
Glucocorticoid Receptor Activation in Lobular Breast Cancer Is Associated with Reduced Cell Proliferation and Promotion of Metastases

 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
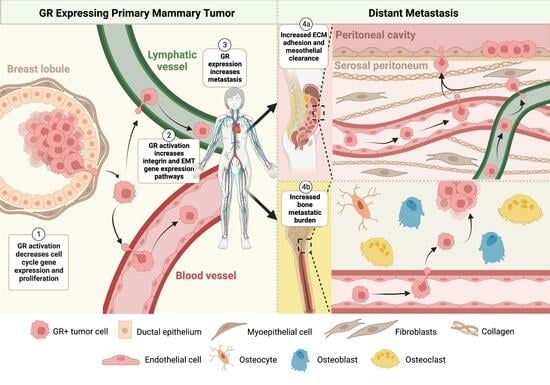
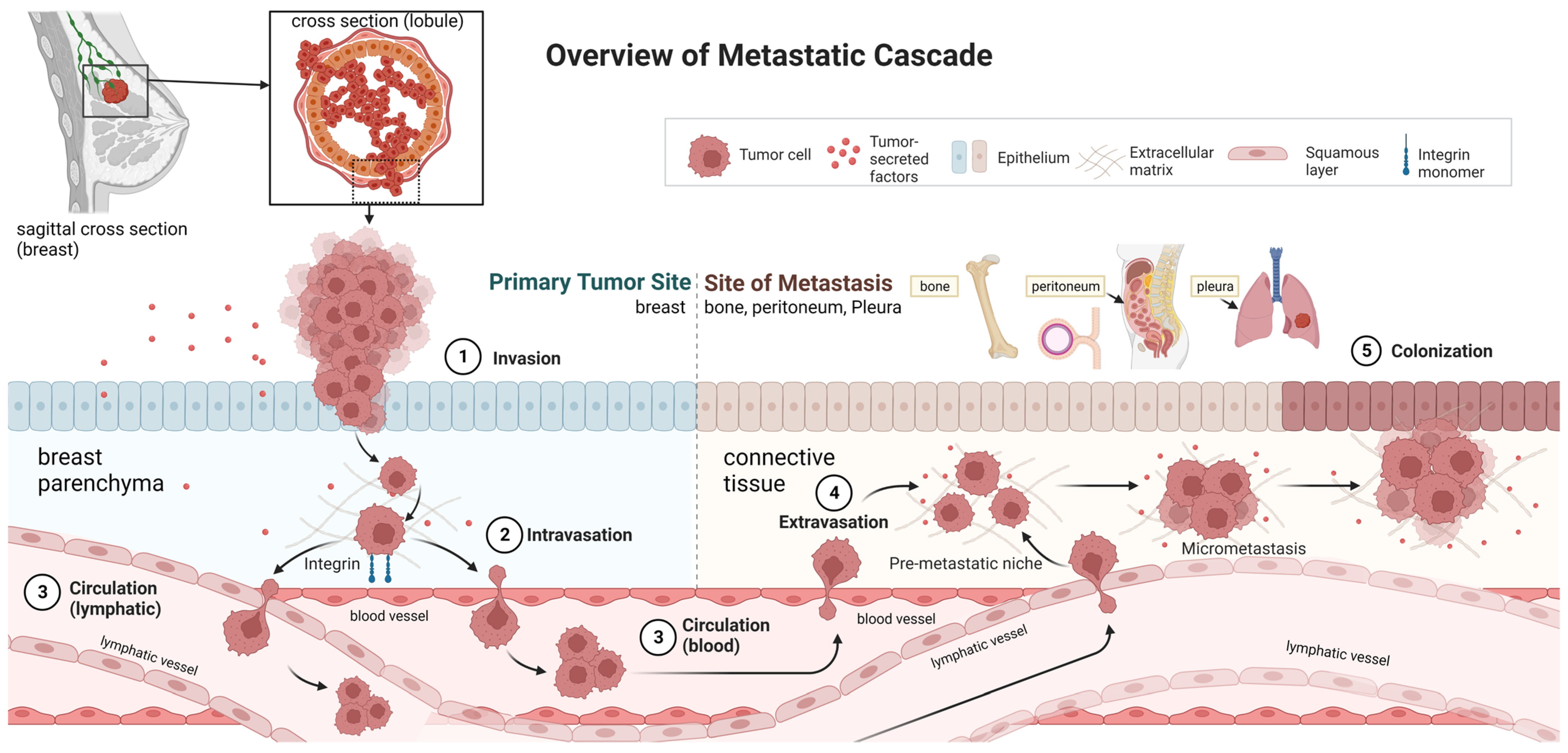
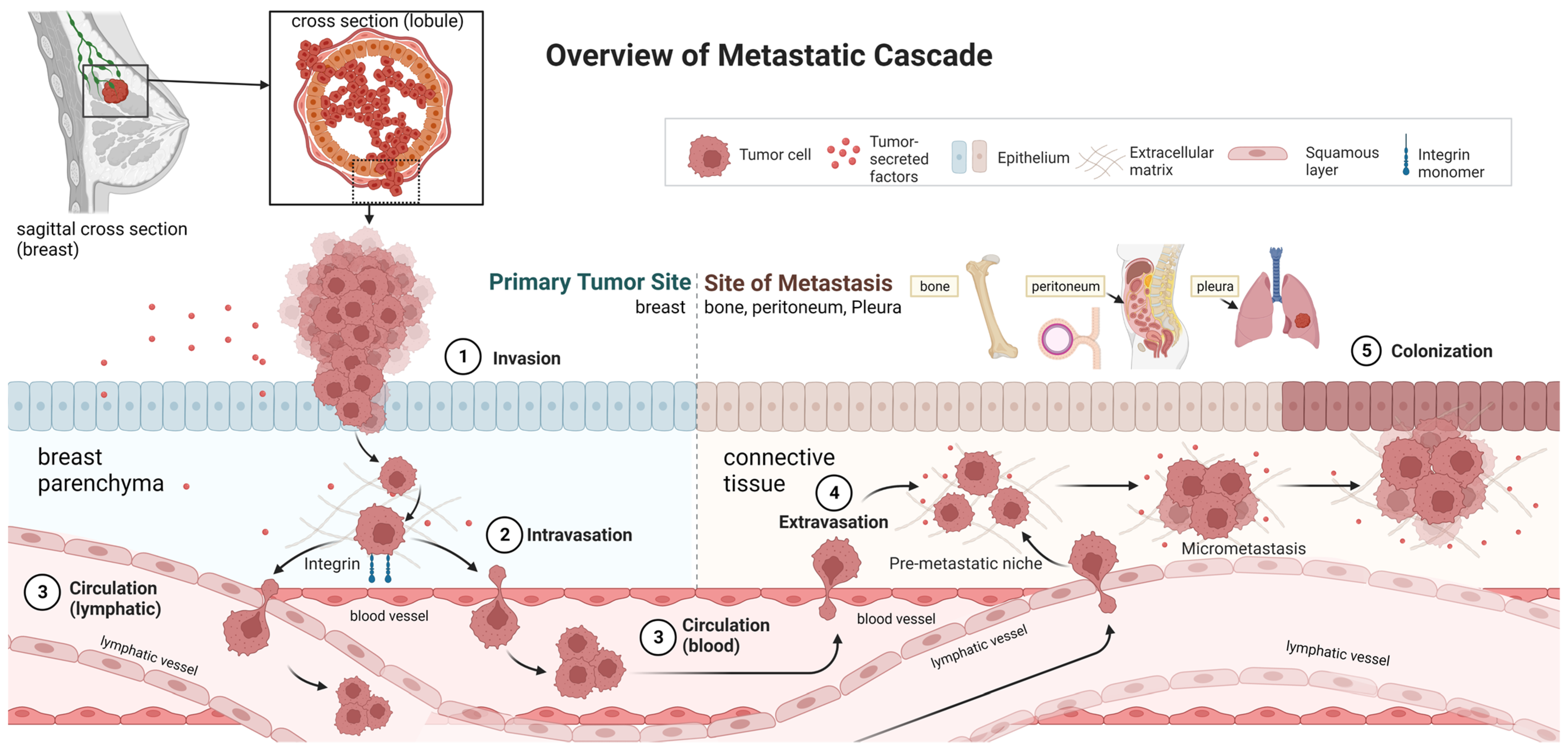
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Recombinant DNA and Stable Cell Line Generation
2.3. Hormones
2.4. ECM Adhesion Assay
2.5. Cell Proliferation Assay
2.6. Mesothelial Clearance Assay
2.7. Immunoblotting
2.8. Immunohistochemistry
2.9. Animal Studies and Xenograft Mammary Intraductal (MIND) Model
2.10. RNA Sequencing
2.11. Quantitative PCR
2.12. Software
3. Results
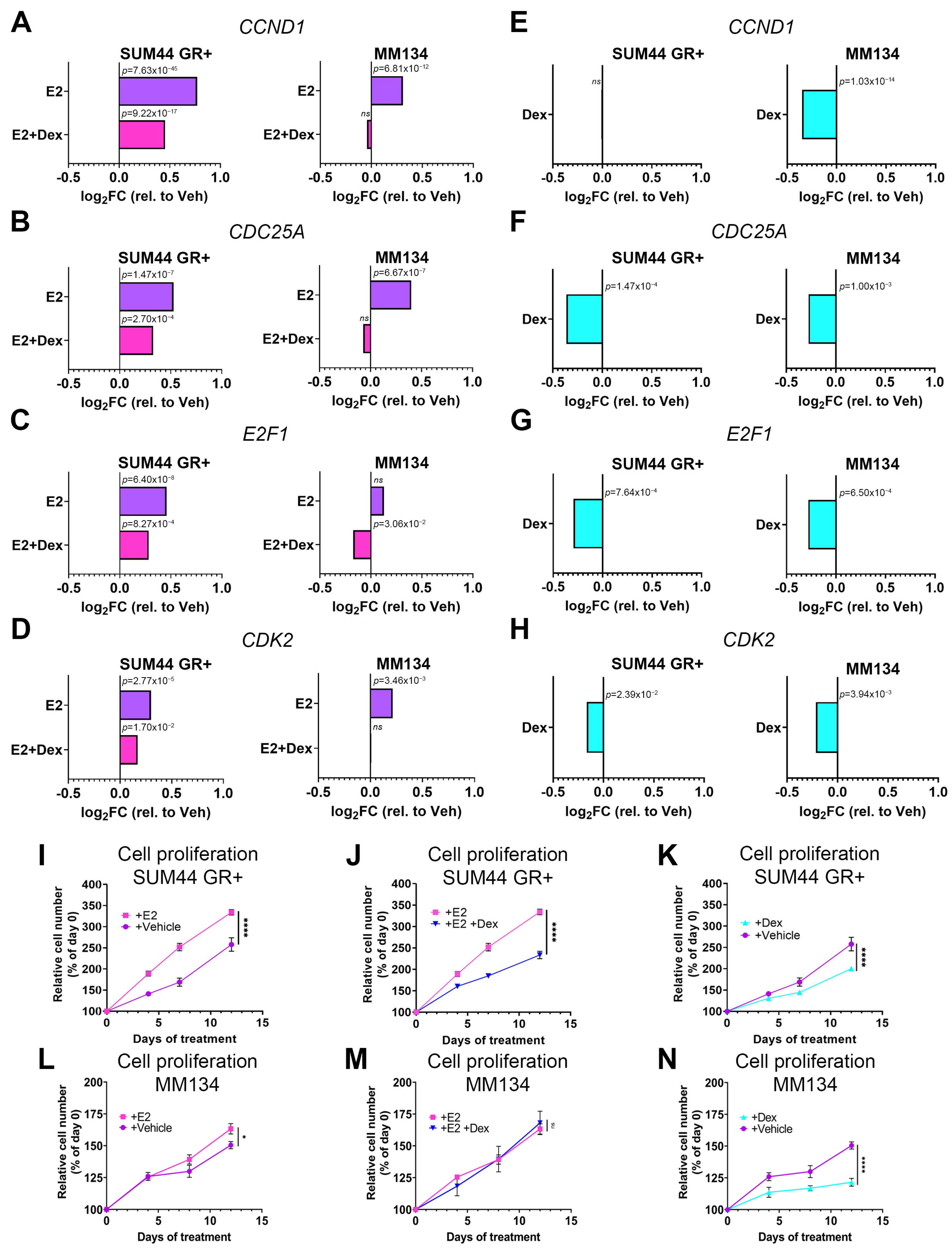
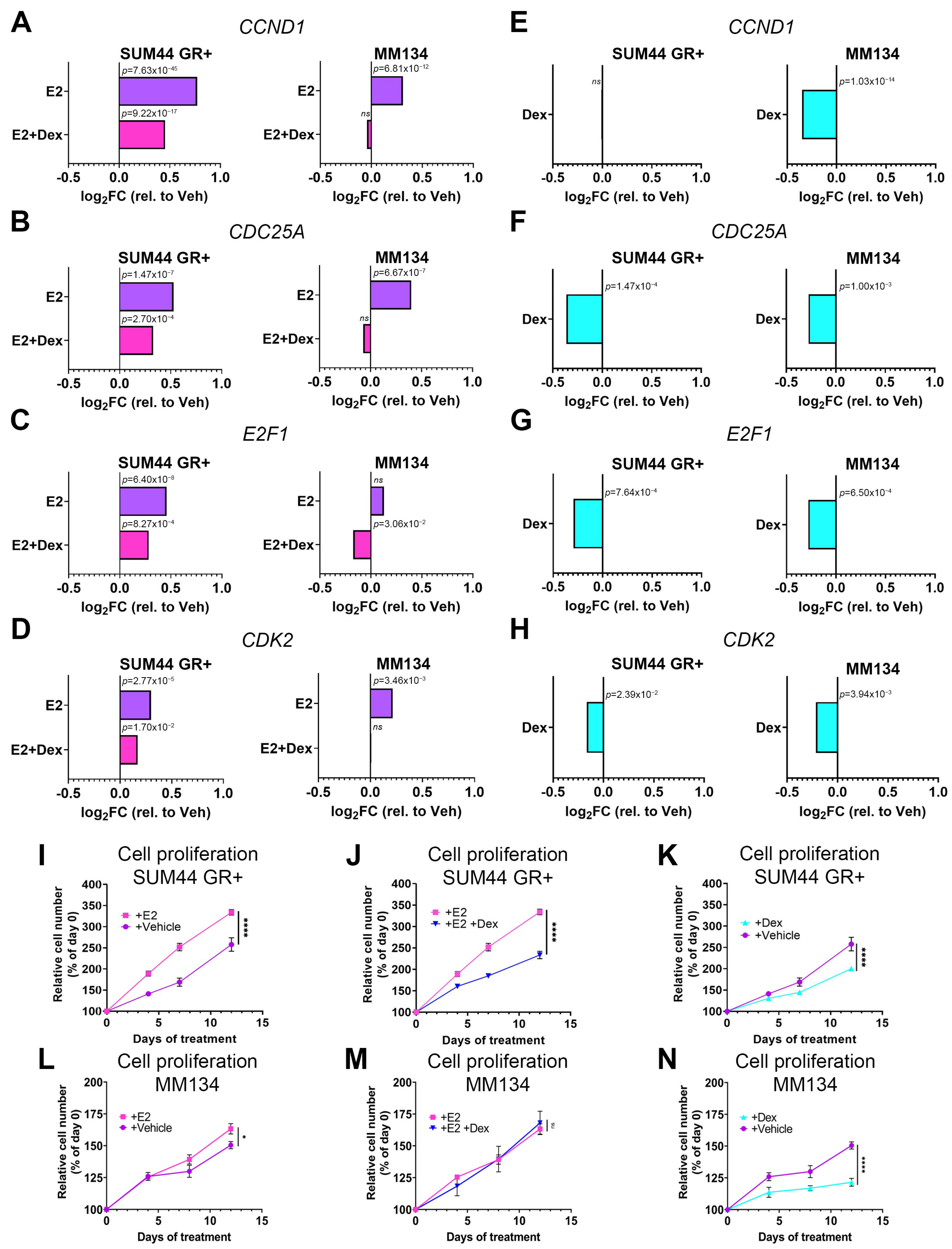
3.1. GR Activation Attenuates ILC Cell Cycle Gene Transcription and Inhibits Cell Proliferation
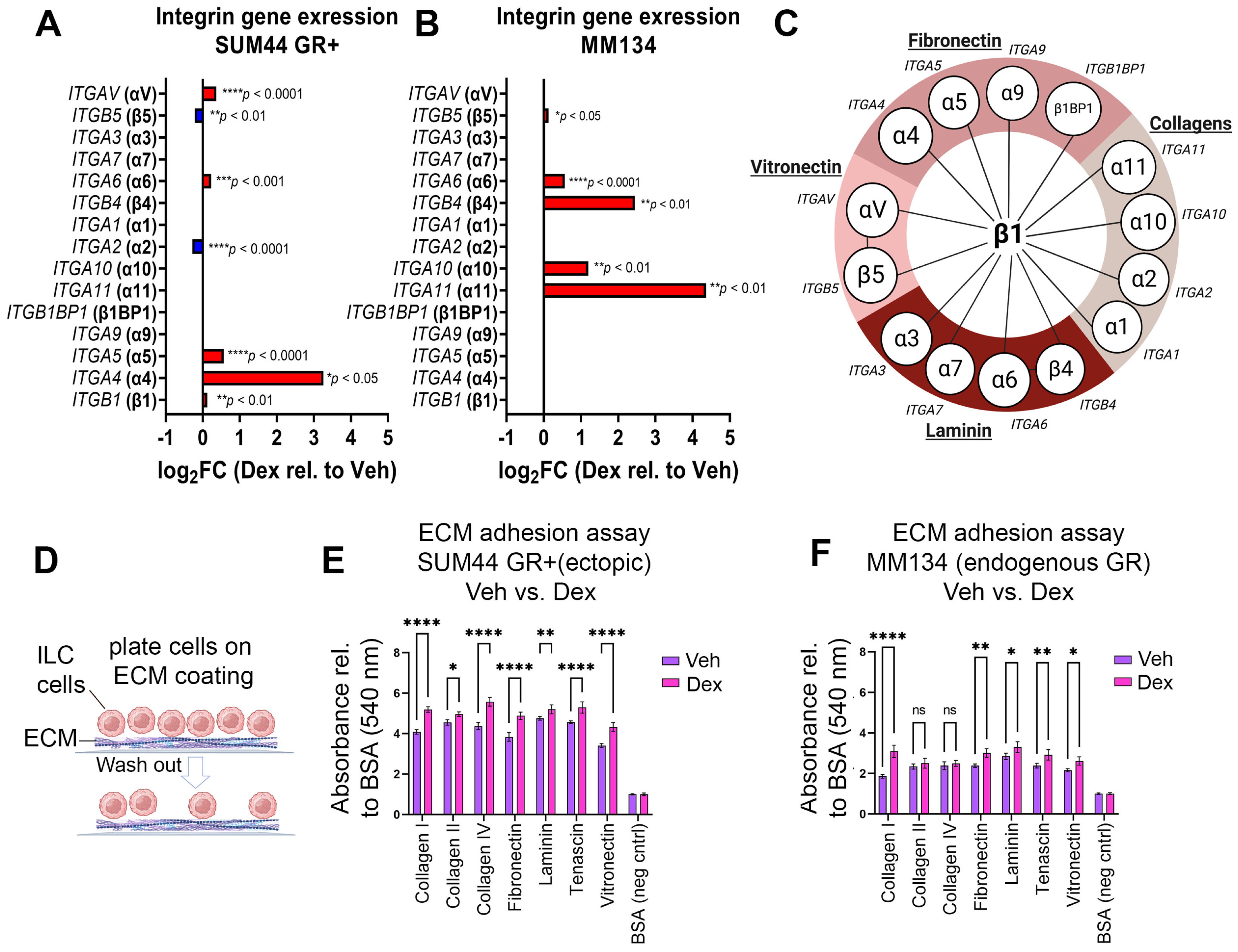
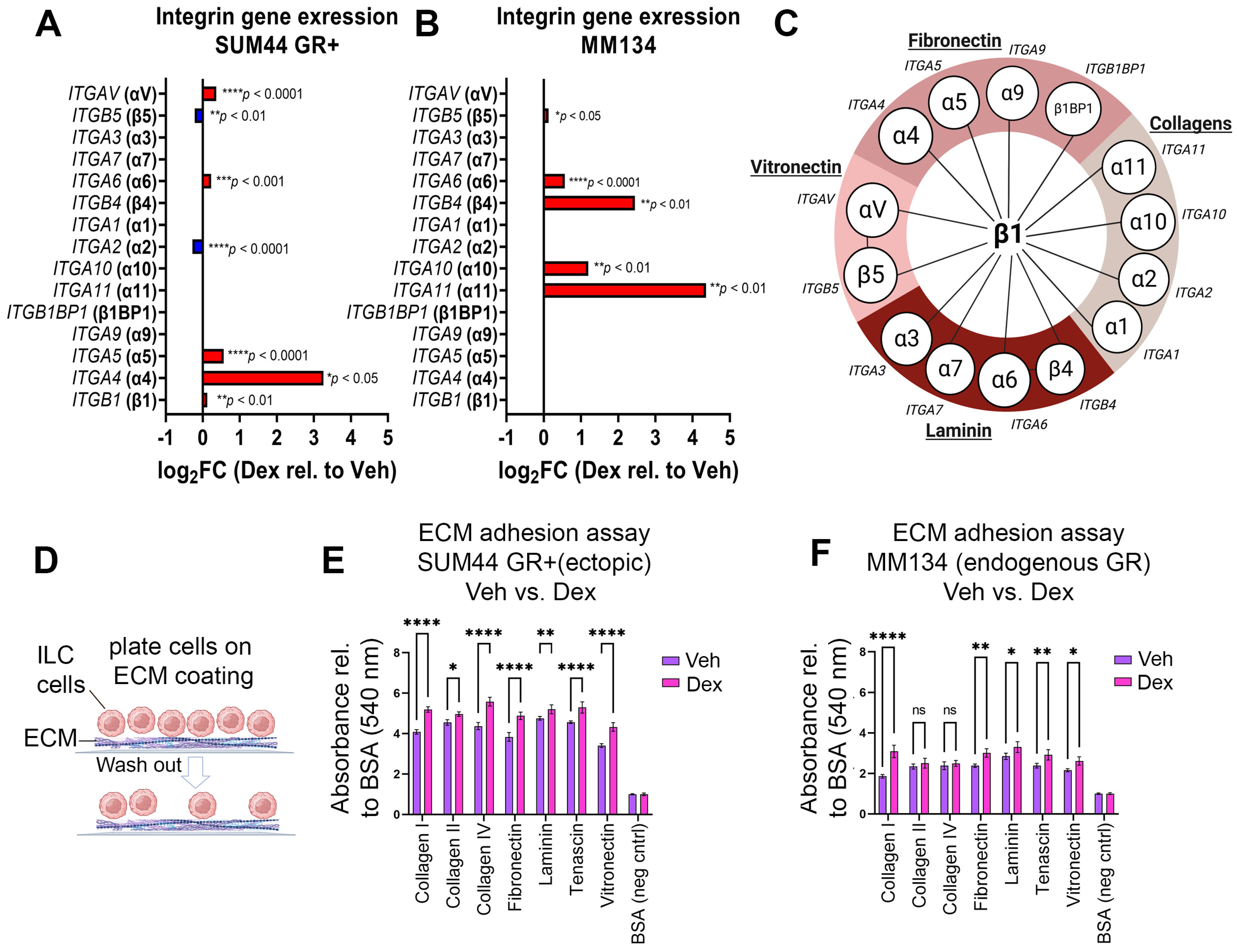
3.2. GR Activation Is Associated with Increased Integrin Gene Expression, EMT Pathway Gene Expression, and Adherence to ECM Proteins in ILC
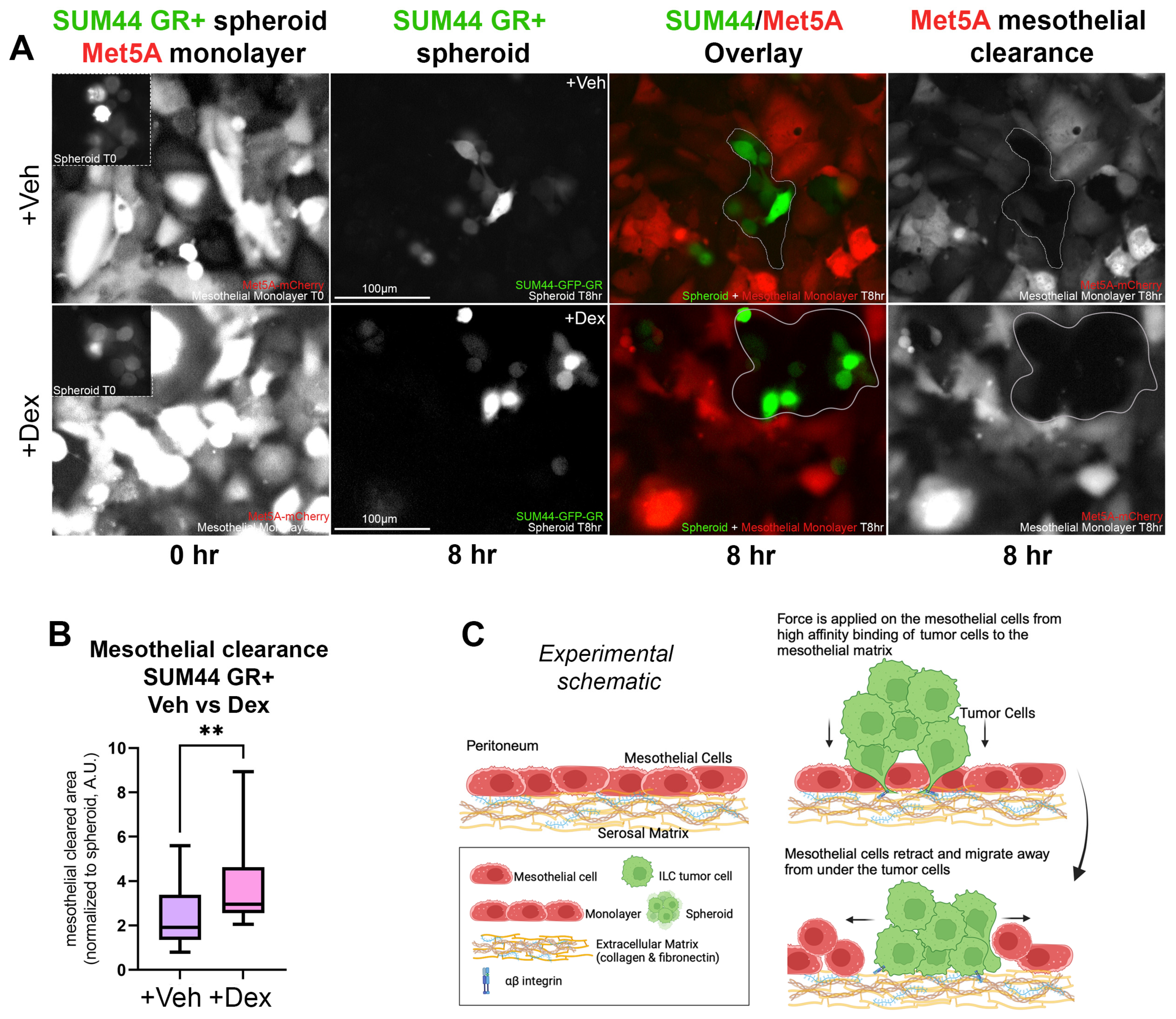
3.3. GR Activation in ILC Cells Increases Disruption of a Co-Cultured Mesothelial Layer
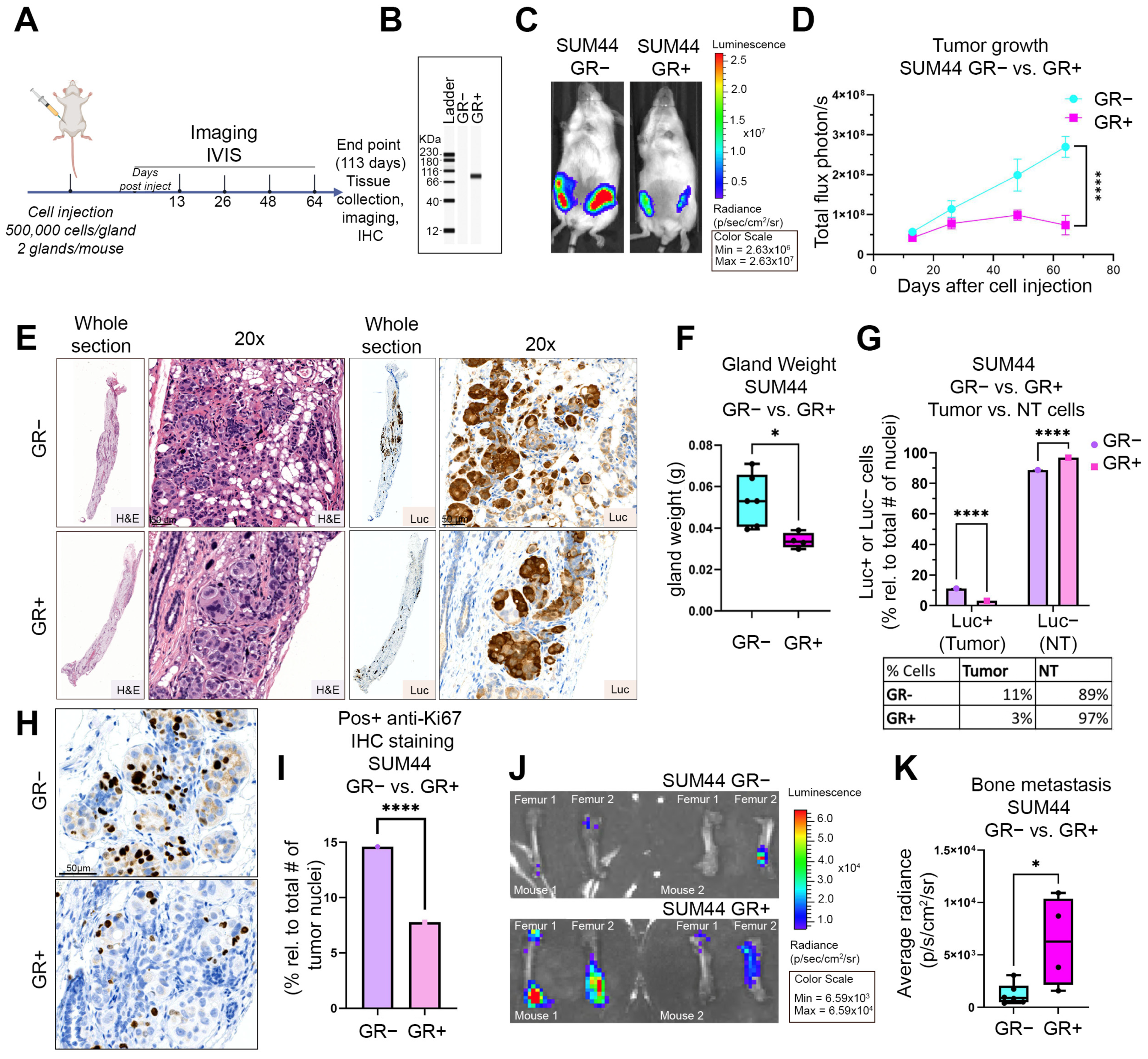
3.4. GR Expression Is Associated with Decreased Primary Tumor Growth and Increased Bone Metastases in an ILC Xenograft MIND Model
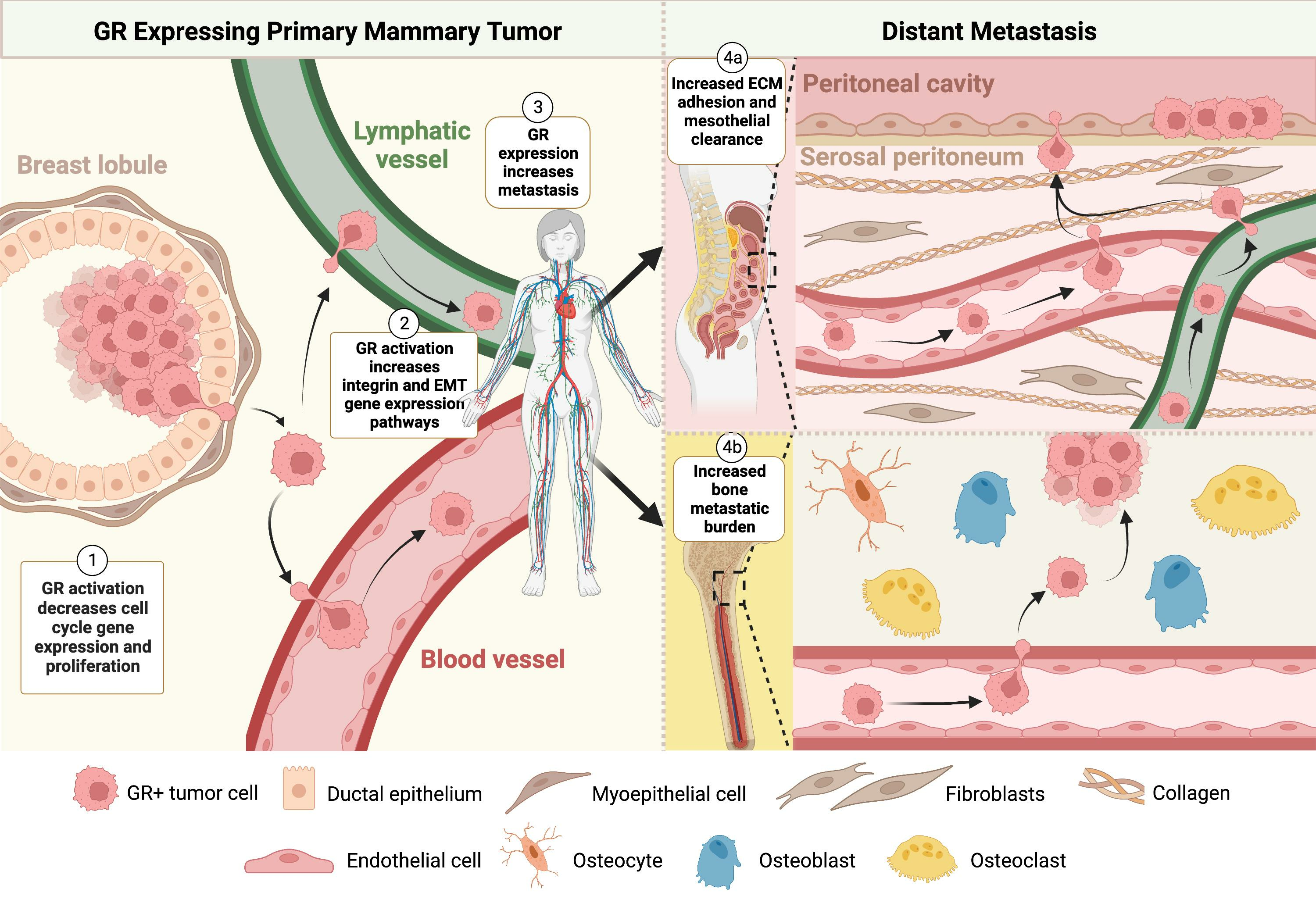
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Definition |
| ER/ER+ | Estrogen receptor (positive) |
| ILC | Invasive or infiltrating lobular carcinoma |
| IDC | Invasive ductal carcinoma |
| GR/GR+/GR− | Glucocorticoid receptor (+ = positive; − = null) |
| PR/PR+ | Progesterone receptor (+ = positive) |
| EMT | Epithelial to mesenchymal transition |
| SUM44 | SUM44-PE, endogenously GR null |
| MM134 | MDA-MB-134-VI, endogenous GR positive |
| ECM | Extracellular matrix |
