
Immune Checkpoint Blockade for Metastatic Uveal Melanoma: Re-Induction following Resistance or Toxicity

 ,
, 
 , , , , , , , , , ,
, , , , , , , , , ,  , , , and
on behalf of the German Dermatologic Cooperative Oncology Group (DeCOG, Committee Ocular Melanoma)add
Show full author list
, , , and
on behalf of the German Dermatologic Cooperative Oncology Group (DeCOG, Committee Ocular Melanoma)add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Population and Study Design
2.2. Data Collection and Treatment Outcomes
2.3. Statistical Analyses
3. Results
3.1. Baseline Patient Characteristics
3.2. Response Rates to ICB
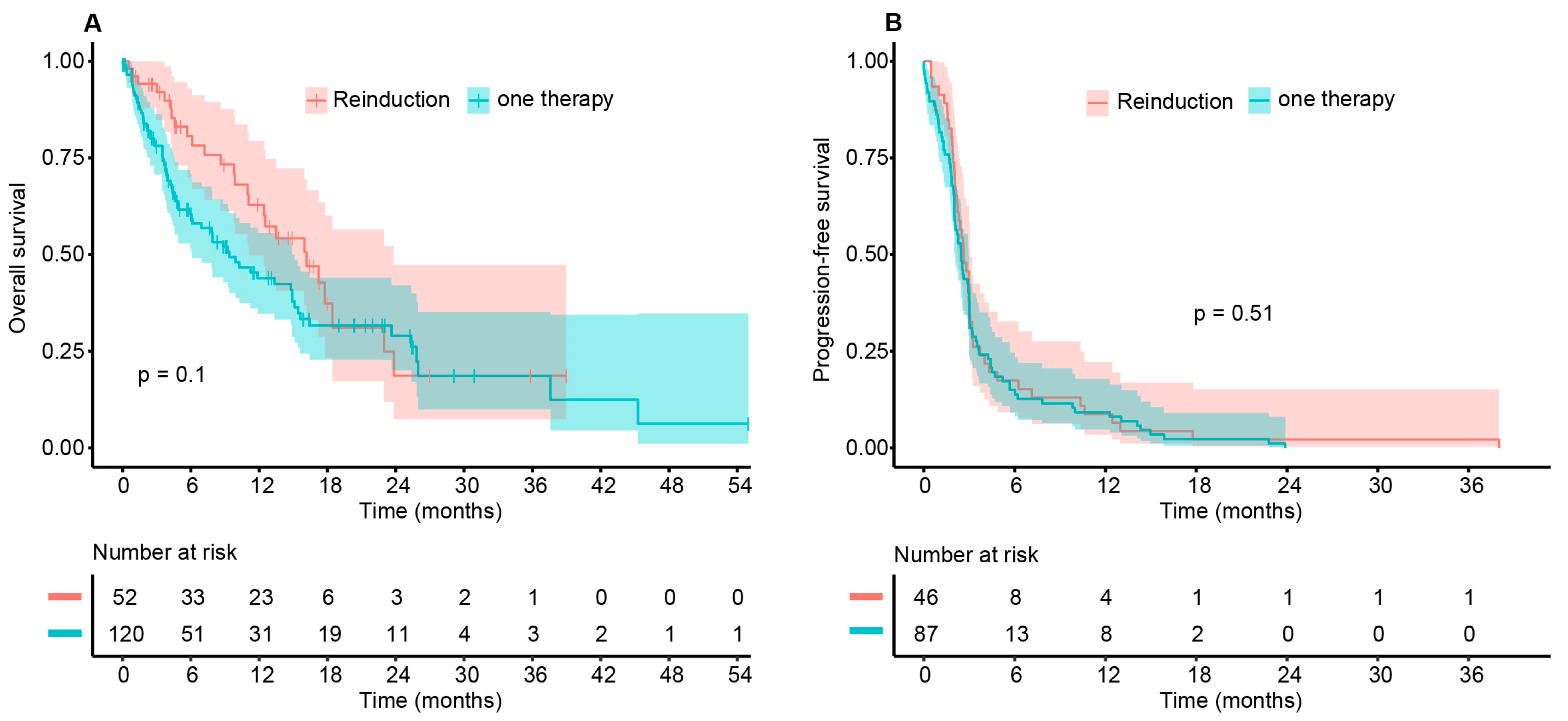
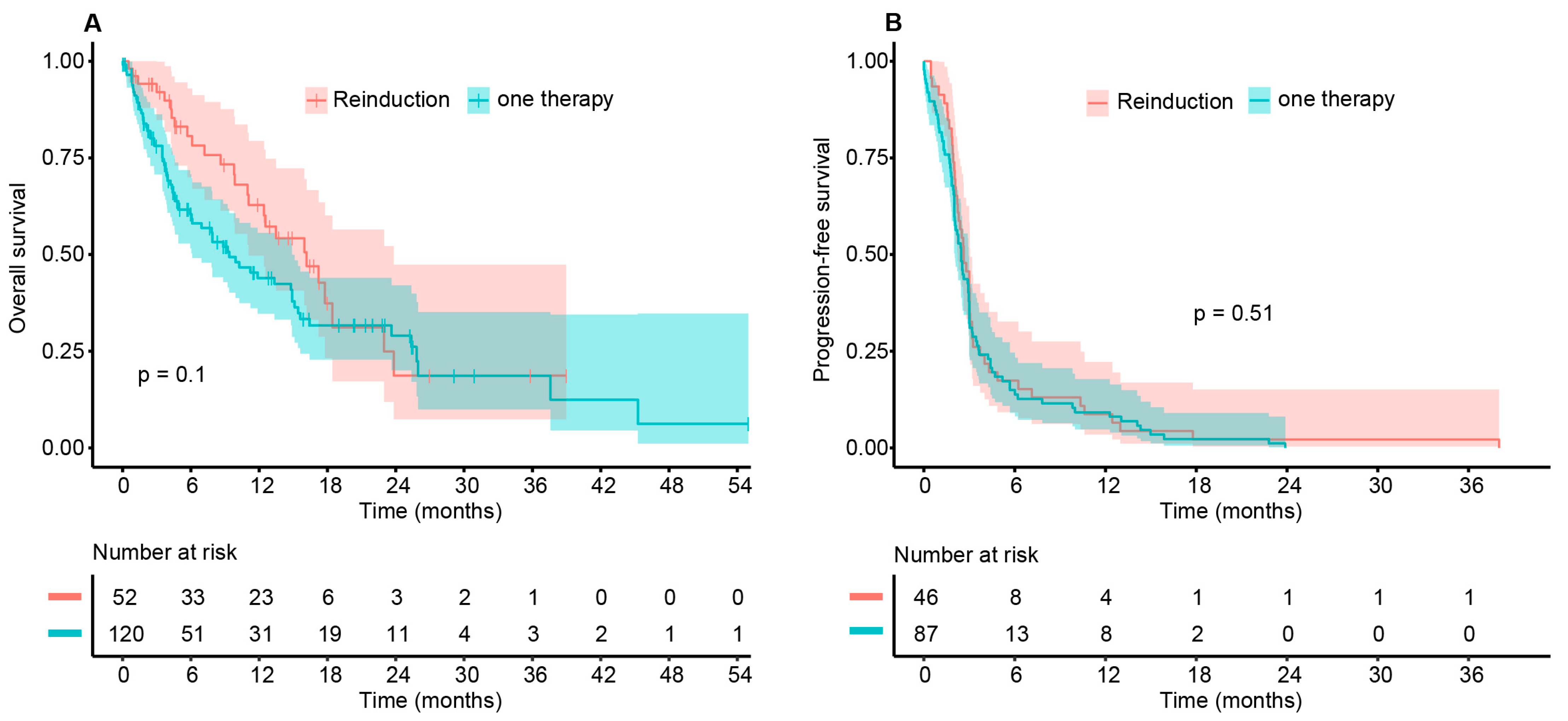
3.3. Survival Data
3.4. Adverse Events (AE)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smit, K.N.; Jager, M.J.; de Klein, A.; Kili, E. Uveal melanoma: Towards a molecular understanding. Prog. Retin. Eye Res. 2020, 75, 100800. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Ocular Melanoma Study Group. Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study (COMS): COMS report no. 15. Arch. Ophthalmol. 2001, 119, 670–676. [Google Scholar] [CrossRef]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, E.A.T.; Petzold, A.; Wessely, A.; Dippel, E.; Erdmann, M.; Heinzerling, L.; Hohberger, B.; Knorr, H.; Leiter, U.; Meier, F.; et al. Clinical determinants of long-term survival in metastatic uveal melanoma. Cancer Immunol. Immunother. 2021. online ahead of print. [Google Scholar] [CrossRef]
- Koch, E.A.T.; Petzold, A.; Wessely, A.; Dippel, E.; Gesierich, A.; Gutzmer, R.; Hassel, J.C.; Haferkamp, S.; Hohberger, B.; Kahler, K.C.; et al. Immune Checkpoint Blockade for Metastatic Uveal Melanoma: Patterns of Response and Survival According to the Presence of Hepatic and Extrahepatic Metastasis. Cancers 2021, 13, 3359. [Google Scholar] [CrossRef]
- Koch, E.A.T.; Nickel, F.T.; Heinzerling, L.; Schulz, Y.K.; Berking, C.; Erdmann, M. Immune Checkpoint Inhibitor-induced Bilateral Vestibulopathy. J. Immunother. 2021, 44, 114–117. [Google Scholar] [CrossRef]
