
Molecular Targets of Triple-Negative Breast Cancer: Where Do We Stand?

 , ,
, ,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Chemotherapy
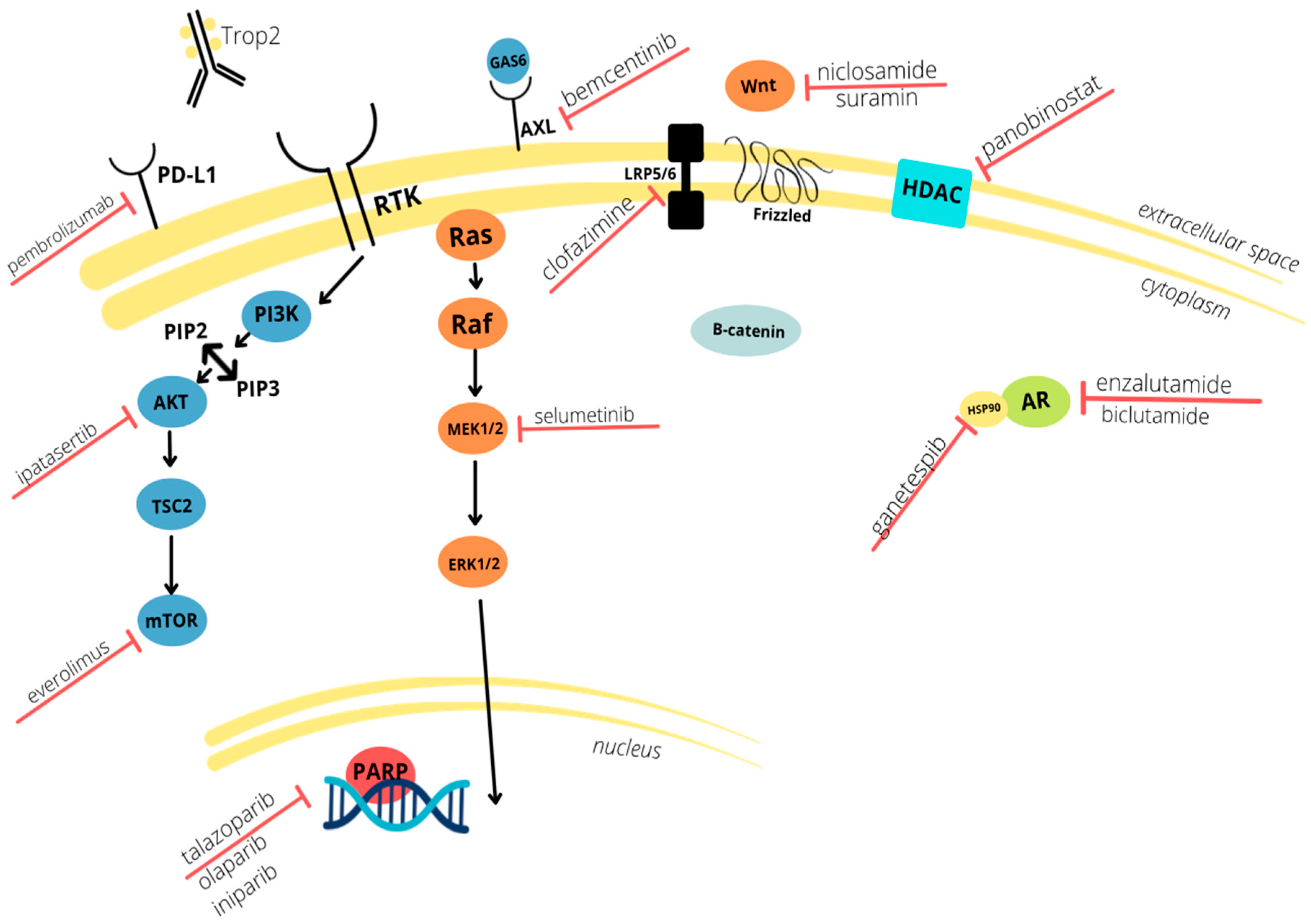
3. Tumor Markers and Potential Therapeutic Targets
3.1. Immune Checkpoint Inhibitors
3.2. Antibody Drug Conjugates
3.3. Receptor Tyrosine Kinase Pathways
3.4. Androgen Receptor and Coordinating Pathways
3.5. Wnt/β-Catenin
3.6. DNA Damage Repair
3.7. EMT and Associated Pathways
3.8. Protein Stability Agents
4. Tumor Markers and Potential Targets
5. Resistance to Treatment
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, Z.H.; Hu, P.H.; Tu, J.H.; Yu, N.S. Luminal B breast cancer: Patterns of recurrence and clinical outcome. Oncotarget 2016, 7, 65024–65033. [Google Scholar] [CrossRef]
- Van Swearingen, A.E.D.; Sambade, M.J.; Siegel, M.B.; Sud, S.; McNeill, R.S.; Bevill, S.M.; Chen, X.; Bash, R.E.; Mounsey, L.; Golitz, B.T.; et al. Combined kinase inhibitors of MEK1/2 and either PI3K or PDGFR are efficacious in intracranial triple-negative breast cancer. Neuro Oncol. 2017, 19, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.R.; Park, T.; Saridakis, A.; Golshan, M.; Greenup, R.A.; Ahuja, N. Immunotherapy Treatment for Triple Negative Breast Cancer. Pharmaceuticals 2021, 14, 763. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Auricchio, F.; Castoria, G.; Migliaccio, A. Androgens Induce Invasiveness of Triple Negative Breast Cancer Cells Through AR/Src/PI3-K Complex Assembly. Sci. Rep. 2019, 9, 4490. [Google Scholar] [CrossRef]
- Neophytou, C.; Boutsikos, P.; Papageorgis, P. Molecular Mechanisms and Emerging Therapeutic Targets of Triple-Negative Breast Cancer Metastasis. Front. Oncol. 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Carey, L.A. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Cancer 2009, 9 (Suppl. 2), S73–S81. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Ryu, W.J.; Sohn, J.H. Molecular Targets and Promising Therapeutics of Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 1008. [Google Scholar] [CrossRef]
- Chen, H.; Wu, J.; Zhang, Z.; Tang, Y.; Li, X.; Liu, S.; Cao, S.; Li, X. Association Between BRCA Status and Triple-Negative Breast Cancer: A Meta-Analysis. Front. Pharmacol. 2018, 9, 909. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Niwinska, A.; Sarnowska, E.; Nowakowska, D.; Jagiello-Gruszfeld, A.; Siedlecki, J.; Nowecki, Z. Effects of BRCA Germline Mutations on Triple-Negative Breast Cancer Prognosis. J. Oncol. 2020, 2020, 8545643. [Google Scholar] [CrossRef]
- Diana, A.; Carlino, F.; Franzese, E.; Oikonomidou, O.; Criscitiello, C.; De Vita, F.; Ciardiello, F.; Orditura, M. Early Triple Negative Breast Cancer: Conventional Treatment and Emerging Therapeutic Landscapes. Cancers 2020, 12, 819. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Azim, H.A.; Ghosn, M.; Oualla, K.; Kassem, L. Personalized treatment in metastatic triple-negative breast cancer: The outlook in 2020. Breast J. 2020, 26, 69–80. [Google Scholar] [CrossRef]
