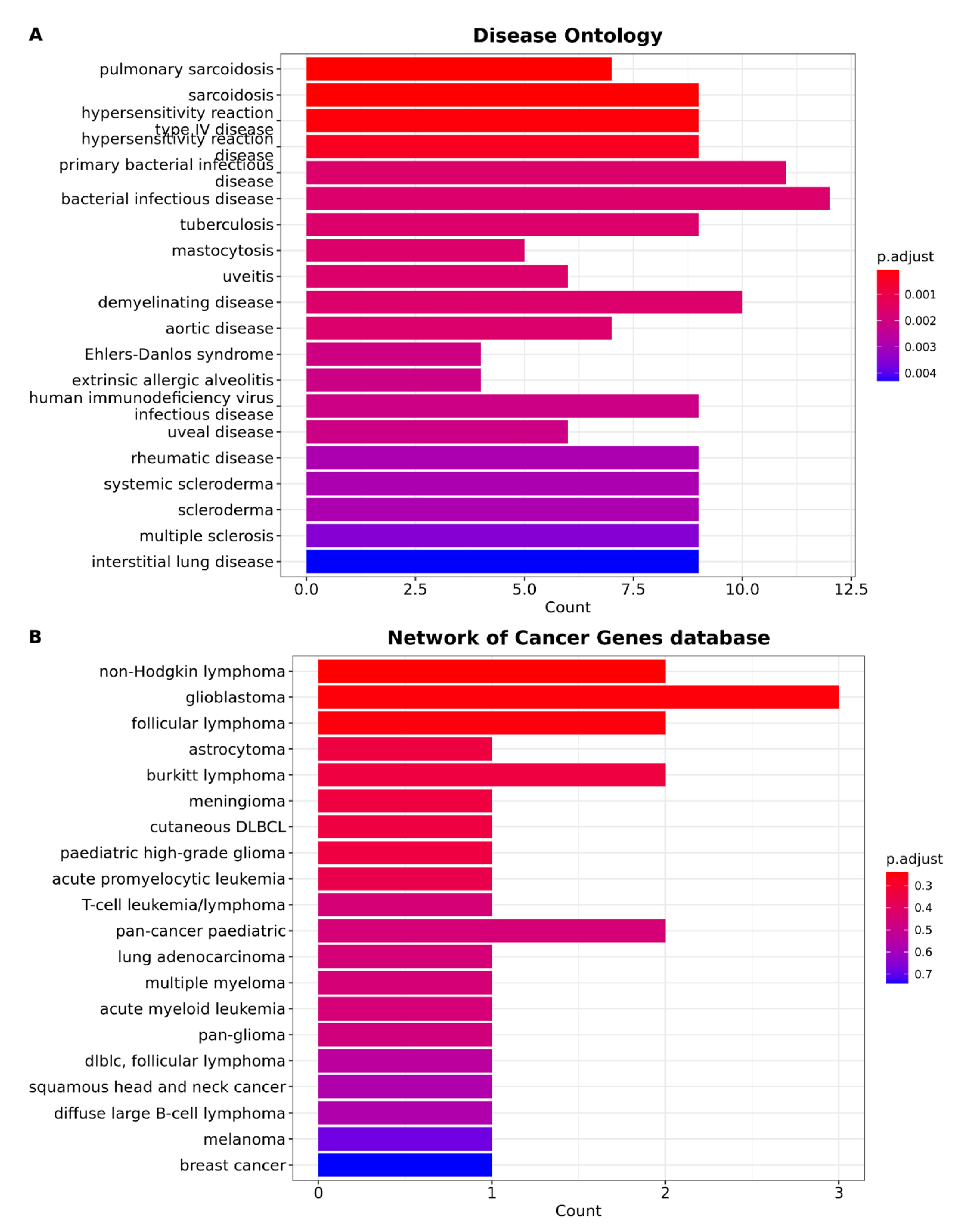
3.4. Brain Metastasis from Lung Adenocarcinoma Exhibits Distinctive Characteristics That Distinguish It from All Other Types of Cancer
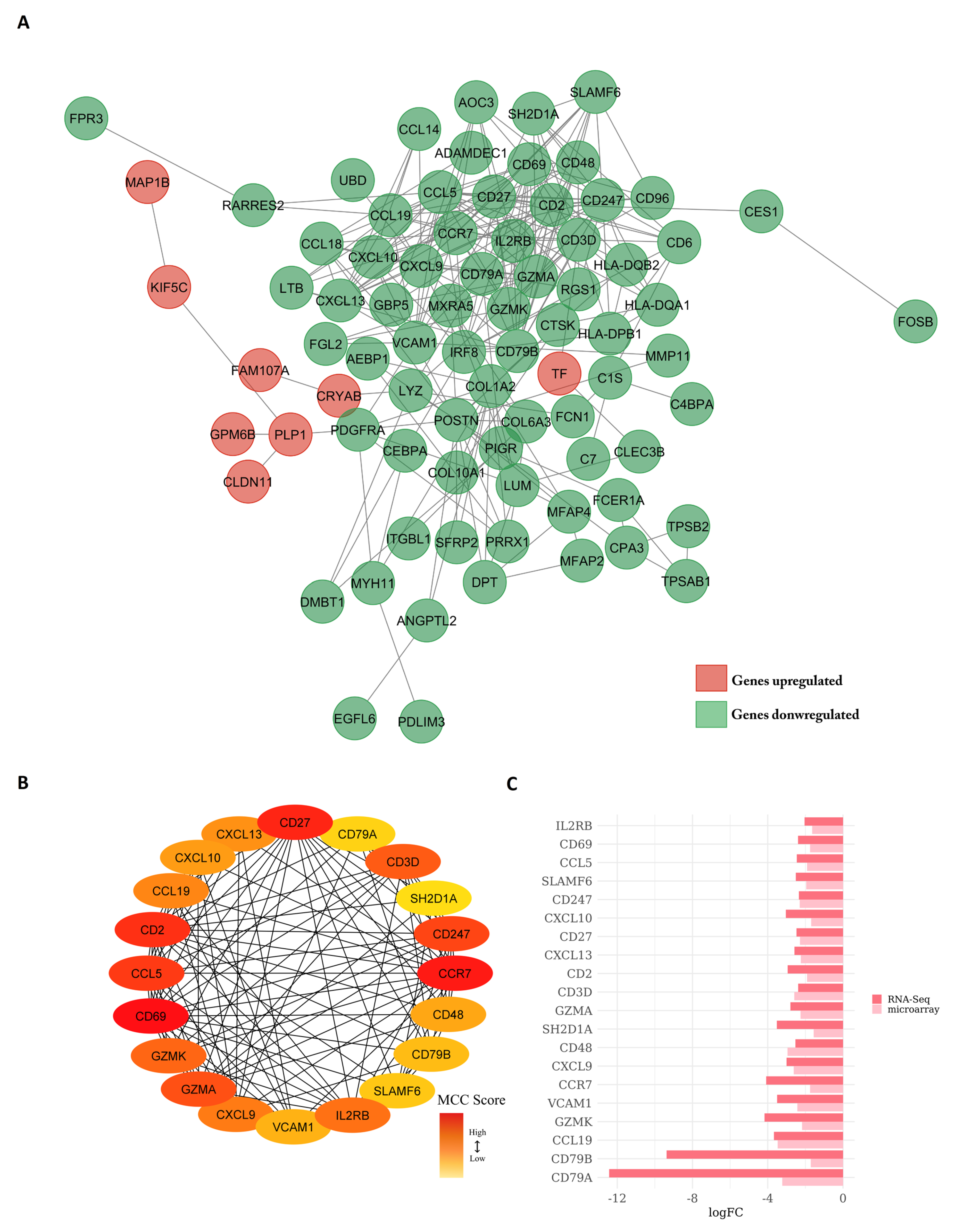
3.5. Protein–Protein Interaction Network Constructed from DEGs Reveals the Biological Network of Brain Metastasis from Lung Adenocarcinoma Is Associated with the Immune System
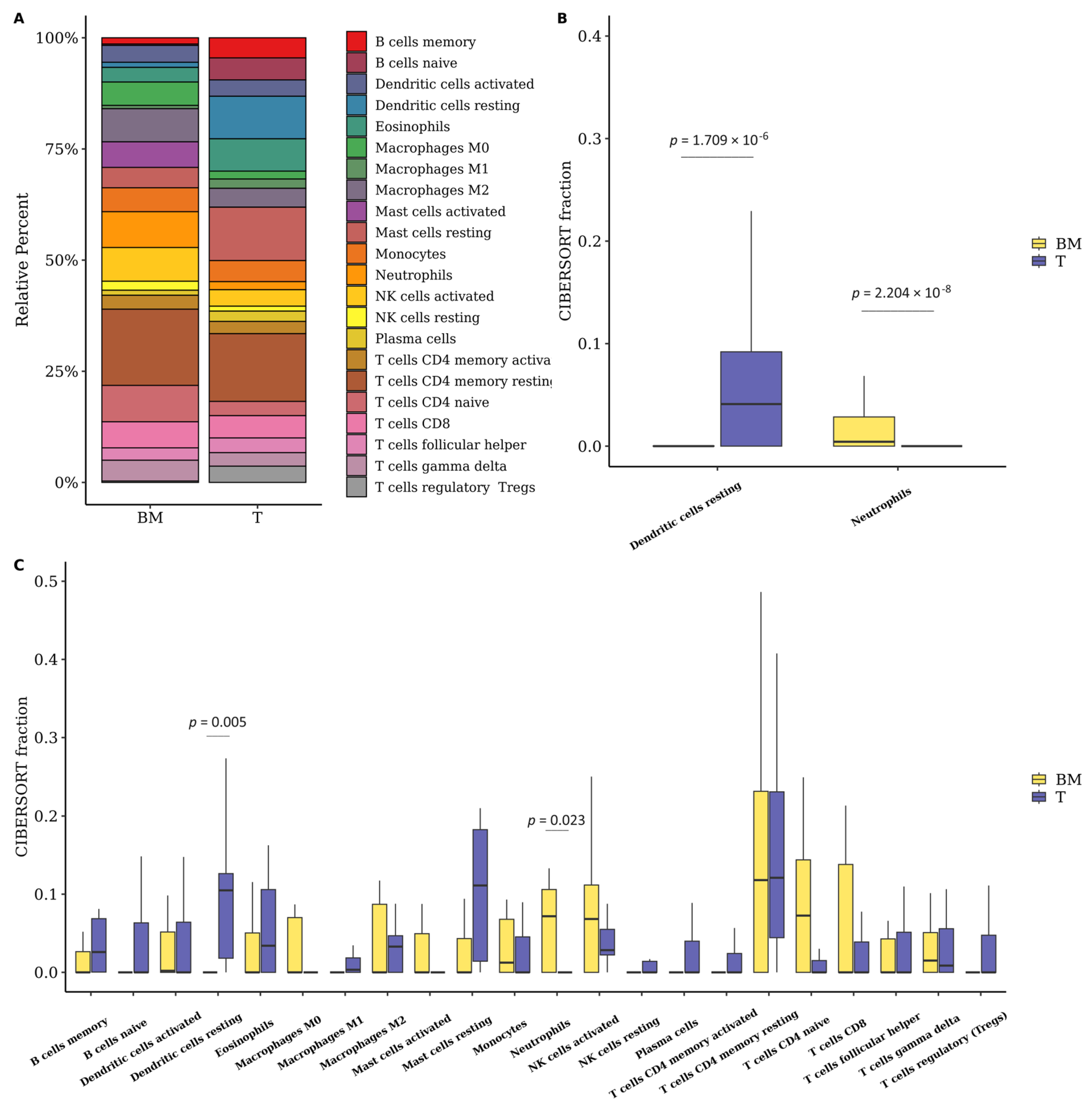
3.6. The Fraction of Neutrophils Is Greater in Brain Metastasis Compared to the Primary Tumor
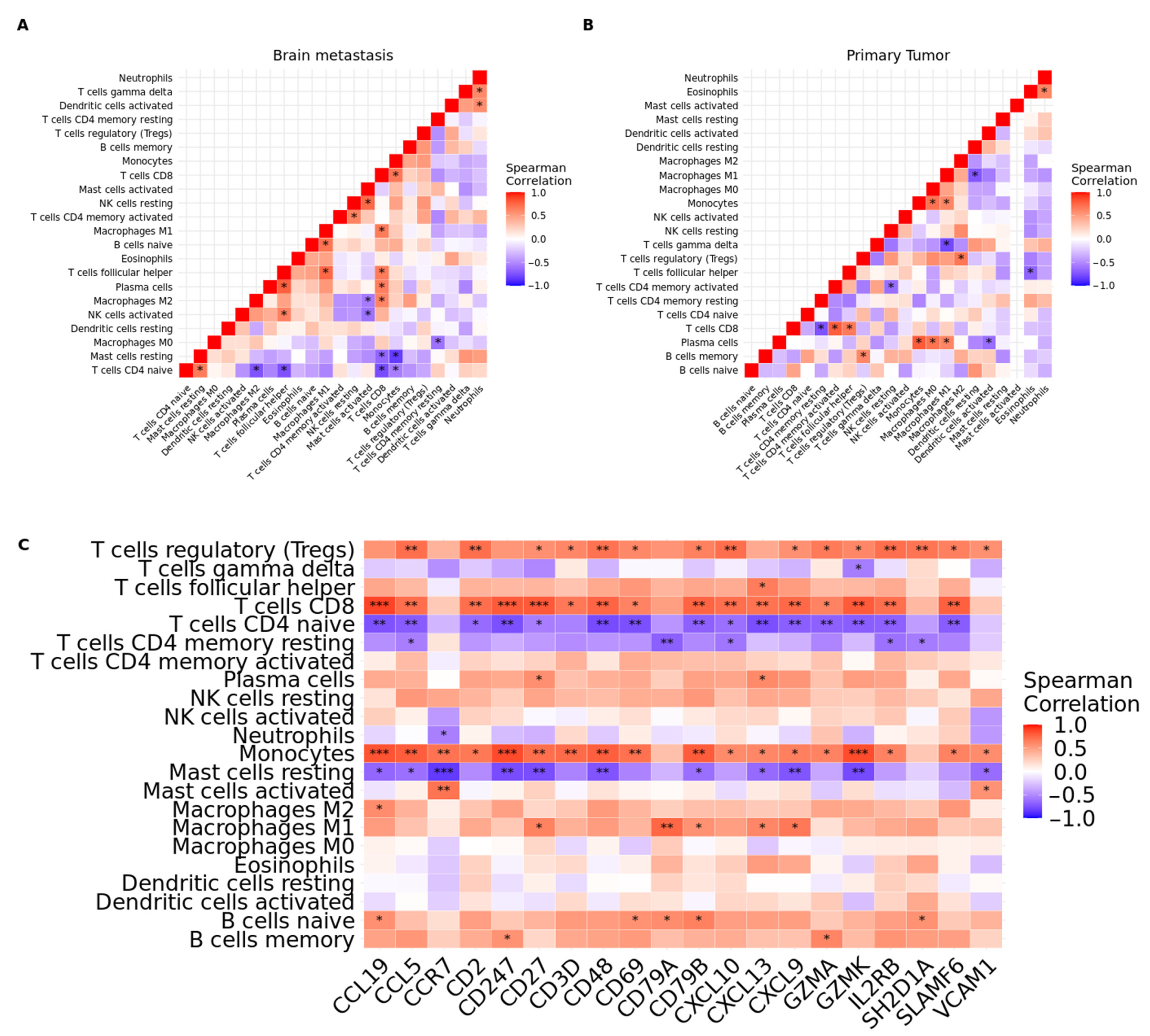
3.7. scRNA-Seq Data Reveals an Immunosuppressed Tumor Microenvironment in BM from Lung Adenocarcinoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schouten, L.J.; Rutten, J.; Huveneers, H.A.M.; Twijnstra, A. Incidence of Brain Metastases in a Cohort of Patients with Carcinoma of the Breast, Colon, Kidney, and Lung and Melanoma. Cancer 2002, 94, 2698–2705. [Google Scholar] [CrossRef] [PubMed]
- Sacks, P.; Rahman, M. Epidemiology of Brain Metastases. Neurosurg. Clin. N. Am. 2020, 31, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.; Lee, E.Q.; Wen, P.Y. Epidemiology of Brain Metastases. Curr. Oncol. Rep. 2012, 14, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Barnholtz-Sloan, J.S.; Sloan, A.E.; Davis, F.G.; Vigneau, F.D.; Lai, P.; Sawaya, R.E. Incidence Proportions of Brain Metastases in Patients Diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 2865–2872. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Schur, S.; Füreder, L.M.; Gatterbauer, B.; Dieckmann, K.; Widhalm, G.; Hainfellner, J.; Zielinski, C.C.; Birner, P.; Bartsch, R.; et al. Descriptive Statistical Analysis of a Real Life Cohort of 2419 Patients with Brain Metastases of Solid Cancers. ESMO Open 2016, 1, e000024. [Google Scholar] [CrossRef] [PubMed]
- Moro-Sibilot, D.; Smit, E.; de Castro Carpeño, J.; Lesniewski-Kmak, K.; Aerts, J.G.; Villatoro, R.; Kraaij, K.; Nacerddine, K.; Dyachkova, Y.; Smith, K.T.; et al. Non-Small Cell Lung Cancer Patients with Brain Metastases Treated with First-Line Platinum-Doublet Chemotherapy: Analysis from the European FRAME Study. Lung Cancer Amst. Neth. 2015, 90, 427–432. [Google Scholar] [CrossRef]
