
Identification of Early-Onset Metastasis in SF3B1 Mutated Uveal Melanoma

 , , , , , , ,
, , , , , , ,  ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of a Uniform Clinical Dataset of UM Patients
2.2. Mutation Analysis
2.3. Survival Analysis
2.4. Processing and Analysis of Whole-Transcriptome Data
2.5. Differential Gene-Expression Analysis
2.6. Differential Gene-Set Enrichment Analysis (GSEA)
2.7. Detection of Aberrant Splicing Patterns
2.8. Validation of In Silico Results
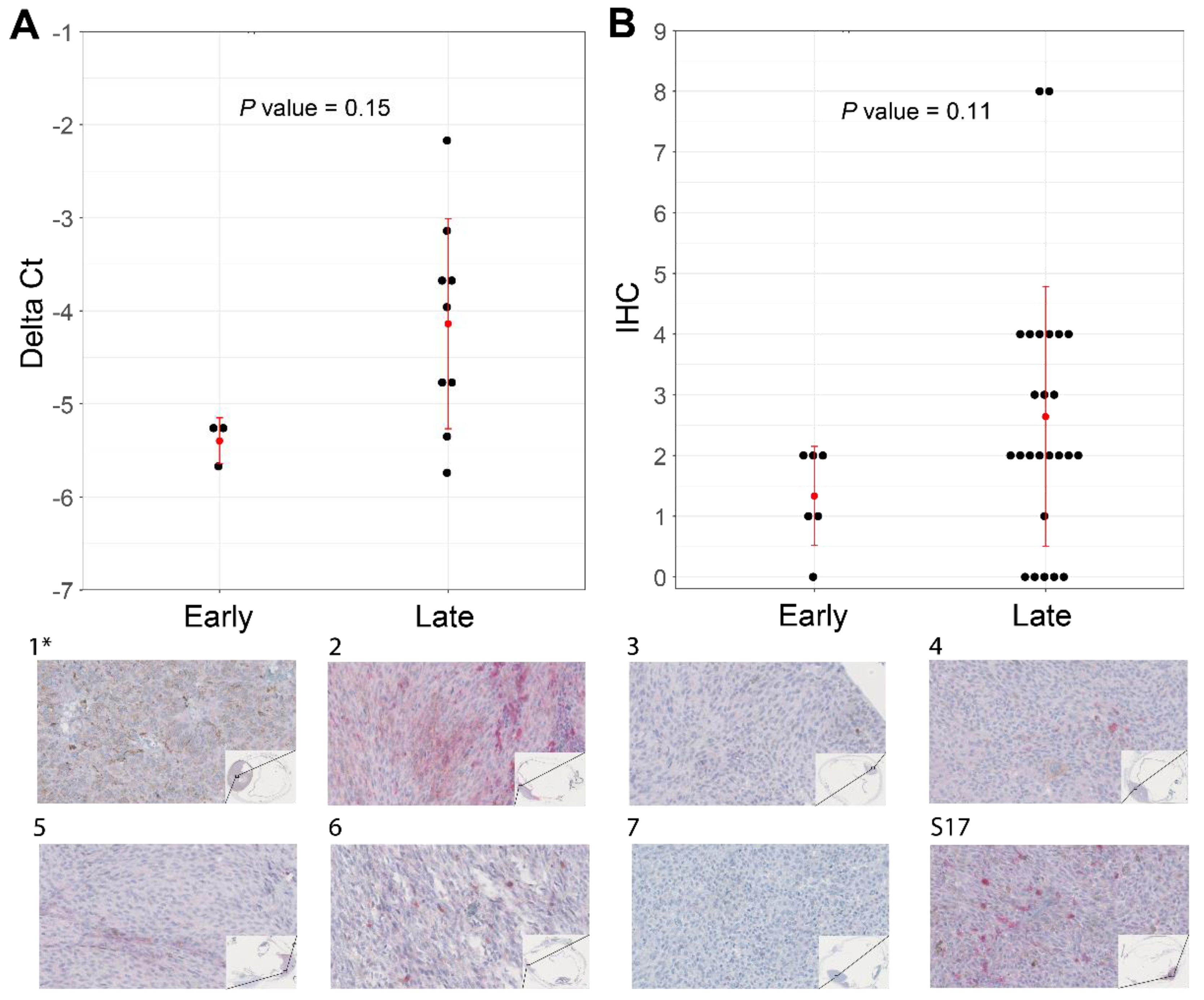
2.9. ABHD6 Immunohistochemistry
2.10. Statistical Analysis and Code Availability
3. Results
3.1. Establishing a Uniform Clinical Dataset of UM from Various Studies
3.2. Overview of the Clinical Parameters of the ROMS Cohort
3.3. Stratification of All SF3B1mut UM
3.4. Metastatic Location of SF3B1mut UM
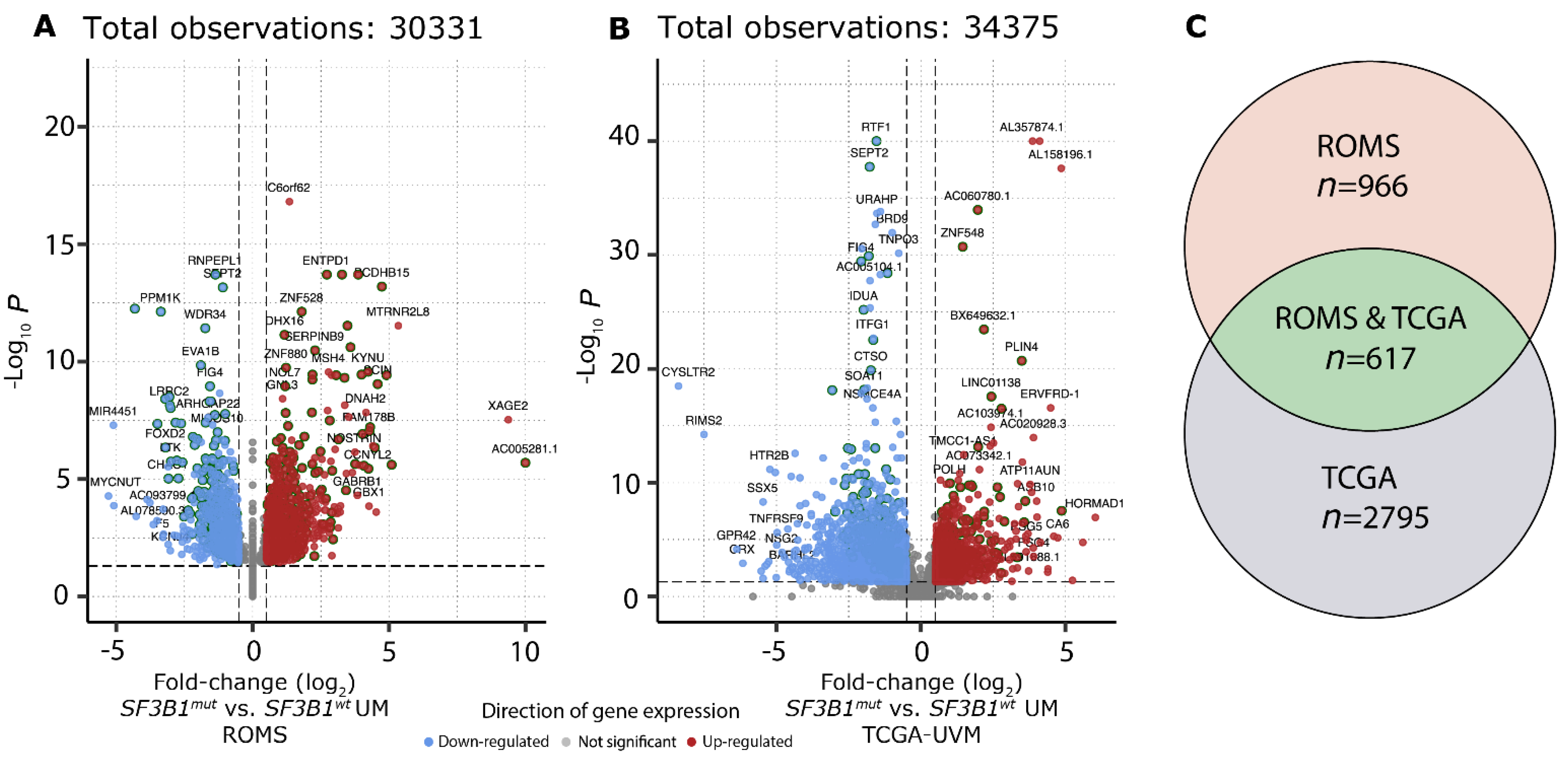
3.5. Differential Analysis of Canonical Transcripts Reveals SF3B1mut-Exclusive Transcripts
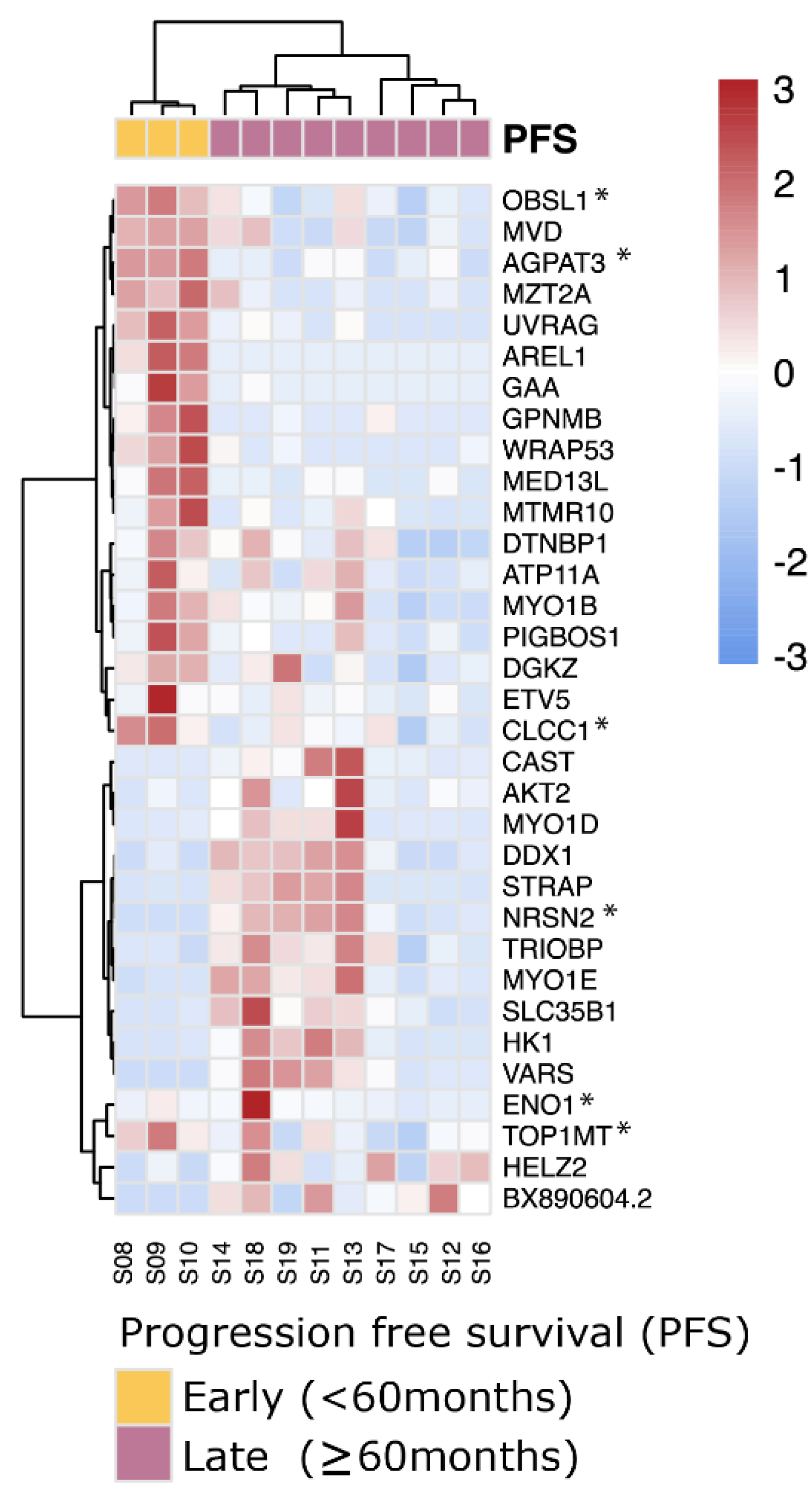
3.6. SF3B1mut UM Can Be Stratified Using Differentially Expressed Canonical Transcripts in PFS < 60 and PFS ≥ 60 Months
3.7. Differential Analysis of Aberrant-Splicing Reveals SF3B1mut-Exclusive Transcripts
3.8. SF3B1mut UM Can Be Stratified Using Differentially Expressed Aberrant Transcripts in PFS < 60 and PFS ≥ 60 Months
4. Discussion
4.1. Unbiased Detection of Pathogenic SF3B1mut UM
4.2. Potential Biomarkers Capable of Distinguishing between Early- and Late-Onset SF3B1mut UM
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shields, C.L.; Kaliki, S.; Furuta, M.; Fulco, E.; Alarcon, C.; Shields, J.A. American joint committee on cancer classification of posterior uveal melanoma (tumor size category) predicts prognosis in 7731 patients. Ophthalmology 2013, 120, 2066–2071. [Google Scholar] [CrossRef]