- Pandy, J.G.P.; Balolong-Garcia, J.C.; Cruz-Ordinario, M.V.B.; Que, F.V.F. Triple negative breast cancer and platinum-based systemic treatment: A meta-analysis and systematic review. BMC Cancer 2019, 19, 1065. [Google Scholar] [CrossRef] [PubMed]
- Elghazaly, H.; Rugo, H.S.; Azim, H.A.; Swain, S.M.; Arun, B.; Aapro, M.; Perez, E.A.; Anderson, B.O.; Penault-Llorca, F.; Conte, P.; et al. Breast-Gynaecological & Immuno-Oncology International Cancer Conference (BGICC) Consensus and Recommendations for the Management of Triple-Negative Breast Cancer. Cancers 2021, 13, 2262. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Carey, L.A. ER/PR negative, HER2-negative (triple-negative) breast cancer. In UpToDate; Vora, S.R., Ed.; Wolters Kluwer: Waltham, MA, USA, 2021. [Google Scholar]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Nasser, N.J.; Gorenberg, M.; Agbarya, A. First line Immunotherapy for Non-Small Cell Lung Cancer. Pharmaceuticals 2020, 13, 373. [Google Scholar] [CrossRef]
- Larkin, J.; Hodi, F.S.; Wolchok, J.D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Oualla, K.; Kassem, L.; Nouiakh, L.; Amaadour, L.; Benbrahim, Z.; Arifi, S.; Mellas, N. Immunotherapeutic Approaches in Triple-Negative Breast Cancer: State of the Art and Future Perspectives. Int. J. Breast Cancer 2020, 2020, 8209173. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.E.; Tolaney, S.M. Role of Immunotherapy in Triple-Negative Breast Cancer. J. Natl. Compr. Cancer Netw. 2020, 18, 479–489. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef]
- Miyashita, M.; Sasano, H.; Tamaki, K.; Chan, M.; Hirakawa, H.; Suzuki, A.; Tada, H.; Watanabe, G.; Nemoto, N.; Nakagawa, S.; et al. Tumor-infiltrating CD8+ and FOXP3+ lymphocytes in triple-negative breast cancer: Its correlation with pathological complete response to neoadjuvant chemotherapy. Breast Cancer Res. Treat. 2014, 148, 525–534. [Google Scholar] [CrossRef]
- Baker, K.; Lachapelle, J.; Zlobec, I.; Bismar, T.A.; Terracciano, L.; Foulkes, W.D. Prognostic significance of CD8+ T lymphocytes in breast cancer depends upon both oestrogen receptor status and histological grade. Histopathology 2011, 58, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Nanda, R.; Liu, M.C.; Yau, C.; Shatsky, R.; Pusztai, L.; Wallace, A.; Chien, A.J.; Forero-Torres, A.; Ellis, E.; Han, H.; et al. Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer: An Analysis of the Ongoing Phase 2 Adaptively Randomized I-SPY2 Trial. JAMA Oncol. 2020, 6, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Kulangara, K.; Zhang, N.; Corigliano, E.; Guerrero, L.; Waldroup, S.; Jaiswal, D.; Ms, M.J.; Shah, S.; Hanks, D.; Wang, J.; et al. Clinical Utility of the Combined Positive Score for Programmed Death Ligand-1 Expression and the Approval of Pembrolizumab for Treatment of Gastric Cancer. Arch. Pathol. Lab. Med. 2019, 143, 330–337. [Google Scholar] [CrossRef]