- Hubbs, J.L.; Boyd, J.A.; Hollis, D.; Chino, J.P.; Saynak, M.; Kelsey, C.R. Factors Associated with the Development of Brain Metastases: Analysis of 975 Patients with Early Stage Nonsmall Cell Lung Cancer. Cancer 2010, 116, 5038–5046. [Google Scholar] [CrossRef]
- Riihimäki, M.; Hemminki, A.; Fallah, M.; Thomsen, H.; Sundquist, K.; Sundquist, J.; Hemminki, K. Metastatic Sites and Survival in Lung Cancer. Lung Cancer Amst. Neth. 2014, 86, 78–84. [Google Scholar] [CrossRef]
- Achrol, A.S.; Rennert, R.C.; Anders, C.; Soffietti, R.; Ahluwalia, M.S.; Nayak, L.; Peters, S.; Arvold, N.D.; Harsh, G.R.; Steeg, P.S.; et al. Brain Metastases. Nat. Rev. Dis. Primer 2019, 5, 5. [Google Scholar] [CrossRef]
- Sperduto, P.W.; Mesko, S.; Li, J.; Cagney, D.; Aizer, A.; Lin, N.U.; Nesbit, E.; Kruser, T.J.; Chan, J.; Braunstein, S.; et al. Survival in Patients With Brain Metastases: Summary Report on the Updated Diagnosis-Specific Graded Prognostic Assessment and Definition of the Eligibility Quotient. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 3773–3784. [Google Scholar] [CrossRef]
- Souza, V.G.P.; de Araújo, R.P.; Santesso, M.R.; Seneda, A.L.; Minutentag, I.W.; Felix, T.F.; Hamamoto Filho, P.T.; Pewarchuk, M.E.; Brockley, L.J.; Marchi, F.A.; et al. Advances in the Molecular Landscape of Lung Cancer Brain Metastasis. Cancers 2023, 15, 722. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-W.; Xu, J.; Zhou, J.; Liu, W.-J. Targeted Drugs for Systemic Therapy of Lung Cancer with Brain Metastases. Oncotarget 2018, 9, 5459–5472. [Google Scholar] [CrossRef]
- Di Lorenzo, R.; Ahluwalia, M.S. Targeted Therapy of Brain Metastases: Latest Evidence and Clinical Implications. Ther. Adv. Med. Oncol. 2017, 9, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Tsui, D.C.C.; Camidge, D.R.; Rusthoven, C.G. Managing Central Nervous System Spread of Lung Cancer: The State of the Art. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 40, 642–660. [Google Scholar] [CrossRef] [PubMed]
- Buriolla, S.; Pelizzari, G.; Corvaja, C.; Alberti, M.; Targato, G.; Bortolot, M.; Torresan, S.; Cortiula, F.; Fasola, G.; Follador, A. Immunotherapy in NSCLC Patients with Brain Metastases. Int. J. Mol. Sci. 2022, 23, 7068. [Google Scholar] [CrossRef]