- Pelster, M.S.; Gruschkus, S.K.; Bassett, R.; Gombos, D.S.; Shephard, M.; Posada, L.; Glover, M.S.; Simien, R.; Diab, A.; Hwu, P.; et al. Nivolumab and Ipilimumab in Metastatic Uveal Melanoma: Results From a Single-Arm Phase II Study. J. Clin. Oncol. 2020, 39, 599–607. [Google Scholar] [CrossRef]
- Piulats, J.M.; Espinosa, E.; de la Cruz Merino, L.; Varela, M.; Alonso Carrion, L.; Martin-Algarra, S.; Lopez Castro, R.; Curiel, T.; Rodriguez-Abreu, D.; Redrado, M.; et al. Nivolumab Plus Ipilimumab for Treatment-Naive Metastatic Uveal Melanoma: An Open-Label, Multicenter, Phase II Trial by the Spanish Multidisciplinary Melanoma Group (GEM-1402). J. Clin. Oncol. 2021, 39, 586–598. [Google Scholar] [CrossRef]
- Heppt, M.V.; Amaral, T.; Kahler, K.C.; Heinzerling, L.; Hassel, J.C.; Meissner, M.; Kreuzberg, N.; Loquai, C.; Reinhardt, L.; Utikal, J.; et al. Combined immune checkpoint blockade for metastatic uveal melanoma: A retrospective, multi-center study. J. Immunother. Cancer 2019, 7, 299. [Google Scholar] [CrossRef]
- Bossi, G.; Buisson, S.; Oates, J.; Jakobsen, B.K.; Hassan, N.J. ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol. Immunother. 2014, 63, 437–448. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Hassel, J.C.; Rutkowski, P.; Baurain, J.-F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Abstract CT002: Phase 3 randomized trial comparing tebentafusp with investigator’s choice in first line metastatic uveal melanoma. Cancer Res. 2021, 81, CT002. [Google Scholar] [CrossRef]
- Hepner, A.; Atkinson, V.G.; Larkin, J.; Burrell, R.A.; Carlino, M.S.; Johnson, D.B.; Zimmer, L.; Tsai, K.K.; Klein, O.; Lo, S.N.; et al. Re-induction ipilimumab following acquired resistance to combination ipilimumab and anti-PD-1 therapy. Eur. J. Cancer 2021, 153, 213–222. [Google Scholar] [CrossRef]
- Wessely, A.; Steeb, T.; Erdmann, M.; Heinzerling, L.; Vera, J.; Schlaak, M.; Berking, C.; Heppt, M.V. The Role of Immune Checkpoint Blockade in Uveal Melanoma. Int. J. Mol. Sci. 2020, 21, 879. [Google Scholar] [CrossRef] [Green Version]
- Tietze, J.K.; Forschner, A.; Loquai, C.; Mitzel-Rink, H.; Zimmer, L.; Meiss, F.; Rafei-Shamsabadi, D.; Utikal, J.; Bergmann, M.; Meier, F.; et al. The efficacy of re-challenge with BRAF inhibitors after previous progression to BRAF inhibitors in melanoma: A retrospective multicenter study. Oncotarget 2018, 9, 34336–34346. [Google Scholar] [CrossRef]