- Winer, E.P.; Lipatov, O.; Im, S.A.; Goncalves, A.; Munoz-Couselo, E.; Lee, K.S.; Schmid, P.; Tamura, K.; Testa, L.; Witzel, I.; et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): A randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 499–511. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Emens, L.A.; Cruz, C.; Eder, J.P.; Braiteh, F.; Chung, C.; Tolaney, S.M.; Kuter, I.; Nanda, R.; Cassier, P.A.; Delord, J.P.; et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol. 2019, 5, 74–82. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): A randomised, double-blind, phase 3 trial. Lancet 2020, 396, 1090–1100. [Google Scholar] [CrossRef]
- Suppan, C.; Balic, M. Treatment options in early triple-negative breast cancer. Memo 2020, 13, 346–348. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M.; Cardillo, T.M.; Govindan, S.V.; Rossi, E.A.; Sharkey, R.M. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget 2015, 6, 22496–22512. [Google Scholar] [CrossRef] [PubMed]
- Cubas, R.; Zhang, S.; Li, M.; Chen, C.; Yao, Q. Trop2 expression contributes to tumor pathogenesis by activating the ERK MAPK pathway. Mol. Cancer 2010, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Vallone, V.; Leprovots, M.; Strollo, S.; Vasile, G.; Lefort, A.; Libert, F.; Vassart, G.; Garcia, M.I. Trop2 marks transient gastric fetal epithelium and adult regenerating cells after epithelial damage. Development 2016, 143, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Diamond, J.R.; Moroose, R.L.; Isakoff, S.J.; Starodub, A.N.; Shah, N.C.; O’Shaughnessy, J.; Kalinsky, K.; Guarino, M.; et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients With Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2017, 35, 2141–2148. [Google Scholar] [CrossRef]
- Modi, S.; Park, H.; Murthy, R.K.; Iwata, H.; Tamura, K.; Tsurutani, J.; Moreno-Aspitia, A.; Doi, T.; Sagara, Y.; Redfern, C.; et al. Antitumor Activity and Safety of Trastuzumab Deruxtecan in Patients With HER2-Low-Expressing Advanced Breast Cancer: Results From a Phase Ib Study. J. Clin. Oncol. 2020, 38, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lin, S.; Tseng, K.F.; Han, K.; Wang, Y.; Gan, Z.H.; Min, D.L.; Hu, H.Y. Selumetinib suppresses cell proliferation, migration and trigger apoptosis, G1 arrest in triple-negative breast cancer cells. BMC Cancer 2016, 16, 818. [Google Scholar] [CrossRef]
- Bartholomeusz, C.; Gonzalez-Angulo, A.M.; Liu, P.; Hayashi, N.; Lluch, A.; Ferrer-Lozano, J.; Hortobagyi, G.N. High ERK protein expression levels correlate with shorter survival in triple-negative breast cancer patients. Oncologist 2012, 17, 766–774. [Google Scholar] [CrossRef]
