
Stratification of Tamoxifen Synergistic Combinations for the Treatment of ER+ Breast Cancer

 , , , , and
, , , , and
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. ER+ Breast Cancer and Standard of Care
1.2. Etiology of Endocrine Resistance
1.3. PDX Models/Models of ER+ BC
1.4. Approach
2. Materials and Methods
2.1. Patient-Derived Xenograft (PDX) Models
Development of Endocrine-Resistant PDX Models
2.2. Cell Lines
2.3. Bulk RNA Sequencing
2.3.1. Bulk RNA Sequencing Analysis
2.3.2. Quality Control and Pre-Processing
2.3.3. Gene Signatures and Clustering
2.4. Targeted Mutational Profiling
2.5. Single-Cell RNA Sequencing
Single-Cell RNA Sequencing Quality Control and Preprocessing
2.6. Drug Screening
2.6.1. In Vitro
2.6.2. In Vivo
2.7. Western Blot
2.8. Immunohistochemistry (IHC)
2.9. Quantification and Statistical Analysis
3. Results
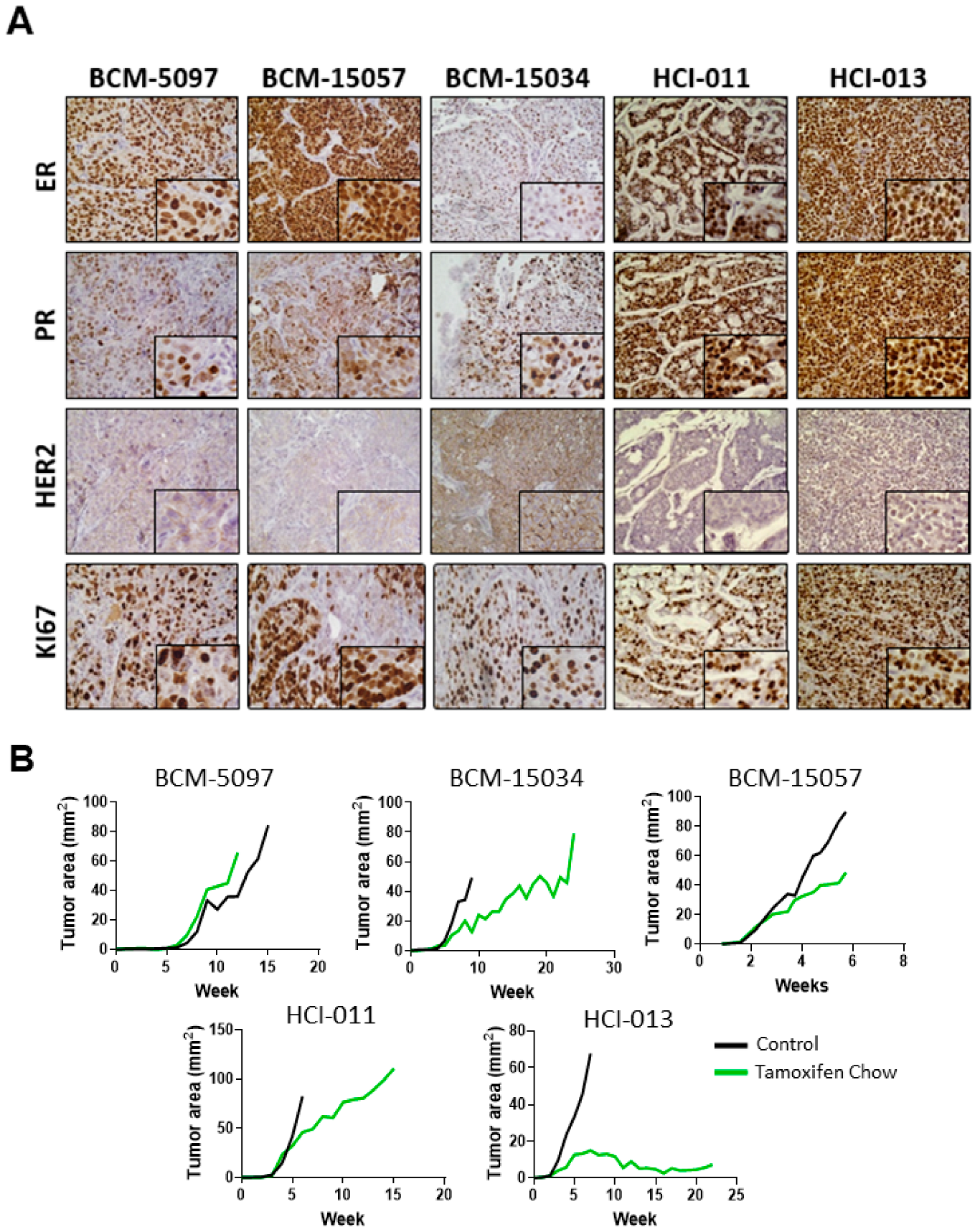
3.1. Tamoxifen Treatment Response Was Classified in Five ER+ PDX Models In Vivo
3.2. Tumor Heterogeneity Is Conserved in PDX Models
3.3. Genes Associated with Proliferation and ER Signaling Were Profiled at the Single-Cell Level in ER+ PDX Models
3.4. Tamoxifen Treatment Upregulated Genes Associated with Hormone Therapy Resistance
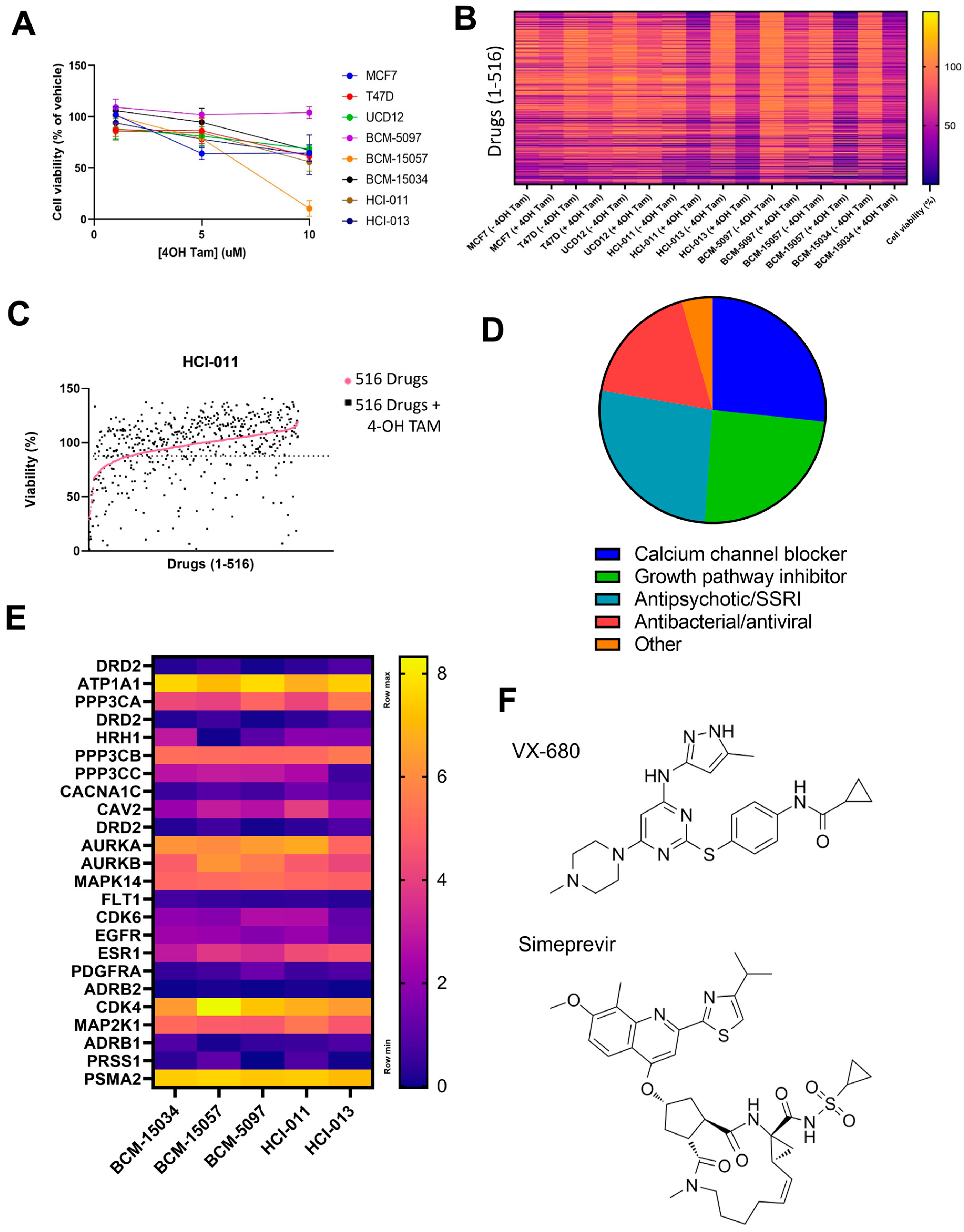
3.5. HTS Identified VX-680 and Simeprevir as Additively Effective Compounds with Tamoxifen
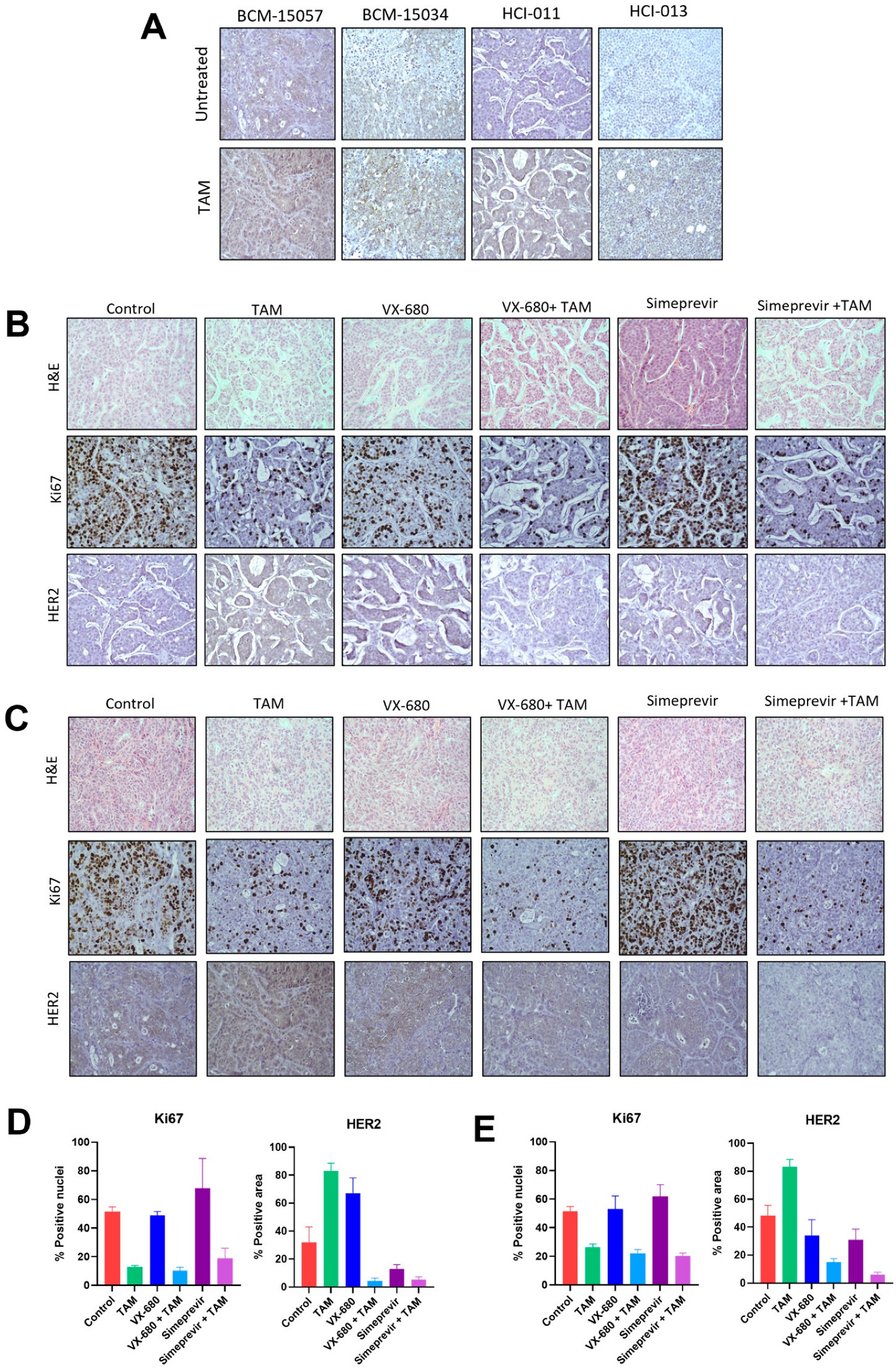