- Shih, D.J.H.; Nayyar, N.; Bihun, I.; Dagogo-Jack, I.; Gill, C.M.; Aquilanti, E.; Bertalan, M.; Kaplan, A.; D’Andrea, M.R.; Chukwueke, U.; et al. Genomic Characterization of Human Brain Metastases Identifies Drivers of Metastatic Lung Adenocarcinoma. Nat. Genet. 2020, 52, 371–377. [Google Scholar] [CrossRef]
- Nicoś, M.; Harbers, L.; Patrucco, E.; Kramer-Drauberg, M.; Zhang, X.; Voena, C.; Kowalczyk, A.; Bożyk, A.; Pęksa, R.; Jarosz, B.; et al. Genomic Profiling Identifies Putative Pathogenic Alterations in NSCLC Brain Metastases. JTO Clin. Res. Rep. 2022, 3, 100435. [Google Scholar] [CrossRef]
- Liu, Z.; Zheng, M.; Lei, B.; Zhou, Z.; Huang, Y.; Li, W.; Chen, Q.; Li, P.; Deng, Y. Whole-Exome Sequencing Identifies Somatic Mutations Associated with Lung Cancer Metastasis to the Brain. Ann. Transl. Med. 2021, 9, 694. [Google Scholar] [CrossRef]
- Paik, P.K.; Shen, R.; Won, H.; Rekhtman, N.; Wang, L.; Sima, C.S.; Arora, A.; Seshan, V.; Ladanyi, M.; Berger, M.F.; et al. Next-Generation Sequencing of Stage IV Squamous Cell Lung Cancers Reveals an Association of PI3K Aberrations and Evidence of Clonal Heterogeneity in Patients with Brain Metastases. Cancer Discov. 2015, 5, 610–621. [Google Scholar] [CrossRef]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef]
- Han, C.H.; Brastianos, P.K. Genetic Characterization of Brain Metastases in the Era of Targeted Therapy. Front. Oncol. 2017, 7, 230. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining Cell Type Abundance and Expression from Bulk Tissues with Digital Cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Ma, Y.; Zou, Y.; Yin, A.; Li, W.; Dong, D. HCMDB: The Human Cancer Metastasis Database. Nucleic Acids Res. 2018, 46, D950–D955. [Google Scholar] [CrossRef] [PubMed]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress Update—From Bulk to Single-Cell Expression Data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ramírez, J.C.; Deng, N.; Qiu, X.; Wu, C.; Zheng, W.J.; Wu, H. Restructured GEO: Restructuring Gene Expression Omnibus Metadata for Genome Dynamics Analysis. Database J. Biol. Databases Curation 2019, 2019, bay145. [Google Scholar] [CrossRef]