- Kim, S.B.; Dent, R.; Im, S.A.; Espie, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wongchenko, M.J.; et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017, 18, 1360–1372. [Google Scholar] [CrossRef]
- Ibrahim, Y.H.; Garcia-Garcia, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzman, M.; Grueso, J.; et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef]
- Leconet, W.; Chentouf, M.; du Manoir, S.; Chevalier, C.; Sirvent, A.; Ait-Arsa, I.; Busson, M.; Jarlier, M.; Radosevic-Robin, N.; Theillet, C.; et al. Therapeutic Activi.ity of Anti-AXL Antibody against Triple-Negative Breast Cancer Patient-Derived Xenografts and Metastasis. Clin. Cancer Res. 2017, 23, 2806–2816. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Zajac, O.; Leclere, R.; Nicolas, A.; Meseure, D.; Marchio, C.; Vincent-Salomon, A.; Roman-Roman, S.; Schoumacher, M.; Dubois, T. AXL Controls Directed Migration of Mesenchymal Triple-Negative Breast Cancer Cells. Cells 2020, 9, 247. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, Z.; Miao, L.; Yang, Z.; Yang, J.; Wang, Y.; Qian, D.; Cai, H.; Wang, Y. Anexelekto (AXL) Increases Resistance to EGFR-TKI and Activation of AKT and ERK1/2 in Non-Small Cell Lung Cancer Cells. Oncol. Res. 2016, 24, 295–303. [Google Scholar] [CrossRef]
- Gomes, A.M.; Carron, E.C.; Mills, K.L.; Dow, A.M.; Gray, Z.; Fecca, C.R.; Lakey, M.A.; Carmeliet, P.; Kittrell, F.; Medina, D.; et al. Stromal Gas6 promotes the progression of premalignant mammary cells. Oncogene 2019, 38, 2437–2450. [Google Scholar] [CrossRef]
- Ibrahim, A.M.; Gray, Z.; Gomes, A.M.; Myers, L.; Behbod, F.; Machado, H.L. Gas6 expression is reduced in advanced breast cancers. NPJ Precis Oncol. 2020, 4, 9. [Google Scholar] [CrossRef]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 153. [Google Scholar] [CrossRef]
- Yule, M.; Davidsen, K.; Bloe, M.; Hodneland, L.; Engelsen, A.; Lie, M.; Bougnaud, S.; D’Mello, S.; Aguilera, K.; Ahmed, L.; et al. Combination of bemcentinib (BGB324): A first-in-class selective oral AXL inhibitor, with pembrolizumab in patients with triple negative breast cancer and adenocarcinoma of the lung. J. Clin. Oncol. 2018, 36, TPS43. [Google Scholar] [CrossRef]
- Wu, N.; Zhang, J.; Zhao, J.; Mu, K.; Zhang, J.; Jin, Z.; Yu, J.; Liu, J. Precision medicine based on tumorigenic signaling pathways for triple-negative breast cancer. Oncol. Lett. 2018, 16, 4984–4996. [Google Scholar] [CrossRef]
- Kim, H.R.; Jung, K.H.; Im, S.A.; Im, Y.H.; Kang, S.Y.; Park, K.H.; Lee, S.; Kim, S.B.; Lee, K.H.; Ahn, J.S.; et al. Multicentre phase II trial of bevacizumab combined with docetaxel-carboplatin for the neoadjuvant treatment of triple-negative breast cancer (KCSG BR-0905). Ann. Oncol. 2013, 24, 1485–1490. [Google Scholar] [CrossRef]
- Li, Y.; Yang, D.; Chen, P.; Yin, X.; Sun, J.; Li, H.; Ren, G. Efficacy and safety of neoadjuvant chemotherapy regimens for triple-negative breast cancer: A network meta-analysis. Aging 2019, 11, 6286–6311. [Google Scholar] [CrossRef] [PubMed]