| E2 | Estradiol, used at 10 nM is an estrogen receptor ligand which induces estrogen receptor transcriptional function |
| Dex | Dexamethasone, used at 100 nM. Is a high affinity synthetic glucocorticoid receptor ligand which induces glucocorticoid receptor transcriptional function |
| E2/Dex | Combination of 100 nM dexamethasone treatment and 10 nM Estradiol treatment to induce concurrent GR and ER transcriptional programs |
| VC | Vehicle control, 100% ETOH |
| CSS | Charcoal stripped FBS used to deplete cell media of hormone mediated activation |
| FBS | Fetal bovine serum |
| GFP | Green fluorescent protein with excitation at 488 nm |
| mCherry | Monomeric red fluorescent protein derived from Discosoma sp. with excitation at 587 nm |
| IPA | Ingenuity pathway analysis |
| T | Time |
| hr | hours |
| Luc+/− | Anti-luciferase antibody positive (+) or (−) staining |
| FC | Final concentration |
References
- Noureddine, L.M.; Trédan, O.; Hussein, N.; Badran, B.; Le Romancer, M.; Poulard, C. Glucocorticoid Receptor: A Multifaceted Actor in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 4446. [Google Scholar] [CrossRef] [PubMed]
- Butz, H.; Patócs, A. Mechanisms behind context-dependent role of glucocorticoids in breast cancer progression. Cancer Metastasis Rev. 2022, 41, 803–832. [Google Scholar] [CrossRef] [PubMed]
- Zhidkova, E.M.; Lylova, E.S.; Savinkova, A.V.; Mertsalov, S.A.; Kirsanov, K.I.; Belitsky, G.A.; Yakubovskaya, M.G.; Lesovaya, E.A. A Brief Overview of the Paradoxical Role of Glucocorticoids in Breast Cancer. Breast Cancer Basic Clin. Res. 2020, 14, 1178223420974667. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.; Ironside, A.; Diana, A.; Oikonomidou, O. Lobular Breast Cancer: A Review. Front. Oncol. 2021, 10, 591399. [Google Scholar] [CrossRef]
- Reed, A.E.M.; Kutasovic, J.R.; Lakhani, S.R.; Simpson, P.T. Invasive lobular carcinoma of the breast: Morphology, biomarkers and ’omics. Breast Cancer Res. 2015, 17, 12. [Google Scholar] [CrossRef]