- Lappalainen, I.; Almeida-King, J.; Kumanduri, V.; Senf, A.; Spalding, J.D.; Ur-Rehman, S.; Saunders, G.; Kandasamy, J.; Caccamo, M.; Leinonen, R.; et al. The European Genome-Phenome Archive of Human Data Consented for Biomedical Research. Nat. Genet. 2015, 47, 692–695. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M.; International Nucleotide Sequence Database Collaboration. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef]
- Mailman, M.D.; Feolo, M.; Jin, Y.; Kimura, M.; Tryka, K.; Bagoutdinov, R.; Hao, L.; Kiang, A.; Paschall, J.; Phan, L.; et al. The NCBI dbGaP Database of Genotypes and Phenotypes. Nat. Genet. 2007, 39, 1181–1186. [Google Scholar] [CrossRef]
- Tryka, K.A.; Hao, L.; Sturcke, A.; Jin, Y.; Wang, Z.Y.; Ziyabari, L.; Lee, M.; Popova, N.; Sharopova, N.; Kimura, M.; et al. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2014, 42, D975–D979. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- GitHub—EGA-Archive/Ega-Download-Client: A Python-Based EGA Download Client. Available online: https://github.com/EGA-archive/ega-download-client (accessed on 8 August 2023).
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 March 2023).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinforma. Oxf. Engl. 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- GitHub—Ibest/Seqyclean. Available online: https://github.com/ibest/seqyclean (accessed on 9 August 2023).
- The UniVec Database. Available online: https://www.ncbi.nlm.nih.gov/tools/vecscreen/univec/ (accessed on 8 August 2023).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinforma. Oxf. Engl. 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python Framework to Work with High-Throughput Sequencing Data. Bioinforma. Oxf. Engl. 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinforma. Oxf. Engl. 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-Seq: Batch Effect Adjustment for RNA-Seq Count Data. NAR Genom. Bioinforma. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The Sva Package for Removing Batch Effects and Other Unwanted Variation in High-Throughput Experiments. Bioinforma. Oxf. Engl. 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2023. [Google Scholar]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. Gplots: Various R Programming Tools for Plotting Data. Available online: https://github.com/talgalili/gplots (accessed on 1 October 2021).