- El Guerrab, A.; Bamdad, M.; Bignon, Y.J.; Penault-Llorca, F.; Aubel, C. Co-targeting EGFR and mTOR with gefitinib and everolimus in triple-negative breast cancer cells. Sci. Rep. 2020, 10, 6367. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.; Gradishar, W.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the Treatment of Androgen Receptor-Expressing Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Abramson, V.G.; Sanders, M.E.; Mayer, E.L.; Haddad, T.C.; Nanda, R.; Van Poznak, C.; Storniolo, A.M.; Nangia, J.R.; Gonzalez-Ericsson, P.I.; et al. TBCRC 032 IB/II Multicenter Study: Molecular Insights to AR Antagonist and PI3K Inhibitor Efficacy in Patients with AR(+) Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2020, 26, 2111–2123. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef]
- Dey, N.; Young, B.; Abramovitz, M.; Bouzyk, M.; Barwick, B.; De, P.; Leyland-Jones, B. Differential activation of Wnt-beta-catenin pathway in triple negative breast cancer increases MMP7 in a PTEN dependent manner. PLoS ONE 2013, 8, e77425. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, X.; Ward, T.; Pegram, M.; Shen, K. Combined niclosamide with cisplatin inhibits epithelial-mesenchymal transition and tumor growth in cisplatin-resistant triple-negative breast cancer. Tumour. Biol. 2016, 37, 9825–9835. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Koval, A.; Xu, J.; Bodmer, A.; Katanaev, V.L. Towards the first targeted therapy for triple-negative breast cancer: Repositioning of clofazimine as a chemotherapy-compatible selective Wnt pathway inhibitor. Cancer Lett. 2019, 449, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Alexey Koval, K.A.; Vladimir, K. Inhibition of Wnt signalling and breast tumour growth by the multi-purpose drug suramin through suppression of heterotrimeric G proteins and Wnt endocytosis. Biochem. J. 2016, 473, 371–381. [Google Scholar] [CrossRef]
- Litton, J.K.; Scoggins, M.E.; Hess, K.R.; Adrada, B.E.; Murthy, R.K.; Damodaran, S.; DeSnyder, S.M.; Brewster, A.M.; Barcenas, C.H.; Valero, V.; et al. Neoadjuvant Talazoparib for Patients With Operable Breast Cancer With a Germline BRCA Pathogenic Variant. J. Clin. Oncol. 2020, 38, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Hurvitz, S.A.; Mina, L.A.; Rugo, H.S.; Lee, K.H.; Goncalves, A.; Diab, S.; Woodward, N.; Goodwin, A.; Yerushalmi, R.; et al. Talazoparib versus chemotherapy in patients with germline BRCA1/2-mutated HER2-negative advanced breast cancer: Final overall survival results from the EMBRACA trial. Ann. Oncol. 2020, 31, 1526–1535. [Google Scholar] [CrossRef]
- Bergin, A.R.T.; Loi, S. Triple-negative breast cancer: Recent treatment advances. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Thomas, R.; Al-Khadairi, G.; Decock, J. Immune Checkpoint Inhibitors in Triple Negative Breast Cancer Treatment: Promising Future Prospects. Front. Oncol. 2020, 10, 600573. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed]