3.6. Tamoxifen Combination Therapy Was Effective in PDX Models of ER+ Breast Cancer
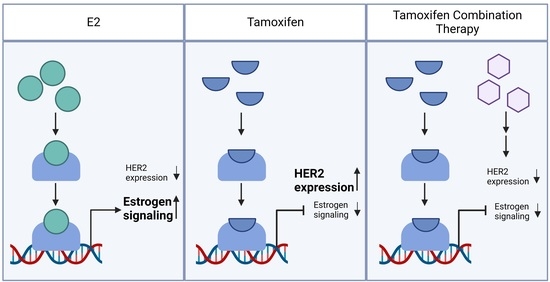
3.7. Tamoxifen Combination Therapy Abrogated Tamoxifen-Treatment-Induced Upregulation of HER2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Łukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanisławek, A. Breast Cancer-Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies—An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.B.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.; Miller, K.; Sauer, A.G.; Jemal, A.; Siegel, R. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Burguin, A.; Diorio, C.; Durocher, F. Breast Cancer Treatments: Updates and New Challenges. J. Pers. Med. 2021, 11, 808. [Google Scholar] [CrossRef]
- Haines, C.N.; Wardell, S.E.; McDonnell, D.P. Current and emerging estrogen receptor-targeted therapies for the treatment of breast cancer. Essays Biochem. 2021, 65, 985–1001. [Google Scholar] [CrossRef]
- Pistilli, B.; Lohrisch, C.; Sheade, J.; Fleming, G.F. Personalizing Adjuvant Endocrine Therapy for Early-Stage Hormone Receptor–Positive Breast Cancer. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 60–72. [Google Scholar] [CrossRef]
- Fabian, C.J. The what, why and how of aromatase inhibitors: Hormonal agents for treatment and prevention of breast cancer. Int. J. Clin. Pract. 2007, 61, 2051–2063. [Google Scholar] [CrossRef]
- Blondon, M.; Bodmer, A.; Thouvenin, L.; Lecompte, T.; Righini, M.; Fontana, P.; Casini, A. Differential impact of tamoxifen and aromatase inhibitors on thrombin generation: The prospective HEMOBREAST cohort. Blood Adv. 2022, 6, 2884–2892. [Google Scholar] [CrossRef]
- O’Brien, N.A.; McDermott, M.S.J.; Conklin, D.; Luo, T.; Ayala, R.; Salgar, S.; Chau, K.; DiTomaso, E.; Babbar, N.; Su, F.; et al. Targeting activated PI3K/mTOR signaling overcomes acquired resistance to CDK4/6-based therapies in preclinical models of hormone receptor-positive breast cancer. Breast Cancer Res. 2020, 22, 89. [Google Scholar] [CrossRef]
- Achinger-Kawecka, J.; Valdes-Mora, F.; Luu, P.-L.; Giles, K.A.; Caldon, C.E.; Qu, W.; Nair, S.; Soto, S.; Locke, W.J.; Yeo-The, N.S.; et al. Epigenetic reprogramming at estrogen-receptor binding sites alters 3D chromatin landscape in endocrine-resistant breast cancer. Nat. Commun. 2020, 11, 320. [Google Scholar] [CrossRef] [PubMed]
- Shiino, S.; Kinoshita, T.; Yoshida, M.; Jimbo, K.; Asaga, S.; Takayama, S.; Tsuda, H. Prognostic Impact of Discordance in Hormone Receptor Status Between Primary and Recurrent Sites in Patients With Recurrent Breast Cancer. Clin. Breast Cancer 2016, 16, e133–e140. [Google Scholar] [CrossRef] [PubMed]
- Hosford, S.R.; Miller, T.W. Clinical potential of novel therapeutic targets in breast cancer: CDK4/6, Src, JAK/STAT, PARP, HDAC, and PI3K/AKT/mTOR pathways. Pharm. Pers. Med. 2014, 7, 203–215. [Google Scholar]