- Khoja, L.; Atenafu, E.G.; Suciu, S.; Leyvraz, S.; Sato, T.; Marshall, E.; Keilholz, U.; Zimmer, L.; Patel, S.P.; Piperno-Neumann, S.; et al. Meta-analysis in metastatic uveal melanoma to determine progression free and overall survival benchmarks: An international rare cancers initiative (IRCI) ocular melanoma study. Ann. Oncol. 2019, 30, 1370–1380. [Google Scholar] [CrossRef]
- Rantala, E.S.; Hernberg, M.; Kivela, T.T. Overall survival after treatment for metastatic uveal melanoma: A systematic review and meta-analysis. Melanoma. Res. 2019, 29, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, A.; Eggermont, A.M.M.; Robert, C.; Dummer, R.; Ugurel, S.; Livingstone, E.; Ascierto, P.A.; Long, G.V.; Schadendorf, D.; Zimmer, L. The concepts of rechallenge and retreatment with immune checkpoint blockade in melanoma patients. Eur. J. Cancer 2021, 155, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Blasig, H.; Bender, C.; Hassel, J.C.; Eigentler, T.K.; Sachse, M.M.; Hiernickel, J.; Koop, A.; Satzger, I.; Gutzmer, R. Reinduction of PD1-inhibitor therapy: First experience in eight patients with metastatic melanoma. Melanoma. Res. 2017, 27, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Otsuka, A.; Kondo, T.; Nagai, H.; Nonomura, Y.; Kaku, Y.; Matsumoto, S.; Muto, M. Efficacy and safety of retreatment with nivolumab in metastatic melanoma patients previously treated with nivolumab. Cancer Chemother. Pharmacol. 2017, 80, 999–1004. [Google Scholar] [CrossRef]
- Hoefsmit, E.P.; Rozeman, E.A.; Van, T.M.; Dimitriadis, P.; Krijgsman, O.; Conway, J.W.; Pires da Silva, I.; van der Wal, J.E.; Ketelaars, S.L.C.; Bresser, K.; et al. Comprehensive analysis of cutaneous and uveal melanoma liver metastases. J. Immunother. Cancer 2020, 8, e001501. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Hashimoto, K.; Nishimura, S.; Ito, T.; Akagi, M. Characterization of PD-1/PD-L1 immune checkpoint expression in soft tissue sarcomas. Eur. J. Histochem. 2021, 65, 3203. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Nishimura, S.; Sakata, N.; Inoue, M.; Sawada, A.; Akagi, M. Characterization of PD-1/PD-L1 immune checkpoint expression in the pathogenesis of musculoskeletal Langerhans cell histiocytosis: A retrospective study. Medicine 2021, 100, e27650. [Google Scholar] [CrossRef] [PubMed]