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor Package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A Powerful Link between Biological Databases and Microarray Data Analysis. Bioinforma. Oxf. Engl. 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Tweedie, S.; Braschi, B.; Gray, K.; Jones, T.E.M.; Seal, R.L.; Yates, B.; Bruford, E.A. Genenames.Org: The HGNC and VGNC Resources in 2021. Nucleic Acids Res. 2021, 49, D939–D946. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A Package for the Generation of Highly-Customizable Venn and Euler Diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innov. Camb. Mass 2021, 2, 100141. [Google Scholar] [CrossRef]
- Gene Ontology Consortium. The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Schriml, L.M.; Arze, C.; Nadendla, S.; Chang, Y.-W.W.; Mazaitis, M.; Felix, V.; Feng, G.; Kibbe, W.A. Disease Ontology: A Backbone for Disease Semantic Integration. Nucleic Acids Res. 2012, 40, D940–D946. [Google Scholar] [CrossRef]
- Repana, D.; Nulsen, J.; Dressler, L.; Bortolomeazzi, M.; Venkata, S.K.; Tourna, A.; Yakovleva, A.; Palmieri, T.; Ciccarelli, F.D. The Network of Cancer Genes (NCG): A Comprehensive Catalogue of Known and Candidate Cancer Genes from Cancer Sequencing Screens. Genome Biol. 2019, 20, 1. [Google Scholar] [CrossRef]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R Package for Visually Combining Expression Data with Functional Analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Carlson, M. org.Hs.eg.db: Genome Wide Annotation for Human. R Package Version 3.8.2. 2019. Available online: https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html (accessed on 1 October 2021).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying Hub Objects and Sub-Networks from Complex Interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. Clifton NJ 2018, 1711, 243–259. [Google Scholar] [CrossRef]
- Thompson, J.; Brett, C.; Neuhaus, I.; Thompson, R. DGEobj.Utils: Differential Gene Expression (DGE) Analysis Utility Toolkit. Available online: https://cran.r-project.org/web/packages/DGEobj.utils/index.html (accessed on 1 October 2021).
- R Core Team R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 14 August 2023).
- Hmisc. Available online: https://hbiostat.org/R/Hmisc/ (accessed on 8 August 2023).