- Arpino, G.; Bardou, V.J.; Clark, G.M.; Elledge, R.M. Infiltrating lobular carcinoma of the breast: Tumor characteristics and clinical outcome. Breast Cancer Res. 2004, 6, R149–R156. [Google Scholar] [CrossRef]
- Sflomos, G.; Schipper, K.; Koorman, T.; Fitzpatrick, A.; Oesterreich, S.; Lee, A.V.; Jonkers, J.; Brunton, V.G.; Christgen, M.; Isacke, C.; et al. Atlas of Lobular Breast Cancer Models: Challenges and Strategic Directions. Cancers 2021, 13, 5396. [Google Scholar] [CrossRef]
- Desmedt, C.; Zoppoli, G.; Sotiriou, C.; Salgado, R. Transcriptomic and genomic features of invasive lobular breast cancer. Semin. Cancer Biol. 2017, 44, 98–105. [Google Scholar] [CrossRef]
- Cosar, R.; Sut, N.; Topaloglu, S.; Tastekin, E.; Nurlu, D.; Ozler, T.; Şenödeyici, E.; Dedeli, M.; Chousein, M.; Cicin, I. Classifying invasive lobular carcinoma as special type breast cancer may be reducing its treatment success: A comparison of survival among invasive lobular carcinoma, invasive ductal carcinoma, and no-lobular special type breast cancer. PLoS ONE 2023, 18, e0283445. [Google Scholar] [CrossRef]
- Sflomos, G.; Schaumann, N.; Christgen, M.; Christgen, H.; Bartels, S.; Kreipe, H.; Battista, L.; Brisken, C. Optimized Modeling of Metastatic Triple-Negative Invasive Lobular Breast Carcinoma. Cancers 2023, 15, 3299. [Google Scholar] [CrossRef]
- Jain, S.; Fisher, C.; Smith, P.; Millis, R.R.; Rubens, R.D. Patterns of metastatic breast cancer in relation to histological type. Eur. J. Cancer 1993, 29, 2155–2157. [Google Scholar] [CrossRef]
- Borst, M.J.; Ingold, J.A. Metastatic patterns of invasive lobular versus invasive ductal carcinoma of the breast. Surgery 1993, 114, 637–641; discussion 641–642. [Google Scholar]
- Lamovec, J.; Bračkko, M. Metastatic pattern of infiltrating lobular carcinoma of the breast: An autopsy study. J. Surg. Oncol. 1991, 48, 28–33. [Google Scholar] [CrossRef]
- Winters, N.I.; Bader, D.M. Development of the Serosal Mesothelium. J. Dev. Biol. 2013, 1, 64–81. [Google Scholar] [CrossRef]
- Mouabbi, J.A.; Hassan, A.; Lim, B.; Hortobagyi, G.N.; Tripathy, D.; Layman, R.M. Invasive lobular carcinoma: An understudied emergent subtype of breast cancer. Breast Cancer Res. Treat. 2022, 193, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Tonsing-Carter, E.; Hernandez, K.M.; Kim, C.R.; Harkless, R.V.; Oh, A.; Bowie, K.R.; West-Szymanski, D.C.; Betancourt-Ponce, M.A.; Green, B.D.; Lastra, R.R.; et al. Glucocorticoid receptor modulation decreases ER-positive breast cancer cell proliferation and suppresses wild-type and mutant ER chromatin association. Breast Cancer Res. 2019, 21, 82. [Google Scholar] [CrossRef] [PubMed]
- West, D.C.; Kocherginsky, M.; Tonsing-Carter, E.Y.; Dolcen, D.N.; Hosfield, D.J.; Lastra, R.R.; Sinnwell, J.P.; Thompson, K.J.; Bowie, K.R.; Harkless, R.V.; et al. Discovery of a Glucocorticoid Receptor (GR) Activity Signature Using Selective GR Antagonism in ER-Negative Breast Cancer. Clin. Cancer Res. 2018, 24, 3433–3446. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Davidowitz, R.A.; Iwanicki, M.P.; Brugge, J.S. In vitro mesothelial clearance assay that models the early steps of ovarian cancer metastasis. J. Vis. Exp. 2012, 60, 3888. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Liu, H.; Patel, M.R.; Prescher, J.A.; Patsialou, A.; Qian, D.; Lin, J.; Wen, S.; Chang, Y.-F.; Bachmann, M.H.; Shimono, Y.; et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl. Acad. Sci. USA 2010, 107, 18115–18120. [Google Scholar] [CrossRef] [PubMed]
- Behbod, F.; Kittrell, F.S.; LaMarca, H.; Edwards, D.; Kerbawy, S.; Heestand, J.C.; Young, E.; Mukhopadhyay, P.; Yeh, H.-W.; Allred, D.C.; et al. An intraductal human-in-mouse transplantation model mimics the subtypes of ductal carcinoma in situ. Breast Cancer Res. 2009, 11, R66. [Google Scholar] [CrossRef] [PubMed]
- Lainé, M.; Fanning, S.W.; Chang, Y.-F.; Green, B.; Greene, M.E.; Komm, B.; Kurleto, J.D.; Phung, L.; Greene, G.L. Lasofoxifene as a potential treatment for therapy-resistant ER-positive metastatic breast cancer. Breast Cancer Res. 2021, 23, 54. [Google Scholar] [CrossRef] [PubMed]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 21 August 2023).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Cheng, N.; Li, G.; Kanchwala, M.; Evers, B.M.; Xing, C.; Yu, H. STAG2 promotes the myelination transcriptional program in oligodendrocytes. eLife 2022, 11, e77848. [Google Scholar] [CrossRef]
- Stringer-Reasor, E.M.; Baker, G.M.; Skor, M.N.; Kocherginsky, M.; Lengyel, E.; Fleming, G.F.; Conzen, S.D. Glucocorticoid receptor activation inhibits chemotherapy-induced cell death in high-grade serous ovarian carcinoma. Gynecol. Oncol. 2015, 138, 656–662. [Google Scholar] [CrossRef]
- Iwanicki, M.P.; Davidowitz, R.A.; Ng, M.R.; Besser, A.; Muranen, T.; Merritt, M.; Danuser, G.; Ince, T.; Brugge, J.S. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011, 1, 144–157. [Google Scholar] [CrossRef]
- Davidowitz, R.A.; Selfors, L.M.; Iwanicki, M.P.; Elias, K.M.; Karst, A.; Piao, H.; Ince, T.A.; Drage, M.G.; Dering, J.; Konecny, G.E.; et al. Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. J. Clin. Investig. 2014, 124, 2611–2625. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Jin, Y.; Nagaich, A.K. Interaction of glucocorticoid receptor (GR) with estrogen receptor (ER) alpha and activator protein 1 (AP1) in dexamethasone-mediated interference of ERalpha activity. J. Biol. Chem. 2013, 288, 24020–24034. [Google Scholar] [CrossRef] [PubMed]
- West, D.C.; Pan, D.; Tonsing-Carter, E.Y.; Hernandez, K.M.; Pierce, C.F.; Styke, S.C.; Bowie, K.R.; Garcia, T.I.; Kocherginsky, M.; Conzen, S.D. GR and ER Coactivation Alters the Expression of Differentiation Genes and Associates with Improved ER+ Breast Cancer Outcome. Mol. Cancer Res. 2016, 14, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Miranda, T.B.; Voss, T.C.; Sung, M.H.; Baek, S.; John, S.; Hawkins, M.; Grøntved, L.; Schiltz, R.L.; Hager, G.L. Reprogramming the chromatin landscape: Interplay of the estrogen and glucocorticoid receptors at the genomic level. Cancer Res. 2013, 73, 5130–5139. [Google Scholar] [CrossRef]
- Yang, F.; Ma, Q.; Liu, Z.; Li, W.; Tan, Y.; Jin, C.; Ma, W.; Hu, Y.; Shen, J.; Ohgi, K.A.; et al. Glucocorticoid Receptor:MegaTrans Switching Mediates the Repression of an ERalpha-Regulated Transcriptional Program. Mol. Cell 2017, 66, 321–331.e6. [Google Scholar] [CrossRef]
- Kittrell, F.; Valdez, K.; Elsarraj, H.; Hong, Y.; Medina, D.; Behbod, F. Mouse Mammary Intraductal (MIND) Method for Transplantation of Patient Derived Primary DCIS Cells and Cell Lines. Bio-Protoc. 2016, 6, e1744. [Google Scholar] [CrossRef]
- Sflomos, G.; Dormoy, V.; Metsalu, T.; Jeitziner, R.; Battista, L.; Scabia, V.; Raffoul, W.; Delaloye, J.F.; Treboux, A.; Fiche, M.; et al. A Preclinical Model for ERalpha-Positive Breast Cancer Points to the Epithelial Microenvironment as Determinant of Luminal Phenotype and Hormone Response. Cancer Cell 2016, 29, 407–422. [Google Scholar] [CrossRef]
- Tabár, L.; Bozó, R.; Dean, P.B.; Ormándi, K.; Puchkova, O.; Oláh-Németh, O.; Németh, I.B.; Veréb, Z.; Yen, M.F.; Chen, L.S.; et al. Does Diffusely Infiltrating Lobular Carcinoma of the Breast Arise from Epithelial-Mesenchymal Hybrid Cells? Int. J. Mol. Sci. 2023, 24, 10752. [Google Scholar] [CrossRef]
- Pang, D.; Kocherginsky, M.; Krausz, T.; Kim, S.Y.; Conzen, S. Dexamethasone decreases xenograft response to Paclitaxel through inhibition of tumor cell apoptosis. Cancer Biol. Ther. 2006, 5, 933–940. [Google Scholar] [CrossRef]
- Rogatsky, I.; Trowbridge, J.M.; Garabedian, M.J. Glucocorticoid Receptor-Mediated Cell Cycle Arrest Is Achieved through Distinct Cell-Specific Transcriptional Regulatory Mechanisms. Mol. Cell. Biol. 1997, 17, 3181–3193. [Google Scholar] [CrossRef]
- Poulsen, R.C.; Watts, A.C.; Murphy, R.J.; Snelling, S.J.; Carr, A.J.; Hulley, P.A. Glucocorticoids induce senescence in primary human tenocytes by inhibition of sirtuin 1 and activation of the p53/p21 pathway: In vivo and in vitro evidence. Ann. Rheum. Dis. 2014, 73, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and cancer—Role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Wang, D.; Yuan, X.; Liu, Y.; Guo, X.; Li, J.; Song, J.-G. Glucocorticoid receptor–IRS-1 axis controls EMT and the metastasis of breast cancers. J. Mol. Cell Biol. 2019, 11, 1042–1055. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Aleksandrowicz, E.; Schönsiegel, F.; Gröner, D.; Bauer, N.; Nwaeburu, C.C.; Zhao, Z.; Gladkich, J.; Hoppe-Tichy, T.; Yefenof, E.; et al. Dexamethasone mediates pancreatic cancer progression by glucocorticoid receptor, TGFβ and JNK/AP-1. Cell Death Dis. 2017, 8, e3064. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, Q.; Song, C.; Mi, B.; Zhang, H.; Kang, H.; Liu, H.; Sun, Y.; Wang, J.; Lei, Z.; et al. Serum- and Glucocorticoid-inducible Kinase 1 is Essential for Osteoclastogenesis and Promotes Breast Cancer Bone Metastasis. Mol. Cancer Ther. 2020, 19, 650–660. [Google Scholar] [CrossRef]
- Orr, W.; Varani, J.; Gondek, M.D.; Ward, P.A.; Mundy, G.R. Chemotactic Responses of Tumor Cells to Products of Resorbing Bone. Science 1979, 203, 176–1799. [Google Scholar] [CrossRef]
- Monteiro, A.C.; Leal, A.C.; Gonçalves-Silva, T.; Mercadante, A.C.T.; Kestelman, F.; Chaves, S.B.; Azevedo, R.B.; Monteiro, J.P.; Bonomo, A. T cells induce pre-metastatic osteolytic disease and help bone metastases establishment in a mouse model of metastatic breast cancer. PLoS ONE 2013, 8, e68171. [Google Scholar] [CrossRef]

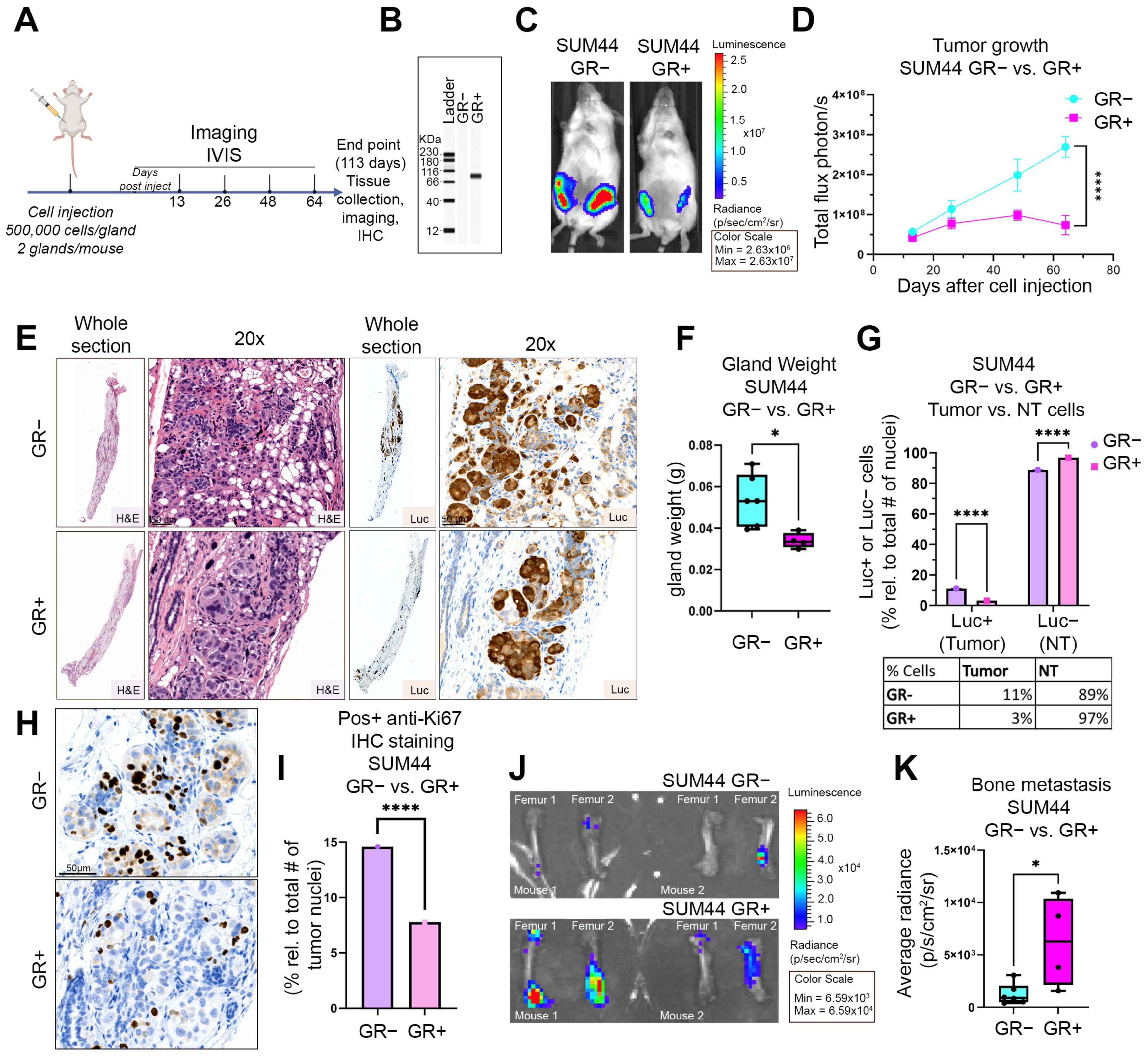

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingenuity Pathway Analysis | Activation z-Score | p-Values | |||
|---|---|---|---|---|---|
| E2 vs. Veh | E2 Plus Dex vs. Veh | Dex vs. Veh | |||
| Cell proliferation | |||||