- Prescher, G.; Bornfeld, N.; Hirche, H.; Horsthemke, B.; Jockel, K.H.; Becher, R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet 1996, 347, 1222–1225. [Google Scholar] [PubMed]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of bap1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [Green Version]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; van Bodegom, A.; Paridaens, D.; Kilic, E.; de Klein, A.; Rotterdam Ocular Melanoma Study, G. Uveal melanomas with sf3b1 mutations: A distinct subclass associated with late-onset metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Ehlers, J.P.; Harbour, J.W. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004, 64, 7205–7209. [Google Scholar] [CrossRef] [Green Version]
- Dogrusoz, M.; Bagger, M.; van Duinen, S.G.; Kroes, W.G.; Ruivenkamp, C.A.; Bohringer, S.; Andersen, K.K.; Luyten, G.P.; Kiilgaard, J.F.; Jager, M.J. The prognostic value of ajcc staging in uveal melanoma is enhanced by adding chromosome 3 and 8q status. Investig. Ophthalmol. Vis. Sci. 2017, 58, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Furney, S.J.; Pedersen, M.; Gentien, D.; Dumont, A.G.; Rapinat, A.; Desjardins, L.; Turajlic, S.; Piperno-Neumann, S.; de la Grange, P.; Roman-Roman, S.; et al. Sf3b1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013, 3, 1122–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated sf3b1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Roberson, E.D.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent mutations at codon 625 of the splicing factor sf3b1 in uveal melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef]
- Martin, M.; Masshofer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; van de Nes, J.; Klein-Hitpass, L.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome sequencing identifies recurrent somatic mutations in eif1ax and sf3b1 in uveal melanoma with disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef] [Green Version]
- Yavuzyigitoglu, S.; Drabarek, W.; Smit, K.N.; van Poppelen, N.; Koopmans, A.E.; Vaarwater, J.; Brands, T.; Eussen, B.; Dubbink, H.J.; van Riet, J.; et al. Correlation of gene mutation status with copy number profile in uveal melanoma. Ophthalmology 2017, 124, 573–575. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220 e215. [Google Scholar] [CrossRef] [Green Version]
- Mazloumi, M.; Vichitvejpaisal, P.; Dalvin, L.A.; Yaghy, A.; Ewens, K.G.; Ganguly, A.; Shields, C.L. Accuracy of the cancer genome atlas classification vs american joint committee on cancer classification for prediction of metastasis in patients with uveal melanoma. JAMA Ophthalmol. 2020, 138, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Martin-Marcos, P.; Zhou, F.; Karunasiri, C.; Zhang, F.; Dong, J.; Nanda, J.; Kulkarni, S.D.; Sen, N.D.; Tamame, M.; Zeschnigk, M.; et al. Eif1a residues implicated in cancer stabilize translation preinitiation complexes and favor suboptimal initiation sites in yeast. Elife 2017, 6. [Google Scholar]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. Cosmic: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-associated sf3b1 hotspot mutations induce cryptic 3’ splice site selection through use of a different branch point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- Drabarek, W.; Yavuzyigitoglu, S.; Obulkasim, A.; van Riet, J.; Smit, K.N.; van Poppelen, N.M.; Vaarwater, J.; Brands, T.; Eussen, B.; Verdijk, R.M.; et al. Multi-modality analysis improves survival prediction in enucleated uveal melanoma patients. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3595–3605. [Google Scholar] [CrossRef]
- Royer-Bertrand, B.; Torsello, M.; Rimoldi, D.; El Zaoui, I.; Cisarova, K.; Pescini-Gobert, R.; Raynaud, F.; Zografos, L.; Schalenbourg, A.; Speiser, D.; et al. Comprehensive genetic landscape of uveal melanoma by whole-genome sequencing. Am. J. Hum. Genet. 2016, 99, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.P.; Kim, I.K.; Esmaeli, B.; Amin-Mansour, A.; Treacy, D.J.; Carter, S.L.; Hodis, E.; Wagle, N.; Seepo, S.; Yu, X.; et al. Systematic genomic and translational efficiency studies of uveal melanoma. PLoS ONE 2017, 12, e0178189. [Google Scholar] [CrossRef] [Green Version]
- Shain, A.H.; Bagger, M.M.; Yu, R.; Chang, D.; Liu, S.; Vemula, S.; Weier, J.F.; Wadt, K.; Heegaard, S.; Bastian, B.C.; et al. The genetic evolution of metastatic uveal melanoma. Nat. Genet. 2019, 51, 1123–1130. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Rodrigues, M.; Mobuchon, L.; Houy, A.; Alsafadi, S.; Baulande, S.; Mariani, O.; Marande, B.; Ait Rais, K.; Van der Kooij, M.K.; Kapiteijn, E.; et al. Evolutionary routes in metastatic uveal melanomas depend on mbd4 alterations. Clin. Cancer Res. 2019, 25, 5513–5524. [Google Scholar] [CrossRef]
- Kivela, T.; Simpson, E.R.; Grossniklaus, H.E.; Jager, M.J.; Singh, A.D.; Caminal, J.M.; Pavlick, A.; Kujala, E.; Coupland, S.E. Uveal melanoma. In Ajcc Cancer Staging Manual, 8th ed.; Amin, M.B., Ed.; Springer: New York, NY, USA, 2017; pp. 805–817. [Google Scholar]
- Szalai, E.; Jiang, Y.; van Poppelen, N.M.; Jager, M.J.; de Klein, A.; Kilic, E.; Grossniklaus, H.E. Association of uveal melanoma metastatic rate with stochastic mutation rate and type of mutation. JAMA Ophthalmol. 2018, 136, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, A.E.; Vaarwater, J.; Paridaens, D.; Naus, N.C.; Kilic, E.; de Klein, A.; Rotterdam Ocular Melanoma Study, g. Patient survival in uveal melanoma is not affected by oncogenic mutations in gnaq and gna11. Br. J. Cancer 2013, 109, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Koopmans, A.E.; Verdijk, R.M.; Brouwer, R.W.; van den Bosch, T.P.; van den Berg, M.M.; Vaarwater, J.; Kockx, C.E.; Paridaens, D.; Naus, N.C.; Nellist, M.; et al. Clinical significance of immunohistochemistry for detection of bap1 mutations in uveal melanoma. Mod. Pathol. 2014, 27, 1321–1330. [Google Scholar] [CrossRef] [Green Version]
- Smit, K.N.; Chang, J.; Derks, K.; Vaarwater, J.; Brands, T.; Verdijk, R.M.; Wiemer, E.A.C.; Mensink, H.W.; Pothof, J.; de Klein, A.; et al. Aberrant microrna expression and its implications for uveal melanoma metastasis. Cancers 2019, 11, 815. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of ngs alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Gingeras, T.R. Mapping rna-seq reads with star. Curr. Protoc. Bioinform. 2015, 51, 11.14.1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. Gencode reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, S.; Li, W. Rseqc: Quality control of rna-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignatiadis, N.; Klaus, B.; Zaugg, J.B.; Huber, W. Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 2016, 13, 577–580. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.; Ibrahim, J.G.; Love, M.I. Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 2019, 35, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- Sergushichev, A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 2016, 60012, 1–9. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from rna-seq data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- van Ipenburg, J.A.; de Waard, N.E.; Naus, N.C.; Jager, M.J.; Paridaens, D.; Verdijk, R.M. Chemokine receptor expression pattern correlates to progression of conjunctival melanocytic lesions. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2950–2957. [Google Scholar] [CrossRef] [Green Version]
- Shields, C.L.; Furuta, M.; Thangappan, A.; Nagori, S.; Mashayekhi, A.; Lally, D.R.; Kelly, C.C.; Rudich, D.S.; Nagori, A.V.; Wakade, O.A.; et al. Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch. Ophthalmol. 2009, 127, 989–998. [Google Scholar] [CrossRef]
- Inoue, D.; Chew, G.L.; Liu, B.; Michel, B.C.; Pangallo, J.; D’Avino, A.R.; Hitchman, T.; North, K.; Lee, S.C.; Bitner, L.; et al. Spliceosomal disruption of the non-canonical baf complex in cancer. Nature 2019, 574, 432–436. [Google Scholar] [CrossRef]
- Gruner, B.M.; Schulze, C.J.; Yang, D.; Ogasawara, D.; Dix, M.M.; Rogers, Z.N.; Chuang, C.H.; McFarland, C.D.; Chiou, S.H.; Brown, J.M.; et al. An in vivo multiplexed small-molecule screening platform. Nat. Methods 2016, 13, 883–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Xie, H.; Heier, C.; Huang, J.; Zheng, Q.; Eichmann, T.O.; Schoiswohl, G.; Ni, J.; Zechner, R.; Ni, S.; et al. Enhanced monoacylglycerol lipolysis by abhd6 promotes nsclc pathogenesis. EBioMedicine 2020, 53, 102696. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fei, X.; Xu, J.; Ji, C. An unannotated alpha/beta hydrolase superfamily member, abhd6 differentially expressed among cancer cell lines. Mol. Biol Rep. 2009, 36, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Max, D.; Hesse, M.; Volkmer, I.; Staege, M.S. High expression of the evolutionarily conserved alpha/beta hydrolase domain containing 6 (abhd6) in ewing tumors. Cancer Sci 2009, 100, 2383–2389. [Google Scholar] [CrossRef]
- Fagone, P.; Caltabiano, R.; Russo, A.; Lupo, G.; Anfuso, C.D.; Basile, M.S.; Longo, A.; Nicoletti, F.; De Pasquale, R.; Libra, M.; et al. Identification of novel chemotherapeutic strategies for metastatic uveal melanoma. Sci. Rep. 2017, 7, 44564. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | PFS < 60 Months (n = 19) | PFS ≥ 60 Months (n = 52) | Overall (n = 71) | PFS < 60 vs. PFS ≥ 60 Months p-Value |
|---|---|---|---|---|
| Largest tumor diameter (millimeter) | 0.001 | |||
| Mean (SD) | 17.7 (±2.8) | 14.7 (±3.7) | 15.4 (±3.7) | |
| Median (Min, Max) | 18 (13.9–24.0) | 15 (9.0–25.0) | 15 (9.0–25.0) | |
| Data not reported | 2 (10.5%) | 3 (5.8%) | 5 (7.0%) | |
| Metastasis (number) | <0.001 * | |||
| Yes | 19 (100%) | 16 (30.8%) | 35 (49.3%) | |
| No | 0 (0%) | 32 (61.5%) | 32 (45.1%) | |
| Data not reported | 0 (0%) | 4 (7.7%) | 4 (5.6%) | |
| Metastatic location (number) | 0.510 * | |||
| Liver | 11 (57.9%) | 9 (17.3%) | 20 (28.2%) | |
| Liver and other site | 2 (10.5%) | 4 (7.7%) | 6 (8.5%) | |
| Ossal | 1 (5.3%) | 0 (0%) | 1 (1.4%) | |
| Data not reported | 5 (26.3%) | 39 (75.0%) | 44 (62.0%) | |
| Progression free survival (months) | <0.001 | |||
| Mean (SD) | 38.9 (±11.5) | 109.2 (±42.2) | 90.4 (±48.1) | |
| Median (Min, Max) | 40.1 (13.3–56.4) | 97.8 (61.0–215.9) | 82.1 (13.3–215.9) | |
| Patient status (number) | <0.001 * | |||
| Alive | 3 (15.8%) | 28 (53.8%) | 31 (43.7%) | |
| Died due to UM | 14 (73.7%) | 10 (19.2%) | 24 (33.8%) | |
| Died of other cause than UM | 0 (0%) | 5 (9.6%) | 5 (7.0%) | |
| Data not reported | 2 (10.5%) | 9 (17.3%) | 11 (15.5%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drabarek, W.; van Riet, J.; Nguyen, J.Q.N.; Smit, K.N.; van Poppelen, N.M.; Jansen, R.; Medico-Salsench, E.; Vaarwater, J.; Magielsen, F.J.; Brands, T.; et al. Identification of Early-Onset Metastasis in SF3B1 Mutated Uveal Melanoma. Cancers 2022, 14, 846. https://doi.org/10.3390/cancers14030846
Drabarek W, van Riet J, Nguyen JQN, Smit KN, van Poppelen NM, Jansen R, Medico-Salsench E, Vaarwater J, Magielsen FJ, Brands T, et al. Identification of Early-Onset Metastasis in SF3B1 Mutated Uveal Melanoma. Cancers. 2022; 14(3):846. https://doi.org/10.3390/cancers14030846
Chicago/Turabian StyleDrabarek, Wojtek, Job van Riet, Josephine Q. N. Nguyen, Kyra N. Smit, Natasha M. van Poppelen, Rick Jansen, Eva Medico-Salsench, Jolanda Vaarwater, Frank J. Magielsen, Tom Brands, and et al. 2022. "Identification of Early-Onset Metastasis in SF3B1 Mutated Uveal Melanoma" Cancers 14, no. 3: 846. https://doi.org/10.3390/cancers14030846
APA StyleDrabarek, W., van Riet, J., Nguyen, J. Q. N., Smit, K. N., van Poppelen, N. M., Jansen, R., Medico-Salsench, E., Vaarwater, J., Magielsen, F. J., Brands, T., Eussen, B., Bosch, T. P. P. v. d., Verdijk, R. M., Naus, N. C., Paridaens, D., de Klein, A., Brosens, E., van de Werken, H. J. G., Kilic, E., & on behalf of the Rotterdam Ocular Melanoma Study Group. (2022). Identification of Early-Onset Metastasis in SF3B1 Mutated Uveal Melanoma. Cancers, 14(3), 846. https://doi.org/10.3390/cancers14030846