- Peterson, B.G.; Carl, P.; Boudt, K.; Bennett, R.; Ulrich, J.; Zivot, E.; Cornilly, D.; Hung, E.; Lestel, M.; Balkissoon, K.; et al. PerformanceAnalytics: Econometric Tools for Performance and Risk Analysis. Available online: https://github.com/braverock/PerformanceAnalytics (accessed on 1 October 2021).
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Lee, W.-C.; Reuben, A.; Hu, X.; McGranahan, N.; Chen, R.; Jalali, A.; Negrao, M.V.; Hubert, S.M.; Tang, C.; Wu, C.-C.; et al. Multiomics Profiling of Primary Lung Cancers and Distant Metastases Reveals Immunosuppression as a Common Characteristic of Tumor Cells with Metastatic Plasticity. Genome Biol. 2020, 21, 271. [Google Scholar] [CrossRef]
- Fu, F.; Zhang, Y.; Gao, Z.; Zhao, Y.; Wen, Z.; Han, H.; Li, Y.; Chen, H. Development and Validation of a Five-Gene Model to Predict Postoperative Brain Metastasis in Operable Lung Adenocarcinoma. Int. J. Cancer 2020, 147, 584–592. [Google Scholar] [CrossRef]
- Chen, H.; Carrot-Zhang, J.; Zhao, Y.; Hu, H.; Freeman, S.S.; Yu, S.; Ha, G.; Taylor, A.M.; Berger, A.C.; Westlake, L.; et al. Genomic and Immune Profiling of Pre-Invasive Lung Adenocarcinoma. Nat. Commun. 2019, 10, 5472. [Google Scholar] [CrossRef]
- Tew, B.Y.; Legendre, C.; Gooden, G.C.; Johnson, K.N.; Martinez, R.A.; Kiefer, J.; Bernstein, M.; Glen, J.; Butry, L.; Hinek, A.; et al. Isolation and Characterization of Patient-Derived CNS Metastasis-Associated Stromal Cell Lines. Oncogene 2019, 38, 4002–4014. [Google Scholar] [CrossRef]
- Karlsson, A.; Jönsson, M.; Lauss, M.; Brunnström, H.; Jönsson, P.; Borg, Å.; Jönsson, G.; Ringnér, M.; Planck, M.; Staaf, J. Genome-Wide DNA Methylation Analysis of Lung Carcinoma Reveals One Neuroendocrine and Four Adenocarcinoma Epitypes Associated with Patient Outcome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6127–6140. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Yan, G.-R.; He, Q.-Y. DOSE: An R/Bioconductor Package for Disease Ontology Semantic and Enrichment Analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Perrot, I.; Blanchard, D.; Freymond, N.; Isaac, S.; Guibert, B.; Pachéco, Y.; Lebecque, S. Dendritic Cells Infiltrating Human Non-Small Cell Lung Cancer Are Blocked at Immature Stage. J. Immunol. Baltim. Md 1950 2007, 178, 2763–2769. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-Cell RNA-Seq Reveals New Types of Human Blood Dendritic Cells, Monocytes, and Progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef]
- Schaller, T.H.; Batich, K.A.; Suryadevara, C.M.; Desai, R.; Sampson, J.H. Chemokines as Adjuvants for Immunotherapy: Implications for Immune Activation with CCL3. Expert Rev. Clin. Immunol. 2017, 13, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Spangler, J.B.; Moraga, I.; Mendoza, J.L.; Garcia, K.C. Insights into Cytokine–Receptor Interactions from Cytokine Engineering. Annu. Rev. Immunol. 2015, 33, 139–167. [Google Scholar] [CrossRef] [PubMed]
- Tsakonas, G.; Lewensohn, R.; Botling, J.; Ortiz-Villalon, C.; Micke, P.; Friesland, S.; Nord, H.; Lindskog, M.; Sandelin, M.; Hydbring, P.; et al. An Immune Gene Expression Signature Distinguishes Central Nervous System Metastases from Primary Tumours in Non-Small-Cell Lung Cancer. Eur. J. Cancer Oxf. Engl. 1990 2020, 132, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Reckamp, K.L.; Strieter, R.M.; Figlin, R.A. Chemokines as Therapeutic Targets in Renal Cell Carcinoma. Expert Rev. Anticancer Ther. 2008, 8, 887–893. [Google Scholar] [CrossRef][Green Version]
- Cheng, Z.-H.; Shi, Y.-X.; Yuan, M.; Xiong, D.; Zheng, J.-H.; Zhang, Z.-Y. Chemokines and Their Receptors in Lung Cancer Progression and Metastasis. J. Zhejiang Univ. Sci. B 2016, 17, 342–351. [Google Scholar] [CrossRef]
- Wang, H.; Ou, Q.; Li, D.; Qin, T.; Bao, H.; Hou, X.; Wang, K.; Wang, F.; Deng, Q.; Liang, J.; et al. Genes Associated with Increased Brain Metastasis Risk in Non-Small Cell Lung Cancer: Comprehensive Genomic Profiling of 61 Resected Brain Metastases versus Primary Non-Small Cell Lung Cancer (Guangdong Association Study of Thoracic Oncology 1036). Cancer 2019, 125, 3535–3544. [Google Scholar] [CrossRef]
- Barber, E.K.; Dasgupta, J.D.; Schlossman, S.F.; Trevillyan, J.M.; Rudd, C.E. The CD4 and CD8 Antigens Are Coupled to a Protein-Tyrosine Kinase (P56lck) That Phosphorylates the CD3 Complex. Proc. Natl. Acad. Sci. USA 1989, 86, 3277–3281. [Google Scholar] [CrossRef] [PubMed]