- Olson, E. Combination Therapies in Advanced, Hormone Receptor-Positive Breast Cancer. J. Adv. Pract. Oncol. 2018, 9, 43–54. [Google Scholar]
- Murayama, T.; Gotoh, N. Patient-Derived Xenograft Models of Breast Cancer and Their Application. Cells 2019, 8, 621. [Google Scholar] [CrossRef]
- Woo, X.Y.; Giordano, J.; Srivastava, A.; Zhao, Z.M.; Lloyd, M.W.; de Bruijn, R.; Suh, Y.S.; Patidar, R.; Chen, L.; Scherer, S.; et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nat. Genet. 2021, 53, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Li, Y.; Jia, R.; Fan, X. The fidelity of cancer cells in PDX models: Characteristics, mechanism and clinical significance. Int. J. Cancer 2020, 146, 2078–2088. [Google Scholar] [CrossRef] [PubMed]
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. Cell Chem. Biol. 2017, 24, 1092–1100. [Google Scholar] [CrossRef]
- Williams, J.A. Using PDX for Preclinical Cancer Drug Discovery: The Evolving Field. J. Clin. Med. 2018, 7, 41. [Google Scholar] [CrossRef]
- Coughlan, A.M.; Harmon, C.; Whelan, S.; O’Brien, E.C.; O’Reilly, V.P.; Crotty, P.; Kelly, P.; Ryan, M.; Hickey, F.B.; O’Farrelly, C.; et al. Myeloid Engraftment in Humanized Mice: Impact of Granulocyte-Colony Stimulating Factor Treatment and Transgenic Mouse Strain. Stem Cells Dev. 2016, 25, 530–541. [Google Scholar] [CrossRef]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [PubMed]
- Kabos, P.; Finlay-Schultz, J.; Li, C.; Kline, E.; Finlayson, C.; Wisell, J.; Manuel, C.A.; Edgerton, S.M.; Harrell, J.C.; Elias, A.; et al. Patient-derived luminal breast cancer xenografts retain hormone receptor heterogeneity and help define unique estrogen-dependent gene signatures. Breast Cancer Res. Treat. 2012, 135, 415–432. [Google Scholar] [CrossRef]
- Boyd, D.C.; Zboril, E.K.; Olex, A.L.; Leftwich, T.J.; Hairr, N.S.; Byers, H.A.; Valentine, A.D.; Altman, J.E.; Alzubi, M.A.; Grible, J.M.; et al. Discovering Synergistic Compounds with BYL-719 in PI3K Overactivated Basal-like PDXs. Cancers 2023, 15, 1582. [Google Scholar] [CrossRef] [PubMed]
- Alzubi, M.A.; Turner, T.H.; Olex, A.L.; Sohal, S.S.; Tobin, N.P.; Recio, S.G.; Bergh, J.; Hatschek, T.; Parker, J.S.; Sartorius, C.A.; et al. Separation of breast cancer and organ microenvironment transcriptomes in metastases. Breast Cancer Res. 2019, 21, 36. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Volume 5, no. 1. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 27 January 2023).
- Martin, M. CUTADAPT removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Genefu|Princess Margaret Bioinformatics and Computational Genomics Laboratory. Available online: https://www.pmgenomics.ca/bhklab/software/genefu (accessed on 27 January 2023).
- Gatza, M.L.; Lucas, J.E.; Barry, W.T.; Kim, J.W.; Wang, Q.; Crawford, M.D.; Datto, M.B.; Kelley, M.; Mathey-Prevot, B.; Potti, A.; et al. A pathway-based classification of human breast cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 6994–6999. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Weigman, V.J.; Fan, C.; Sawyer, L.R.; He, X.; Troester, M.A.; Sartor, C.I.; Rieger-House, T.; Bernard, P.S.; Carey, L.A.; et al. EGFR associated expression profiles vary with breast tumor subtype. BMC Genom. 2007, 8, 258. [Google Scholar] [CrossRef]
- Sheffer, M.; Lowry, E.; Beelen, N.; Borah, M.; Amara, S.N.; Mader, C.C.; Roth, J.A.; Tsherniak, A.; Freeman, S.S.; Dashevsky, O.; et al. Genome-scale screens identify factors regulating tumor cell responses to natural killer cells. Nat. Genet. 2021, 53, 1196–1206. [Google Scholar] [CrossRef]
- Parkes, E.E.; Humphries, M.P.; Gilmore, E.; Sidi, F.A.; Bingham, V.; Phyu, S.M.; Craig, S.; Graham, C.; Miller, J.; Griffin, D.; et al. The clinical and molecular significance associated with STING signaling in breast cancer. NPJ Breast Cancer 2021, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Giraddi, R.R.; Chung, C.-Y.; Heinz, R.E.; Balcioglu, O.; Novotny, M.; Trejo, C.L.; Dravis, C.; Hagos, B.M.; Mehrabad, E.M.; Rodewald, L.W.; et al. Single-Cell Transcriptomes Distinguish Stem Cell State Changes and Lineage Specification Programs in Early Mammary Gland Development. Cell Rep. 2018, 24, 1653–1666.e7. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Balaji, U.; Knudsen, E.S. Systematically defining single-gene determinants of response to neoadjuvant chemotherapy reveals specific biomarkers. Clin. Cancer Res. 2014, 20, 4837–4848. [Google Scholar] [CrossRef] [PubMed]
- Heng, Y.J.; Lester, S.C.; Tse, G.M.; Factor, R.E.; Allison, K.H.; Collins, L.C.; Chen, Y.Y.; Jensen, K.C.; Johnson, N.B.; Jeong, J.C.; et al. The molecular basis of breast cancer pathological phenotypes. J. Pathol. 2017, 241, 375–391. [Google Scholar] [CrossRef]
- Pfefferle, A.D.; Herschkowitz, J.I.; Usary, J.; Harrell, J.C.; Spike, B.T.; Adams, J.R.; Torres-Arzayus, M.I.; Brown, M.; Egan, S.E.; Wahl, G.M.; et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 2013, 14, R125. [Google Scholar] [CrossRef]
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; Rasmussen, K.E.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S.; et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007, 8, R76. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Oh, D.S.; Troester, M.A.; Usary, J.; Hu, Z.; He, X.; Fan, C.; Wu, J.; Carey, L.A.; Perou, C.M. Estrogen-regulated genes predict survival in hormone receptor-positive breast cancers. J. Clin. Oncol. 2006, 24, 1656–1664. [Google Scholar] [CrossRef]
- Herschkowitz, J.I.; He, X.; Fan, C.; Perou, C.M. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008, 10, R75. [Google Scholar] [CrossRef]
- Fan, C.; Prat, A.; Parker, J.S.; Liu, Y.; Carey, L.A.; Troester, M.A.; Perou, C.M. Building prognostic models for breast cancer patients using clinical variables and hundreds of gene expression signatures. BMC Med. Genom. 2011, 4, 3. [Google Scholar] [CrossRef]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Gilcrease, M.Z.; Krishnamurthy, S.; Lee, J.S.; Fridlyand, J.; Sahin, A.; Agarwal, R.; Joy, C.; et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009, 69, 4116–4124. [Google Scholar] [CrossRef] [PubMed]
- Finlay-Schultz, J.; Jacobsen, B.M.; Riley, D.; Paul, K.V.; Turner, S.; Ferreira-Gonzalez, A.; Harrell, J.C.; Kabos, P.; Sartorius, C.A. New generation breast cancer cell lines developed from patient-derived xenografts. Breast Cancer Res. 2020, 22, 68. [Google Scholar] [CrossRef]
- Turner, T.H.; Alzubi, M.A.; Harrell, J.C. Identification of synergistic drug combinations using breast cancer patient-derived xenografts. Sci. Rep. 2020, 10, 1493. [Google Scholar] [CrossRef] [PubMed]
- Rashid, N.S.; Hairr, N.S.; Murray, G.; Olex, A.L.; Leftwich, T.J.; Grible, J.M.; Reed, J.; Dozmorov, M.G.; Harrell, J.C. Identification of nuclear export inhibitor-based combination therapies in preclinical models of triple-negative breast cancer. Transl. Oncol. 2021, 14, 101235. [Google Scholar] [CrossRef]
- Zeng, J.; Edelweiss, M.; Ross, D.S.; Xu, B.; Moo, T.A.; Brogi, E.; D’Alfonso, T.M. Triple-Positive Breast Carcinoma: Histopathologic Features and Response to Neoadjuvant Chemotherapy. Arch. Pathol. Lab Med. 2021, 145, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Guillen, K.P.; Fujita, M.; Butterfield, A.J.; Scherer, S.D.; Bailey, M.H.; Chu, Z.; DeRose, Y.S.; Zhao, L.; Cortes-Sanchez, E.; Yang, C.H.; et al. A human breast cancer-derived xenograft and organoid platform for drug discovery and precision oncology. Nat. Cancer 2022, 3, 232–250. [Google Scholar] [CrossRef]
- Li, L.; Lin, L.; Veeraraghavan, J.; Hu, Y.; Wang, X.; Lee, S.; Tan, Y.; Schiff, R.; Wang, X.-S. Therapeutic role of recurrent ESR1-CCDC170 gene fusions in breast cancer endocrine resistance. Breast Cancer Res. 2020, 22, 84. [Google Scholar] [CrossRef]
- Conte, N.; Delaval, B.; Ginestier, C.; Ferrand, A.; Isnardon, D.; Larroque, C.; Prigent, C.; Séraphin, B.; Jacquemier, J.; Birnbaum, D. TACC1–chTOG–Aurora A protein complex in breast cancer. Oncogene 2003, 22, 8102–8116. [Google Scholar] [CrossRef]
- Viedma-Rodríguez, R.; Baiza-Gutman, L.; Salamanca-Gómez, F.; Diaz-Zaragoza, M.; Martínez-Hernández, G.; Esparza-Garrido, R.R.; Velázquez-Flores, M.A.; Arenas-Aranda, D. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer (review). Oncol. Rep. 2014, 32, 3–15. [Google Scholar] [CrossRef]
- Medchemexpress.com. Tozasertib. Available online: https://www.medchemexpress.com/Tozasertib.html (accessed on 10 March 2023).
- Simeprevir. 2023. Available online: https://www.medchemexpress.com/Simeprevir.html (accessed on 10 March 2023).
- Mazumder, A.; Shiao, S.; Haricharan, S. HER2 Activation and Endocrine Treatment Resistance in HER2-negative Breast Cancer. Endocrinology 2021, 162, bqab153. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, R.N.; Esen, B.; Mellemkjær, L.; Christiansen, P.; Ejlertsen, B.; Lash, T.L.; Nørgaard, M.; Cronin-Fenton, D. The Incidence of Breast Cancer Recurrence 10–32 Years After Primary Diagnosis. J. Natl. Cancer Inst. 2022, 114, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Yde, C.W. Aurora A and Mcl-1: New potential treatment targets in antiestrogen-resistant breast cancer. Mol. Cell. Oncol. 2021, 8, 998898. [Google Scholar] [CrossRef]
- Larsen, S.L.; Yde, C.W.; Laenkholm, A.V.; Rasmussen, B.B.; Duun-Henriksen, A.K.; Bak, M.; Lykkesfeldt, A.E.; Kirkegaard, T. Aurora kinase B is important for antiestrogen resistant cell growth and a potential biomarker for tamoxifen resistant breast cancer. BMC Cancer 2015, 15, 239. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Beeram, M.; Beck, J.T.; Conlin, A.; Dees, E.C.; Puhalla, S.L.; Rexer, B.N.; Burris, H.A.; Jhaveri, K.; Helsten, T.; et al. Abemaciclib in Combination With Endocrine Therapy for Patients With Hormone Receptor-Positive, HER2-Negative Metastatic Breast Cancer: A Phase 1b Study. Front. Oncol. 2021, 11, 810023. [Google Scholar] [CrossRef]
- Hwang, K.T.; Kim, E.K.; Jung, S.H.; Lee, E.S.; Kim, S.I.; Lee, S.; Park, H.K.; Kim, J.; Oh, S.; Kim, Y.A. Tamoxifen therapy improves overall survival in luminal A subtype of ductal carcinoma in situ: A study based on nationwide Korean Breast Cancer Registry database. Breast Cancer Res. Treat. 2018, 169, 311–322. [Google Scholar] [CrossRef]
- Yu, N.Y.; Iftimi, A.; Yau, C.; Tobin, N.P.; van ’t Veer, L.; Hoadley, K.A.; Benz, C.C.; Nordenskjöld, B.; Fornander, T.; Stål, O.; et al. Assessment of Long-term Distant Recurrence-Free Survival Associated with Tamoxifen Therapy in Postmenopausal Patients With Luminal A or Luminal B Breast Cancer. JAMA Oncol. 2019, 5, 1304–1309. [Google Scholar] [CrossRef]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- Hamilton, E.; Cortes, J.; Ozyilkan, O.; Chen, S.-C.; Petrakova, K.; Manikhas, A.; Jerusalem, G.; Hegg, R.; Huober, J.; Chapman, S.C.; et al. nextMONARCH: Abemaciclib Monotherapy or Combined with Tamoxifen for Metastatic Breast Cancer. Clin. Breast Cancer 2021, 21, 181–190.e2. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zboril, E.K.; Grible, J.M.; Boyd, D.C.; Hairr, N.S.; Leftwich, T.J.; Esquivel, M.F.; Duong, A.K.; Turner, S.A.; Ferreira-Gonzalez, A.; Olex, A.L.; et al. Stratification of Tamoxifen Synergistic Combinations for the Treatment of ER+ Breast Cancer. Cancers 2023, 15, 3179. https://doi.org/10.3390/cancers15123179
Zboril EK, Grible JM, Boyd DC, Hairr NS, Leftwich TJ, Esquivel MF, Duong AK, Turner SA, Ferreira-Gonzalez A, Olex AL, et al. Stratification of Tamoxifen Synergistic Combinations for the Treatment of ER+ Breast Cancer. Cancers. 2023; 15(12):3179. https://doi.org/10.3390/cancers15123179
Chicago/Turabian StyleZboril, Emily K., Jacqueline M. Grible, David C. Boyd, Nicole S. Hairr, Tess J. Leftwich, Madelyn F. Esquivel, Alex K. Duong, Scott A. Turner, Andrea Ferreira-Gonzalez, Amy L. Olex, and et al. 2023. "Stratification of Tamoxifen Synergistic Combinations for the Treatment of ER+ Breast Cancer" Cancers 15, no. 12: 3179. https://doi.org/10.3390/cancers15123179
APA StyleZboril, E. K., Grible, J. M., Boyd, D. C., Hairr, N. S., Leftwich, T. J., Esquivel, M. F., Duong, A. K., Turner, S. A., Ferreira-Gonzalez, A., Olex, A. L., Sartorius, C. A., Dozmorov, M. G., & Harrell, J. C. (2023). Stratification of Tamoxifen Synergistic Combinations for the Treatment of ER+ Breast Cancer. Cancers, 15(12), 3179. https://doi.org/10.3390/cancers15123179