- Livraghi, L.; Garber, J.E. PARP inhibitors in the management of breast cancer: Current data and future prospects. BMC Med. 2015, 13, 188. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- McCann, K.E.; Hurvitz, S.A. Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context 2018, 7, 212540. [Google Scholar] [CrossRef]
- Daemen, A.; Wolf, D.M.; Korkola, J.E.; Griffith, O.L.; Frankum, J.R.; Brough, R.; Jakkula, L.R.; Wang, N.J.; Natrajan, R.; Reis-Filho, J.S.; et al. Cross-platform pathway-based analysis identifies markers of response to the PARP inhibitor olaparib. Breast Cancer Res. Treat. 2012, 135, 505–517. [Google Scholar] [CrossRef]
- Hassan, S.; Esch, A.; Liby, T.; Gray, J.W.; Heiser, L.M. Pathway-Enriched Gene Signature Associated with 53BP1 Response to PARP Inhibition in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2017, 16, 2892–2901. [Google Scholar] [CrossRef]
- Bruna, A.; Rueda, O.M.; Greenwood, W.; Batra, A.S.; Callari, M.; Batra, R.N.; Pogrebniak, K.; Sandoval, J.; Cassidy, J.W.; Tufegdzic-Vidakovic, A.; et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell 2016, 167, 260–274.e22. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.W.; Yuca, E.; Akcakanat, A.; Scott, S.M.; Arango, N.P.; Zheng, X.; Chen, K.; Tapia, C.; Tarco, E.; Eterovic, A.K.; et al. A Population of Heterogeneous Breast Cancer Patient-Derived Xenografts Demonstrate Broad Activity of PARP Inhibitor in BRCA1/2 Wild-Type Tumors. Clin. Cancer Res. 2017, 23, 6468–6477. [Google Scholar] [CrossRef] [PubMed]
- Layman, R.M.; Arun, B. PARP Inhibitors in Triple-Negative Breast Cancer Including Those With BRCA Mutations. Cancer J. 2021, 27, 67–75. [Google Scholar] [CrossRef]
- Rhodes, L.V.; Martin, E.C.; Segar, H.C.; Miller, D.F.; Buechlein, A.; Rusch, D.B.; Nephew, K.P.; Burow, M.E.; Collins-Burow, B.M. Dual regulation by microRNA-200b-3p and microRNA-200b-5p in the inhibition of epithelial-to-mesenchymal transition in triple-negative breast cancer. Oncotarget 2015, 6, 16638–16652. [Google Scholar] [CrossRef]
- Tate, C.R.; Rhodes, L.V.; Segar, H.C.; Driver, J.L.; Pounder, F.N.; Burow, M.E.; Collins-Burow, B.M. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res. 2012, 14, R79. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, L.V.; Tate, C.R.; Segar, H.C.; Burks, H.E.; Phamduy, T.B.; Hoang, V.; Elliott, S.; Gilliam, D.; Pounder, F.N.; Anbalagan, M.; et al. Suppression of triple-negative breast cancer metastasis by pan-DAC inhibitor panobinostat via inhibition of ZEB family of EMT master regulators. Breast Cancer Res. Treat. 2014, 145, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Mumin, N.H.; Drobnitzky, N.; Patel, A.; Lourenco, L.M.; Cahill, F.F.; Jiang, Y.; Kong, A.; Ryan, A.J. Overcoming acquired resistance to HSP90 inhibition by targeting JAK-STAT signalling in triple-negative breast cancer. BMC Cancer 2019, 19, 102. [Google Scholar] [CrossRef] [PubMed]
- Anant, S.; Ramamoorthy, P.; Tawfik, O.; Jensen, R.A. Effects of Hsp90 Inhibitors on Patient Derived Triple Negative Breast Cancer (TNBC) Cells: BRCA1 as a Therapeutic Target for TNBC. J. Am. Coll. Surg. 2017, 225, e6. [Google Scholar] [CrossRef][Green Version]
- Friedland, J.C.; Smith, D.L.; Sang, J.; Acquaviva, J.; He, S.; Zhang, C.; Proia, D.A. Targeted inhibition of Hsp90 by ganetespib is effective across a broad spectrum of breast cancer subtypes. Investig. New Drugs 2014, 32, 14–24. [Google Scholar] [CrossRef]
- Jhaveri, K.; Chandarlapaty, S.; Lake, D.; Gilewski, T.; Robson, M.; Goldfarb, S.; Drullinsky, P.; Sugarman, S.; Wasserheit-Leiblich, C.; Fasano, J.; et al. A phase II open-label study of ganetespib, a novel heat shock protein 90 inhibitor for patients with metastatic breast cancer. Clin. Breast Cancer 2014, 14, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D’Andrea, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. HSP90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef]
- Yuan, Z.Y.; Luo, R.Z.; Peng, R.J.; Wang, S.S.; Xue, C. High infiltration of tumor-associated macrophages in triple-negative breast cancer is associated with a higher risk of distant metastasis. Onco Targets Ther. 2014, 7, 1475–1480. [Google Scholar] [CrossRef]
- Qiu, S.Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schroder, C.P. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Ibrahim, A.M.; Moss, M.A.; Gray, Z.; Rojo, M.D.; Burke, C.M.; Schwertfeger, K.L.; Dos Santos, C.O.; Machado, H.L. Diverse Macrophage Populations Contribute to the Inflammatory Microenvironment in Premalignant Lesions During Localized Invasion. Front. Oncol. 2020, 10, 569985. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chaffee, T.S.; LaRue, R.S.; Huggins, D.N.; Witschen, P.M.; Ibrahim, A.M.; Nelson, A.C.; Machado, H.L.; Schwertfeger, K.L. Tissue-resident macrophages promote extracellular matrix homeostasis in the mammary gland stroma of nulliparous mice. Elife 2020, 9, e57438. [Google Scholar] [CrossRef]
- Kim, I.S.; Gao, Y.; Welte, T.; Wang, H.; Liu, J.; Janghorban, M.; Sheng, K.; Niu, Y.; Goldstein, A.; Zhao, N.; et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol. 2019, 21, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Kruger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, A.C.; Zea, A.H.; Hernandez, C.; Rodriguez, P.C. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin. Cancer Res. 2007, 13, 721s–726s. [Google Scholar] [CrossRef]
- Ma, X.; Wang, M.; Yin, T.; Zhao, Y.; Wei, X. Myeloid-Derived Suppressor Cells Promote Metastasis in Breast Cancer After the Stress of Operative Removal of the Primary Cancer. Front. Oncol. 2019, 9, 855. [Google Scholar] [CrossRef]
- Bae, S.J.; Cha, Y.J.; Yoon, C.; Kim, D.; Lee, J.; Park, S.; Cha, C.; Kim, J.Y.; Ahn, S.G.; