
Extracellular Matrix Collagen I Differentially Regulates the Metabolic Plasticity of Pancreatic Ductal Adenocarcinoma Parenchymal Cell and Cancer Stem Cell

 , , ,
, , ,  ,
,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Drugs and Their Application
2.3. Three Dimensional Organotypic Growth
2.4. Cell Viability Assay
2.5. Cell Death Assay
2.6. Extracellular Glucose and Lactate and cell ATP Content Assays
2.7. Quantification of Amino Acids in the Growth Medium by HLPC
2.8. Bioenergetic Modulator (BM) Effect on NAB-PTX Cytotoxicity
2.9. Western Blotting Analysis
2.10. Statistical Analysis
3. Results
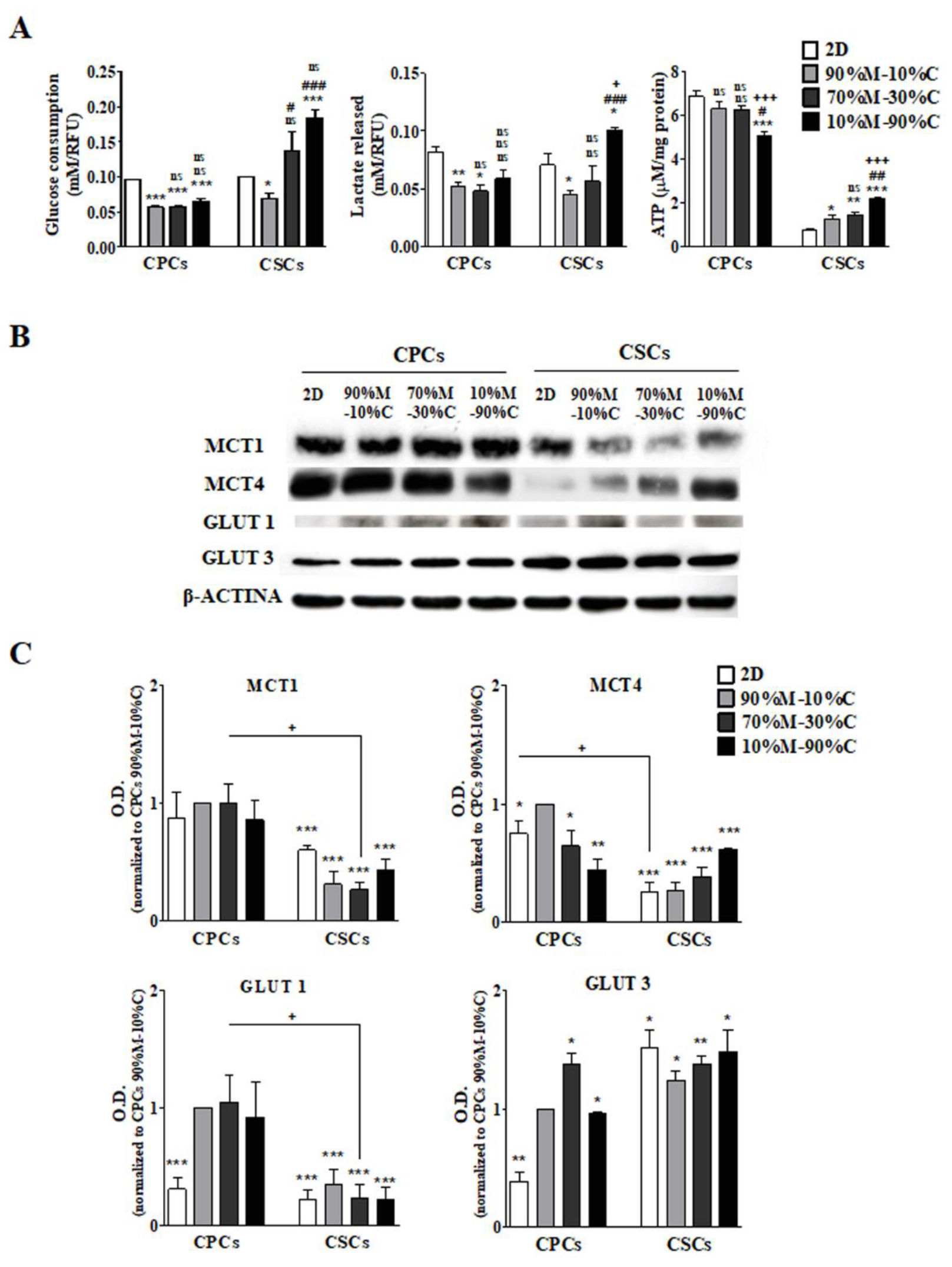
3.1. ECM Composition Is Involved in the Regulation of Metabolic Plasticity, Especially in CSCs
3.1.1. CPCs
3.1.2. CSCs
3.2. High ECM Collagen I Percentage Increases Glutamine Consumption and Glutaminolysis in CSCs
3.3. Treatment with Bioenergetic Modulators (BMs) Affects Cell Growth and Survival of PDAC Cells
3.4. The Disruption of Energetic Pathways Synergistically Potentiates NAB-PTX Action
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated projection of US cancer incidence and death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef]
- Chu, G.C.; Kimmelman, A.C.; Hezel, A.F.; DePinho, R.A. Stromal biology of pancreatic cancer. J. Cell. Biochem. 2007, 101, 887–907. [Google Scholar] [CrossRef]
- Mahadevan, D.; Von Hoff, D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef]
- Hadden, M.; Mittal, A.; Samra, J.; Zreiqat, H.; Sahni, S.; Ramaswamy, Y. Mechanically stressed cancer microenvironment: Role in pancreatic cancer progression. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1874, 188418. [Google Scholar] [CrossRef]
- Carvalho, T.M.; Di Molfetta, D.; Greco, M.R.; Koltai, T.; Alfarouk, K.O.; Reshkin, S.J.; Cardone, R.A. Tumor microenvironment features and chemoresistance in pancreatic ductal adenocarcinoma: Insights into targeting physicochemical barriers and metabolism as therapeutic approaches. Cancers 2021, 13, 6135. [Google Scholar] [CrossRef]
- Ferrara, B.; Pignatelli, C.; Cossutta, M.; Citro, A.; Courty, J.; Piemonti, L. The extracellular matrix in pancreatic cancer: Description of a complex network and promising therapeutic options. Cancers 2021, 13, 4442. [Google Scholar] [CrossRef]
- Perez, V.M.; Kearney, J.F.; Yeh, J.J. The PDAC extracellular matrix: A review of the ECM protein composition, tumor cell interaction, and therapeutic strategies. Front. Oncol. 2021, 11, 751311. [Google Scholar] [CrossRef]
- Ide, T.; Kitajima, Y.; Miyoshi, A.; Ohtsuka, T.; Mitsuno, M.; Ohtaka, K.; Miyazaki, K. The hypoxic environment in tumor-stromal cells accelerates pancreatic cancer progression via the activation of paracrine hepatocyte growth factor/c-Met signaling. Ann. Surg. Oncol. 2007, 14, 2600–2607. [Google Scholar] [CrossRef]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef]
- Chang, Q.; Jurisica, I.; Do, T.; Hedley, D.W. Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res. 2011, 71, 3110–3120. [Google Scholar] [CrossRef]
- Miyake, K.; Yoshizumi, T.; Imura, S.; Sugimoto, K.; Batmunkh, E.; Kanemura, H.; Morine, Y.; Shimada, M. Expression of hypoxia-inducible factor-1α, histone deacetylase 1, and metastasis-associated protein 1 in pancreatic carcinoma: Correlation with poor prognosis with possible regulation. Pancreas 2008, 36, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Liu, Q.; Liao, Q.; Zhao, Y. Chemotherapy and tumor microenvironment of pancreatic cancer. Cancer Cell Int. 2017, 17, 68. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Römmler-Zehrer, J.; Li, J.S.; McGovern, D.; Romano, A.; Stahl, M. Efficacy and safety profile of nab-paclitaxel plus gemcitabine in patients with metastatic pancreatic cancer treated to disease progression: A subanalysis from a phase 3 trial (MPACT). BMC Cancer 2016, 16, 817. [Google Scholar] [CrossRef] [PubMed]
- Vaz, A.P.; Ponnusamy, M.P.; Rachagani, S.; Dey, P.; Ganti, A.K.; Batra, S.K. Novel role of pancreatic differentiation in facilitating self-renewal and drug resistance of pancreatic cancer stem cells. Br. J. Cancer 2014, 111, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Tan, L.; Liu, B. Cancer stem cells: Recent insights and therapies. Biochem. Pharmacol. 2023, 209, 115441. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Yan, X.; Ma, L.; Yi, D.; Yoon, J.-G.; Diercks, A.; Foltz, G.; Price, N.D.; Hood, L.E.; Tian, Q. A CD133-related gene expression signature identifies an aggressive glioblastoma subtype with excessive mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 1591–1596. [Google Scholar] [CrossRef]
- Shmelkov, S.V.; Butler, J.M.; Hooper, A.T.; Hormigo, A.; Kushner, J.; Milde, T.; Clair, R.S.; Baljevic, M.; White, I.; Jin, D.K. CD133 expression is not restricted to stem cells, and both CD133+ and CD133–metastatic colon cancer cells initiate tumors. J. Clin. Investig. 2008, 118, 2111–2120. [Google Scholar] [CrossRef]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 16281–16286. [Google Scholar] [CrossRef]
- Meyer, M.J.; Fleming, J.M.; Lin, A.F.; Hussnain, S.A.; Ginsburg, E.; Vonderhaar, B.K. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor–negative breast cancer. Cancer Res. 2010, 70, 4624–4633. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Sumbly, V.; Landry, I. Understanding pancreatic cancer stem cells and their role in carcinogenesis: A narrative review. Stem Cell Investig. 2022, 9, 1. [Google Scholar] [CrossRef]
- Stoica, A.-F.; Chang, C.-H.; Pauklin, S. Molecular Therapeutics of Pancreatic Ductal Adenocarcinoma: Targeted Pathways and the Role of Cancer Stem Cells. Trends Pharmacol. Sci. 2020, 41, 977–993. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, G.P.; Farran, B.; Luong, T.; El-Rayes, B.F. Understanding the molecular mechanisms that regulate pancreatic cancer stem cell formation, stemness and chemoresistance: A brief overview. Semin. Cancer Biol. 2023, 88, 67–80. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Baek, G.; Yan, F.T.; Hu, Z.; Cox, D.; Buboltz, N.; McCue, P.; Yeo, C.J.; White, M.A.; DeBerardinis, R.J.; Knudsen, E.S. MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 2014, 9, 2233–2249. [Google Scholar] [CrossRef]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 2015, 6, 651. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Dalla Pozza, E.; Biondani, G.; Cordani, M.; Palmieri, M.; Donadelli, M. The metabolic landscape of cancer stem cells. IUBMB Life 2015, 67, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Abrego, J.; Gunda, V.; Vernucci, E.; Shukla, S.K.; King, R.J.; Dasgupta, A.; Goode, G.; Murthy, D.; Yu, F.; Singh, P.K. GOT1-mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett. 2017, 400, 37–46. [Google Scholar] [CrossRef]
- Padinharayil, H.; Rai, V.; George, A. Mitochondrial Metabolism in Pancreatic Ductal Adenocarcinoma: From Mechanism-Based Perspectives to Therapy. Cancers 2023, 15, 1070. [Google Scholar] [CrossRef]
- Roux, C.; Riganti, C.; Borgogno, S.F.; Curto, R.; Curcio, C.; Catanzaro, V.; Digilio, G.; Padovan, S.; Puccinelli, M.P.; Isabello, M. Endogenous glutamine decrease is associated with pancreatic cancer progression. Oncotarget 2017, 8, 95361. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Yang, S.; Hwang, S.; Kim, M.; Seo, S.B.; Lee, J.-H.; Jeong, S.M. Mitochondrial glutamine metabolism via GOT2 supports pancreatic cancer growth through senescence inhibition. Cell Death Dis. 2018, 9, 55. [Google Scholar] [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef]
- Pastò, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef]
- Valle, S.; Alcalá, S.; Martin-Hijano, L.; Cabezas-Sáinz, P.; Navarro, D.; Muñoz, E.R.; Yuste, L.; Tiwary, K.; Walter, K.; Ruiz-Cañas, L. Exploiting oxidative phosphorylation to promote the stem and immunoevasive properties of pancreatic cancer stem cells. Nat. Commun. 2020, 11, 5265. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-M.; Zhang, J.-G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef]
- Dadgar, T.; Ebrahimi, N.; Gholipour, A.R.; Akbari, M.; Khani, L.; Ahmadi, A.; Hamblin, M.R. Targeting the metabolism of cancer stem cells by energy disruptor molecules. Crit. Rev. Oncol. Hematol. 2022, 169, 103545. [Google Scholar] [CrossRef] [PubMed]
- Karp, I.; Lyakhovich, A. Targeting cancer stem cells with antibiotics inducing mitochondrial dysfunction as an alternative anticancer therapy. Biochem. Pharmacol. 2022, 198, 114966. [Google Scholar] [CrossRef]
- Yun, Z.; Lin, Q. Hypoxia and Regulation of Cancer Cell Stemness. In Tumor Microenvironment and Cellular Stress; Springer: New York, NY, USA, 2014; pp. 41–53. [Google Scholar]
- Palorini, R.; Votta, G.; Balestrieri, C.; Monestiroli, A.; Olivieri, S.; Vento, R.; Chiaradonna, F. Energy Metabolism Characterization of a Novel Cancer Stem Cell-L ike Line 3 AB-OS. J. Cell. Biochem. 2014, 115, 368–379. [Google Scholar] [CrossRef]
- Liu, P.; Liao, J.; Tang, Z.; Wu, W.; Yang, J.; Zeng, Z.; Hu, Y.; Wang, P.; Ju, H.; Xu, R. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014, 21, 124–135. [Google Scholar] [CrossRef]
- Emmink, B.L.; Verheem, A.; Van Houdt, W.J.; Steller, E.J.; Govaert, K.M.; Pham, T.V.; Piersma, S.R.; Rinkes, I.H.B.; Jimenez, C.R.; Kranenburg, O. The secretome of colon cancer stem cells contains drug-metabolizing enzymes. J. Proteom. 2013, 91, 84–96. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef]
- Yadav, U.P.; Singh, T.; Kumar, P.; Sharma, P.; Kaur, H.; Sharma, S.; Singh, S.; Kumar, S.; Mehta, K. Metabolic Adaptations in cancer stem cells. Front. Oncol. 2020, 10, 1010. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.-W.; Park, S.-B.; Lee, S.-J.; Seo, M.-S.; Trosko, J.E.; Kang, K.-S. Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression. PLoS ONE 2011, 6, e28068. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Klotz, L.; Venkateswaran, V. Metformin and prostate cancer stem cells: A novel therapeutic target. Prostate Cancer Prostatic Dis. 2015, 18, 303–309. [Google Scholar] [CrossRef]
- Chai, X.; Chu, H.; Yang, X.; Meng, Y.; Shi, P.; Gou, S. Metformin increases sensitivity of pancreatic cancer cells to gemcitabine by reducing CD133+ cell populations and suppressing ERK/P70S6K signaling. Sci. Rep. 2015, 5, 14404. [Google Scholar] [CrossRef]
- Kordes, S.; Pollak, M.N.; Zwinderman, A.H.; Mathôt, R.A.; Weterman, M.J.; Beeker, A.; Punt, C.J.; Richel, D.J.; Wilmink, J.W. Metformin in patients with advanced pancreatic cancer: A double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015, 16, 839–847. [Google Scholar] [CrossRef]
- Reni, M.; Dugnani, E.; Cereda, S.; Belli, C.; Balzano, G.; Nicoletti, R.; Liberati, D.; Pasquale, V.; Scavini, M.; Maggiora, P. (Ir) relevance of metformin treatment in patients with metastatic pancreatic cancer: An open-label, randomized phase II trial. Clin. Cancer Res. 2016, 22, 1076–1085. [Google Scholar] [CrossRef]
- Masoud, R.; Reyes-Castellanos, G.; Lac, S.; Garcia, J.; Dou, S.; Shintu, L.; Hadi, N.A.; Gicquel, T.; El Kaoutari, A.; Die, B.; et al. Targeting Mitochondrial Complex I Overcomes Chemoresistance in High OXPHOS Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100143. [Google Scholar] [CrossRef]
- Blum, R.; Kloog, Y. Metabolism addiction in pancreatic cancer. Cell Death Dis. 2014, 5, e1065. [Google Scholar] [CrossRef]
- Chan, A.K.; Bruce, J.I.; Siriwardena, A.K. Glucose metabolic phenotype of pancreatic cancer. World J. Gastroenterol. 2016, 22, 3471. [Google Scholar] [CrossRef] [PubMed]
- Daemen, A.; Peterson, D.; Sahu, N.; McCord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.; Gao, M. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E4410–E4417. [Google Scholar] [CrossRef] [PubMed]
- Zeeberg, K.; Cardone, R.A.; Greco, M.R.; Saccomano, M.; Nøhr-Nielsen, A.; Alves, F.; Pedersen, S.F.; Reshkin, S.J. Assessment of different 3D culture systems to study tumor phenotype and chemosensitivity in pancreatic ductal adenocarcinoma. Int. J. Oncol. 2016, 49, 243–252. [Google Scholar] [CrossRef] [PubMed][Green Version]
- DeClerck, Y.A. Desmoplasia: A response or a niche? Cancer Discov. 2012, 2, 772–774. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, Y.; Xu, X.-X.; Guo, X.; Sun, G.-W.; Ma, X.-J. Mesenchymal stem cells enhance the metastasis of 3D-cultured hepatocellular carcinoma cells. BMC Cancer 2016, 16, 566. [Google Scholar] [CrossRef]
- Puls, T.; Tan, X.; Whittington, C.F.; Voytik-Harbin, S.L. 3D collagen fibrillar microstructure guides pancreatic cancer cell phenotype and serves as a critical design parameter for phenotypic models of EMT. PLoS ONE 2017, 12, e0188870. [Google Scholar] [CrossRef] [PubMed]
- Biondani, G.; Zeeberg, K.; Greco, M.R.; Cannone, S.; Dando, I.; Dalla Pozza, E.; Mastrodonato, M.; Forciniti, S.; Casavola, V.; Palmieri, M. Extracellular matrix composition modulates PDAC parenchymal and stem cell plasticity and behavior through the secretome. FEBS J. 2018, 285, 2104–2124. [Google Scholar] [CrossRef] [PubMed]
- Forciniti, S.; Dalla Pozza, E.; Greco, M.R.; Amaral Carvalho, T.M.; Rolando, B.; Ambrosini, G.; Carmona-Carmona, C.A.; Pacchiana, R.; Di Molfetta, D.; Donadelli, M. Extracellular Matrix Composition Modulates the Responsiveness of Differentiated and Stem Pancreatic Cancer Cells to Lipophilic Derivate of Gemcitabine. Int. J. Mol. Sci. 2021, 22, 29. [Google Scholar] [CrossRef]
- Azzariti, A.; Brunetti, O.; Porcelli, L.; Graziano, G.; Iacobazzi, R.M.; Signorile, M.; Scarpa, A.; Lorusso, V.; Silvestris, N. Potential predictive role of chemotherapy-induced changes of soluble CD40 ligand in untreated advanced pancreatic ductal adenocarcinoma. Onco Targets Ther. 2016, 9, 4681. [Google Scholar]
- Porcelli, L.; Guida, G.; Tommasi, S.; Guida, M.; Azzariti, A. Metastatic melanoma cells with BRAF G469A mutation: Nab-paclitaxel better than vemurafenib? Cancer Chemother. Pharmacol. 2015, 76, 433–438. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A metabolic driver in the tumour landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Pértega-Gomes, N.; Vizcaíno, J.R.; Attig, J.; Jurmeister, S.; Lopes, C.; Baltazar, F. A lactate shuttle system between tumour and stromal cells is associated with poor prognosis in prostate cancer. BMC Cancer 2014, 14, 352. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Dadhich, R.K.; Dhup, S.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharmacol. 2011, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic Coupling and the Reverse Warburg Effect in Cancer: Implications for Novel Biomarker and Anticancer Agent Development. In Seminars in oncology; Paper presented at the Seminars in oncology; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Baltazar, F.; Afonso, J.; Costa, M.; Granja, S. Lactate beyond a waste metabolite: Metabolic affairs and signaling in malignancy. Front. Oncol. 2020, 10, 231. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Finley, L.W. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019, 1, 177–188. [Google Scholar] [CrossRef]
- Yu, Y.; Newman, H.; Shen, L.; Sharma, D.; Hu, G.; Mirando, A.J.; Zhang, H.; Knudsen, E.; Zhang, G.-F.; Hilton, M.J. Glutamine metabolism regulates proliferation and lineage allocation in skeletal stem cells. Cell Metab. 2019, 29, 966–978.e4. [Google Scholar] [CrossRef]
- Jeong, S.M.; Hwang, S.; Park, K.; Yang, S.; Seong, R.H. Enhanced mitochondrial glutamine anaplerosis suppresses pancreatic cancer growth through autophagy inhibition. Sci. Rep. 2016, 6, 30767. [Google Scholar] [CrossRef]
- Raho, S.; Capobianco, L.; Malivindi, R.; Vozza, A.; Piazzolla, C.; De Leonardis, F.; Gorgoglione, R.; Scarcia, P.; Pezzuto, F.; Agrimi, G. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat. Metab. 2020, 2, 1373–1381. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Juri, A.; Ionescu, C.; Berindan-Neagoe, I.; Braicu, C. The extracellular matrix alteration, implication in modulation of drug resistance mechanism: Friends or foes? J. Exp. Clin. Cancer Res. 2022, 41, 276. [Google Scholar]
- Parker, A.L.; Bowman, E.; Zingone, A.; Ryan, B.M.; Cooper, W.A.; Kohonen, M.; Harris, C.C.; Cox, T.R. Extracellular matrix profiles determine risk and prognosis of the squamous cell carcinoma subtype of non-small cell lung carcinoma. Genome Med. 2022, 14, 126. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Kapoor, A.; Zhang, Q.; Li, J.; Wu, C.-J.; Li, J.; Li, J.; Lan, Z.; Tang, M.; Ma, X.; et al. Tumor microenvironment remodeling enables bypass of oncogenic KRAS dependency in pancreatic cancer. Cancer Discov. 2020, 10, 1058–1077. [Google Scholar] [CrossRef] [PubMed]
- Cannone, S.; Greco, M.R.; Carvalho, T.M.A.; Guizouarn, H.; Soriani, O.; DiMolfetta, D.; Tomasini, R.; Zeeberg, K.; Reshkin, S.J.; Cardone, R.A. Cancer Associated Fibroblast (CAF) Regulation of PDAC Parenchymal (CPC) and CSC Phenotypes Is Modu-lated by ECM Composition. Cancers 2022, 14, 3737. [Google Scholar] [CrossRef] [PubMed]
- Whatcott, C.; Han, H.; Posner, R.G.; Von Hoff, D.D. Tumor-stromal interactions in pancreatic cancer. Crit. Rev. Oncog. 2013, 18, 135–151. [Google Scholar] [CrossRef]
- Penchev, V.R.; Rasheed, Z.A.; Maitra, A.; Matsui, W. Heterogeneity and targeting of pancreatic cancer stem cells. Clin. Cancer Res. 2012, 18, 4277–4284. [Google Scholar] [CrossRef]
- Huang, Y.; Davidson, N.E. Targeting tumorigenicity of breast cancer stem-like cells using combination epigenetic therapy: Something old and something new. J. Thorac. Dis. 2016, 8, 2971. [Google Scholar] [CrossRef]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef]
- Romani, P.; Valcarcel-Jimenez, L.; Frezza, C.; Dupont, S. Crosstalk between mechanotransduction and metabolism. Nat. Rev. Mol. Cell Biol. 2021, 22, 22–38. [Google Scholar] [CrossRef]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’agostino, D. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef]
- Xie, Q.; Wu, Q.; Horbinski, C.M.; Flavahan, W.A.; Yang, K.; Zhou, W.; Dombrowski, S.M.; Huang, Z.; Fang, X.; Shi, Y. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat. Neurosci. 2015, 18, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Fu, Z.; Chen, R.; Zhao, X.; Zhou, Y.; Zeng, B.; Yu, M.; Zhou, Q.; Lin, Q.; Gao, W. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget 2015, 6, 31151. [Google Scholar] [CrossRef]
- Wang, V.M.-Y.; Ferreira, R.M.; Almagro, J.; Evan, T.; Legrave, N.; Thin, M.Z.; Frith, D.; Carvalho, J.; Barry, D.J.; Snijders, A.P. CD9 identifies pancreatic cancer stem cells and modulates glutamine metabolism to fuel tumour growth. Nat. Cell Biol. 2019, 21, 1425–1435. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CPCs | CSCs | |
|---|---|---|
| 2D | 0.86 ± 0.04 | 0.72 ± 0.04 |
| 90%M–10%C | 0.90 ± 0.03 | 0.70 ± 0.07 |
| 70%M–30%C | 0.88 ± 0.05 | 0.64 ± 0.02 * |
| 10%M–90%C | 0.92 ± 0.06 | 0.56 ± 0.03 ** |
| CPCs | CSCs | |
|---|---|---|
| 2D | 1.24 ± 0.72 | 0.53 ± 0.14 |
| 90%M–10%C | 0.94 ± 0.1 | 0.41 ± 0.01 |
| 70%M–30%C | 1.01 ± 1.32 | 0.37 ± 0.01 * |
| 10%M–90%C | 0.76 ± 0.47 | 0.12 ± 0.01 ** |
| CPCs | CSCs | |
|---|---|---|
| 2D | 12.64 ± 0.70 | 10.59 ± 1.44 |
| 90%M–10%C | 7.39 ± 0.38 | 4.13 ± 0.35 |
| 70%M–30%C | 7.94 ± 0.94 | 3.84 ± 0.49 |
| 10%M–90%C | 7.08 ± 0.92 | 2.04 ± 0.15 |
| 90%M–10%C | 10%M–90%C | ||
|---|---|---|---|
| CPCs | 2-DG | <1 (0.58) | <1 (0.66) |
| DCA | <1 (0.65) | <1 (0.52) | |
| Phenformin | <1 (0.79) | <1 (0.86) | |
| CSCs | 2-DG | <1 (0.48) | <1 (0.57) |
| DCA | <1 (0.59) | <1 (0.66) | |
| Phenformin | <1 (0.32) | <1 (0.27) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tavares-Valente, D.; Cannone, S.; Greco, M.R.; Carvalho, T.M.A.; Baltazar, F.; Queirós, O.; Agrimi, G.; Reshkin, S.J.; Cardone, R.A. Extracellular Matrix Collagen I Differentially Regulates the Metabolic Plasticity of Pancreatic Ductal Adenocarcinoma Parenchymal Cell and Cancer Stem Cell. Cancers 2023, 15, 3868. https://doi.org/10.3390/cancers15153868
Tavares-Valente D, Cannone S, Greco MR, Carvalho TMA, Baltazar F, Queirós O, Agrimi G, Reshkin SJ, Cardone RA. Extracellular Matrix Collagen I Differentially Regulates the Metabolic Plasticity of Pancreatic Ductal Adenocarcinoma Parenchymal Cell and Cancer Stem Cell. Cancers. 2023; 15(15):3868. https://doi.org/10.3390/cancers15153868
Chicago/Turabian StyleTavares-Valente, Diana, Stefania Cannone, Maria Raffaella Greco, Tiago Miguel Amaral Carvalho, Fátima Baltazar, Odília Queirós, Gennaro Agrimi, Stephan J. Reshkin, and Rosa Angela Cardone. 2023. "Extracellular Matrix Collagen I Differentially Regulates the Metabolic Plasticity of Pancreatic Ductal Adenocarcinoma Parenchymal Cell and Cancer Stem Cell" Cancers 15, no. 15: 3868. https://doi.org/10.3390/cancers15153868
APA StyleTavares-Valente, D., Cannone, S., Greco, M. R., Carvalho, T. M. A., Baltazar, F., Queirós, O., Agrimi, G., Reshkin, S. J., & Cardone, R. A. (2023). Extracellular Matrix Collagen I Differentially Regulates the Metabolic Plasticity of Pancreatic Ductal Adenocarcinoma Parenchymal Cell and Cancer Stem Cell. Cancers, 15(15), 3868. https://doi.org/10.3390/cancers15153868