- Stege, H.M.; Haist, M.; Schultheis, S.; Fleischer, M.I.; Mohr, P.; Ugurel, S.; Terheyden, P.; Thiem, A.; Kiecker, F.; Leiter, U.; et al. Response durability after cessation of immune checkpoint inhibitors in patients with metastatic Merkel cell carcinoma: A retrospective multicenter DeCOG study. Cancer Immunol. Immunother. 2021, 70, 3313–3322. [Google Scholar] [CrossRef] [PubMed]
- Najjar, Y.G.; Navrazhina, K.; Ding, F.; Bhatia, R.; Tsai, K.; Abbate, K.; Durden, B.; Eroglu, Z.; Bhatia, S.; Park, S.; et al. Ipilimumab plus nivolumab for patients with metastatic uveal melanoma: A multicenter, retrospective study. J. Immunother. Cancer 2020, 8, e000331. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Total | Cohort A | Cohort B | A vs. B | ||
|---|---|---|---|---|---|
| Sex | Women | 89 (50.3%) | 27 (48.1%) | 62 (49.6%) | p = 0.9 |
| Men | 88 (49.7%) | 25 (51.9%) | 63 (50.4%) | ||
| Age | Median in years (range) | 66.2 (17.7–87.6) | 64.2 (31.7–85.7) | 67.0 (17.7–87.6) | p = 0.3 |
| LDH | Not elevated | 40 (22.6%) | 14 (26.9%) | 26 (20.8%) | p = 0.1 |
| Elevated | 90 (50.8%) | 21 (40.4%) | 69 (55.2%) | ||
| NA | 47 (26.6%) | 17 (32.7%) | 30 (24.0%) | ||
| ECOG | ECOG 0 | 84 (47.5%) | 25 (48.1%) | 59 (47.2%) | p = 0.22 |
| ECOG 1 | 19 (10.7%) | 4 (7.7%) | 15 (12.0%) | ||
| ECOG 2 | 4 (2.3%) | 1 (1.9%) | 3 (2.4%) | ||
| ECOG 3 | 2 (1.1%) | 0 (0.0%) | 2 (1.6%) | ||
| ECOG 4 | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | ||
| ECOG 5 | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | ||
| NA | 68 (38.4%) | 22 (42.3%) | 46 (36.8%) | ||
| Number of affected organ systems | Median (range) | 2 (1–8) | 3 (1–7) | 2 (1–8) | p = 0.013 |
| Affected organ systems | Liver | 164 (92.7%) | 50 (96.2%) | 114 (91.2%) | |
| Pulmonary | 87 (49.2%) | 27 (51.9%) | 60 (48.0%) | ||
| Bone | 50 (28.2%) | 19 (36.5%) | 31 (24.8%) | ||
| CNS | 23 (13.0%) | 10 (19.2%) | 13 (10.4%) | ||
| Lymph node | 40 (22.6%) | 15 (28.9%) | 25 (20.0%) | ||
| Connective tissue | 9 (5.1%) | 4 (7.7%) | 5 (4.0%) | ||
| Skin | 24 (13.6%) | 12 (23.1%) | 12 (9.6%) | ||
| Disseminated | 10 (5.6%) | 2 (3.8%) | 8 (6.4%) | ||
| Other | 50 (28.2%) | 20 (38.5%) | 30 (24.0%) | ||
| Treatments other than ICB | Chemotherapy | 46 (25.9%) | 28 (53.8%) | 18 (14.4%) | p < 0.001 |
| MEK inhibitor | 13 (73.4%) | 4 (7.6%) | 9 (7.2%) | p > 0.999 | |
| Sorafenib | 22 (12.4%) | 9 (17.3%) | 13 (10.4%) | p = 0.308 | |
| ICB regimen | Any | 177 (100%) | 52 (100.0%) | 125 (100%) | |
| Anti-PD-1 monotherapy (pembrolizumab, nivolumab) | 53 (29.9%) | 18 (34.6%) | 35 (28.0%) | p = 0.487 | |
| Anti-CTLA-4 monotherapy (ipilimumab) | 5 (2.8%) | 2 (3.8%) | 3 (2.4%) | p = 0.975 | |
| Combined ICB | 119 (67.2%) | 32 (61.5%) | 87 (69.6%) | p = 0.387 |
| Total | Cohort A | Cohort B | Test | ||||
|---|---|---|---|---|---|---|---|
| ICB First Induction (A1) | ICB Re-Induction (A2) | A1 vs. A2 | A1 vs. B | A2 vs. B | |||
| CR | 3/152 = 2.0% | 0/52 = 0.0% | 1/50 = 2.0% | 2/102 = 2.0% | p = 0.98 | p = 0.79 | p > 0.999 |
| PR | 14/152 = 9.2% | 4/52 = 7.7% | 4/50 = 8.0% | 10/102 = 9.8% | p > 0.999 | p = 0.89 | p = 0.94 |
| SD | 26/152 = 17.1% | 13/52 = 25.0% | 5/50 = 10.0% | 21/102 = 20.6% | p = 0.084 | p = 0.68 | p = 0.16 |
| PD | 101/152 = 66.4% | 33/52 = 63.5% | 37/50 = 74.0% | 64/102 = 62.7% | p = 0.35 | p > 0.999 | p = 0.23 |
| ORR | 17/152 = 11.2% | 4/52 = 7.7% | 5/50 = 10.0% | 12/102 = 11.8% | p = 0.95 | p = 0.61 | p = 0.95 |
| DCR | 43/152 = 28.3% | 18/52 = 34.6% | 10/50 = 20.0% | 33/102 = 32.4% | p = 0.15 | p = 0.91 | p = 0.16 |
| Response to First ICB Induction | PD to ICB Re-Induction | SD to ICB Re-Induction | PR to ICB Re-Induction | CR to ICB Re-Induction |
|---|---|---|---|---|
| PD (n = 33/52) | 24 (72.7%) | 3 (9%) | 2 (6%) | 0 (0.0%) |
| SD (n = 13/52) | 9 (69.2%) | 2 (15.3%) | 1 (7.6%) | 0 (0.0%) |
| PR (n = 4/52) | 2 (50%) | 0 (0.0%) | 1 (25%) | 1 (25%) |
| Time in Months | OS (95% CI) Cohort A | OS (95% CI) Cohort B | Difference (95% CI) |
|---|---|---|---|
| 6 | 0.806 (0.6997–0.929) | 0.604 (0.516–0.708) | 0.202 (0.139–0.265) |
| 12 | 0.628 (0.4974–0.794) | 0.439 (0.347–0.556) | 0.189 (0.085–0.293) |
| 18 | 0.374 (0.2325–0.601) | 0.317 (0.228–0.440) | 0.057 (−0.087–0.201) |
| 24 | 0.187 (0.0737–0.474) | 0.290 (0.200–0.420) | −0.103 (−0.240–0.033) |