- Doucey, M.-A.; Goffin, L.; Naeher, D.; Michielin, O.; Baumgärtner, P.; Guillaume, P.; Palmer, E.; Luescher, I.F. CD3 Delta Establishes a Functional Link between the T Cell Receptor and CD8. J. Biol. Chem. 2003, 278, 3257–3264. [Google Scholar] [CrossRef] [PubMed]
- Menon, A.P.; Moreno, B.; Meraviglia-Crivelli, D.; Nonatelli, F.; Villanueva, H.; Barainka, M.; Zheleva, A.; van Santen, H.M.; Pastor, F. Modulating T Cell Responses by Targeting CD3. Cancers 2023, 15, 1189. [Google Scholar] [CrossRef] [PubMed]
- Cibrián, D.; Sánchez-Madrid, F. CD69: From Activation Marker to Metabolic Gatekeeper. Eur. J. Immunol. 2017, 47, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Leibold, A.T.; Monaco, G.N.; Dey, M. The Role of the Immune System in Brain Metastasis. Curr. Neurobiol. 2019, 10, 33–48. [Google Scholar]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Janssen, L.M.E.; Ramsay, E.E.; Logsdon, C.D.; Overwijk, W.W. The Immune System in Cancer Metastasis: Friend or Foe? J. Immunother. Cancer 2017, 5, 79. [Google Scholar] [CrossRef]
- Qiao, S.; Qian, Y.; Xu, G.; Luo, Q.; Zhang, Z. Long-Term Characterization of Activated Microglia/Macrophages Facilitating the Development of Experimental Brain Metastasis through Intravital Microscopic Imaging. J. Neuroinflammation 2019, 16, 4. [Google Scholar] [CrossRef]
- Marin, J.; Journe, F.; Ghanem, G.E.; Awada, A.; Kindt, N. Cytokine Landscape in Central Nervous System Metastases. Biomedicines 2022, 10, 1537. [Google Scholar] [CrossRef]
- Kudo, Y.; Haymaker, C.; Zhang, J.; Reuben, A.; Duose, D.Y.; Fujimoto, J.; Roy-Chowdhuri, S.; Solis Soto, L.M.; Dejima, H.; Parra, E.R.; et al. Suppressed Immune Microenvironment and Repertoire in Brain Metastases from Patients with Resected Non-Small-Cell Lung Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1521–1530. [Google Scholar] [CrossRef]
- Zhang, Q.; Abdo, R.; Iosef, C.; Kaneko, T.; Cecchini, M.; Han, V.K.; Li, S.S.-C. The Spatial Transcriptomic Landscape of Non-Small Cell Lung Cancer Brain Metastasis. Nat. Commun. 2022, 13, 5983. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Lassmann, H.; Preusser, M.; Höftberger, R. Characterization of the Inflammatory Response to Solid Cancer Metastases in the Human Brain. Clin. Exp. Metastasis 2013, 30, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Song, S.G.; Kim, S.; Koh, J.; Yim, J.; Han, B.; Kim, Y.A.; Jeon, Y.K.; Chung, D.H. Comparative Analysis of the Tumor Immune-Microenvironment of Primary and Brain Metastases of Non-Small-Cell Lung Cancer Reveals Organ-Specific and EGFR Mutation-Dependent Unique Immune Landscape. Cancer Immunol. Immunother. CII 2021, 70, 2035–2048. [Google Scholar] [CrossRef] [PubMed]
- Najjary, S.; Kros, J.M.; de Koning, W.; Vadgama, D.; Lila, K.; Wolf, J.; Mustafa, D.A.M. Tumor Lineage-Specific Immune Response in Brain Metastatic Disease: Opportunities for Targeted Immunotherapy Regimen? Acta Neuropathol. Commun. 2023, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Ewen, C.L.; Kane, K.P.; Bleackley, R.C. A Quarter Century of Granzymes. Cell Death Differ. 2012, 19, 28–35. [Google Scholar] [CrossRef]
- Kam, C.M.; Hudig, D.; Powers, J.C. Granzymes (Lymphocyte Serine Proteases): Characterization with Natural and Synthetic Substrates and Inhibitors. Biochim. Biophys. Acta 2000, 1477, 307–323. [Google Scholar] [CrossRef]
- Masson, D.; Tschopp, J. A Family of Serine Esterases in Lytic Granules of Cytolytic T Lymphocytes. Cell 1987, 49, 679–685. [Google Scholar] [CrossRef]
- Cachot, A.; Bilous, M.; Liu, Y.-C.; Li, X.; Saillard, M.; Cenerenti, M.; Rockinger, G.A.; Wyss, T.; Guillaume, P.; Schmidt, J.; et al. Tumor-Specific Cytolytic CD4 T Cells Mediate Immunity against Human Cancer. Sci. Adv. 2021, 7, eabe3348. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global Characterization of T Cells in Non-Small-Cell Lung Cancer by Single-Cell Sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef]
- Shimizu, K.; Yamasaki, S.; Sakurai, M.; Yumoto, N.; Ikeda, M.; Mishima-Tsumagari, C.; Kukimoto-Niino, M.; Watanabe, T.; Kawamura, M.; Shirouzu, M.; et al. Granzyme A Stimulates pDCs to Promote Adaptive Immunity via Induction of Type I IFN. Front. Immunol. 2019, 10, 1450. [Google Scholar] [CrossRef]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from Cytotoxic Lymphocytes Cleaves GSDMB to Trigger Pyroptosis in Target Cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef]
- Huo, Q.; Ning, L.; Xie, N. Identification of GZMA as a Potential Therapeutic Target Involved in Immune Infiltration in Breast Cancer by Integrated Bioinformatical Analysis. Breast Cancer Dove Med. Press 2023, 15, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Giles, D.A.; Duncker, P.C.; Wilkinson, N.M.; Washnock-Schmid, J.M.; Segal, B.M. CNS-Resident Classical DCs Play a Critical Role in CNS Autoimmune Disease. J. Clin. Invest. 2018, 128, 5322–5334. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.-I.; Suh, Y.-L.; Ku, B.M.; Eum, H.H.; et al. Single-Cell RNA Sequencing Demonstrates the Molecular and Cellular Reprogramming of Metastatic Lung Adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef]
- Bousso, P. T-Cell Activation by Dendritic Cells in the Lymph Node: Lessons from the Movies. Nat. Rev. Immunol. 2008, 8, 675–684. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A. The Instructive Role of Dendritic Cells on T-Cell Responses. Arthritis Res. 2002, 4 (Suppl. 3), S127–S132. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, C.-A.; Becht, E.; Irac, S.E.; Khalilnezhad, A.; Narang, V.; Khalilnezhad, S.; Ng, P.Y.; van den Hoogen, L.L.; Leong, J.Y.; Lee, B.; et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 2019, 51, 573–589. [Google Scholar] [CrossRef]
- Zhou, J.; Nefedova, Y.; Lei, A.; Gabrilovich, D. Neutrophils and PMN-MDSC: Their Biological Role and Interaction with Stromal Cells. Semin. Immunol. 2018, 35, 19–28. [Google Scholar] [CrossRef]
- Sun, H.-F.; Li, L.-D.; Lao, I.-W.; Li, X.; Xu, B.-J.; Cao, Y.-Q.; Jin, W. Single-Cell RNA Sequencing Reveals Cellular and Molecular Reprograming Landscape of Gliomas and Lung Cancer Brain Metastases. Clin. Transl. Med. 2022, 12, e1101. [Google Scholar] [CrossRef]
- Rahman, I.; Collado Sánchez, A.; Davies, J.; Rzeniewicz, K.; Abukscem, S.; Joachim, J.; Hoskins Green, H.L.; Killock, D.; Sanz, M.J.; Charras, G.; et al. L-Selectin Regulates Human Neutrophil Transendothelial Migration. J. Cell Sci. 2021, 134, jcs250340. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in Brain Tumors. Glia 2002, 40, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The Brain Tumor Microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils Promote the Malignant Glioma Phenotype through S100A4. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 Proteins in Cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kosaka, A.; Ikeura, M.; Kohanbash, G.; Fellows-Mayle, W.; Snyder, L.A.; Okada, H. Premetastatic Soil and Prevention of Breast Cancer Brain Metastasis. Neuro-Oncol. 2013, 15, 891–903. [Google Scholar] [CrossRef]
- Ahmad, A.; Khan, P.; Rehman, A.U.; Batra, S.K.; Nasser, M.W. Immunotherapy: An Emerging Modality to Checkmate Brain Metastasis. Mol. Cancer 2023, 22, 111. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Database | Access | Technology | Platform | Tissue | Number of Samples | Reference |
|---|---|---|---|---|---|---|---|
| 1 | EGA | EGAS00001004078 | RNA-seq | Illumina HiSeq 2000 | BM/T | 5 BM 4 T | [68] |
| 2 | EGA | EGAS00001004006 | RNA-seq | Illumina HiSeq X Ten | T | 7 | [69,70] |
| 3 | SRA | PRJNA510710 | RNA-seq | Illumina HiSeq 2500 | BM | 2 | [71] |
| 4 | GEO | GSE141685 | RNA-seq | Illumina HiSeq X Ten | BM | 6 | NA |
| 5 | ArrayExpress | E-MTAB-8659 | microarray | Illumina HumanHT-12 V4.0 expression beadchip | BM | 63 | NA |
| 6 | GEO | GSE60645 | microarray | Illumina HumanHT-12 V4.0 expression beadchip | T | 77 | [72] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souza, V.G.P.; Forder, A.; Telkar, N.; Stewart, G.L.; Carvalho, R.F.; Mur, L.A.J.; Lam, W.L.; Reis, P.P. Identifying New Contributors to Brain Metastasis in Lung Adenocarcinoma: A Transcriptomic Meta-Analysis. Cancers 2023, 15, 4526. https://doi.org/10.3390/cancers15184526
Souza VGP, Forder A, Telkar N, Stewart GL, Carvalho RF, Mur LAJ, Lam WL, Reis PP. Identifying New Contributors to Brain Metastasis in Lung Adenocarcinoma: A Transcriptomic Meta-Analysis. Cancers. 2023; 15(18):4526. https://doi.org/10.3390/cancers15184526
Chicago/Turabian StyleSouza, Vanessa G. P., Aisling Forder, Nikita Telkar, Greg L. Stewart, Robson F. Carvalho, Luis A. J. Mur, Wan L. Lam, and Patricia P. Reis. 2023. "Identifying New Contributors to Brain Metastasis in Lung Adenocarcinoma: A Transcriptomic Meta-Analysis" Cancers 15, no. 18: 4526. https://doi.org/10.3390/cancers15184526
APA StyleSouza, V. G. P., Forder, A., Telkar, N., Stewart, G. L., Carvalho, R. F., Mur, L. A. J., Lam, W. L., & Reis, P. P. (2023). Identifying New Contributors to Brain Metastasis in Lung Adenocarcinoma: A Transcriptomic Meta-Analysis. Cancers, 15(18), 4526. https://doi.org/10.3390/cancers15184526