| Estrogen-mediated S phase entry | SUM44 | +2.3 | +1.4 | −1.6 | 4.79 × 10−6, 4.47 × 10−3, 3.09 × 10−4 |
| MM134 | +2.0 | +0.8 | −1.6 | 4.79 × 10−3, 4.23 × 10−2, 1.02 × 10−4 | |
| ECM adhesion (metastasis) | |||||
| Integrin signaling | SUM44 | −1.5 | −0.2 | +1.6 | 1.15 × 10−4, 5.13 × 10−6, 5.75 × 10−3 |
| MM134 | ns | +0.2 | ns | 1.7410 × 10−5 | |
| Epithelial Mesenchymal Transition | SUM44 | −1.4 | +0.5 | +0.7 | 1.05 × 10−5, 5.89 × 10−5, 1.02 × 10−4 |
| MM134 | +1.1 | +1.6 | +1.7 | 2.29 × 10−4, 1.82 × 10−3, 1.41 × 10−2 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porter, B.A.; Frerich, C.; Lainé, M.; Clark, A.B.; Durdana, I.; Lee, J.; Taya, M.; Sahoo, S.; Greene, G.L.; Bennett, L.; et al. Glucocorticoid Receptor Activation in Lobular Breast Cancer Is Associated with Reduced Cell Proliferation and Promotion of Metastases. Cancers 2023, 15, 4679. https://doi.org/10.3390/cancers15194679
Porter BA, Frerich C, Lainé M, Clark AB, Durdana I, Lee J, Taya M, Sahoo S, Greene GL, Bennett L, et al. Glucocorticoid Receptor Activation in Lobular Breast Cancer Is Associated with Reduced Cell Proliferation and Promotion of Metastases. Cancers. 2023; 15(19):4679. https://doi.org/10.3390/cancers15194679
Chicago/Turabian StylePorter, Baylee A., Candace Frerich, Muriel Lainé, Abigail B. Clark, Ishrat Durdana, Jeon Lee, Manisha Taya, Sunati Sahoo, Geoffrey L. Greene, Lynda Bennett, and et al. 2023. "Glucocorticoid Receptor Activation in Lobular Breast Cancer Is Associated with Reduced Cell Proliferation and Promotion of Metastases" Cancers 15, no. 19: 4679. https://doi.org/10.3390/cancers15194679
APA StylePorter, B. A., Frerich, C., Lainé, M., Clark, A. B., Durdana, I., Lee, J., Taya, M., Sahoo, S., Greene, G. L., Bennett, L., & Conzen, S. D. (2023). Glucocorticoid Receptor Activation in Lobular Breast Cancer Is Associated with Reduced Cell Proliferation and Promotion of Metastases. Cancers, 15(19), 4679. https://doi.org/10.3390/cancers15194679