| Total | Cohort A1 | Cohort A2 | Cohort B | p-Value (A1 vs. B) | |
|---|---|---|---|---|---|
| Number of patients with any AE | 74/177 (41.8%) | 24/52 (46.2%) | 21/52 (40.4%) | 50/125 (40.0%) | p = 0.55 |
| Number of patients with severe AE | 44/177 (24.8%) | 14/52 (26.9%) | 10/52 (19.1%) | 30/125 (24.0%) | p = 0.82 |
| Toxicity of Re-Induction | Re-Induction after Resistance to First ICB (n = 30) | Re-Induction after Toxicity to First ICB (n = 13) | p-Value |
|---|---|---|---|
| Number of patients with any AE | 6 (20.0%) | 13 (100.0%) | p < 0.0001 |
| Number of patients with severe AE | 3 (10.0%) | 5 (38.5%) | p = 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koch, E.A.T.; Petzold, A.; Wessely, A.; Dippel, E.; Gesierich, A.; Gutzmer, R.; Hassel, J.C.; Haferkamp, S.; Kähler, K.C.; Knorr, H.; et al. Immune Checkpoint Blockade for Metastatic Uveal Melanoma: Re-Induction following Resistance or Toxicity. Cancers 2022, 14, 518. https://doi.org/10.3390/cancers14030518
Koch EAT, Petzold A, Wessely A, Dippel E, Gesierich A, Gutzmer R, Hassel JC, Haferkamp S, Kähler KC, Knorr H, et al. Immune Checkpoint Blockade for Metastatic Uveal Melanoma: Re-Induction following Resistance or Toxicity. Cancers. 2022; 14(3):518. https://doi.org/10.3390/cancers14030518
Chicago/Turabian StyleKoch, Elias A. T., Anne Petzold, Anja Wessely, Edgar Dippel, Anja Gesierich, Ralf Gutzmer, Jessica C. Hassel, Sebastian Haferkamp, Katharina C. Kähler, Harald Knorr, and et al. 2022. "Immune Checkpoint Blockade for Metastatic Uveal Melanoma: Re-Induction following Resistance or Toxicity" Cancers 14, no. 3: 518. https://doi.org/10.3390/cancers14030518
APA StyleKoch, E. A. T., Petzold, A., Wessely, A., Dippel, E., Gesierich, A., Gutzmer, R., Hassel, J. C., Haferkamp, S., Kähler, K. C., Knorr, H., Kreuzberg, N., Leiter, U., Loquai, C., Meier, F., Meissner, M., Mohr, P., Pföhler, C., Rahimi, F., Schadendorf, D., ... Heppt, M. V., on behalf of the German Dermatologic Cooperative Oncology Group (DeCOG, Committee Ocular Melanoma). (2022). Immune Checkpoint Blockade for Metastatic Uveal Melanoma: Re-Induction following Resistance or Toxicity. Cancers, 14(3), 518. https://doi.org/10.3390/cancers14030518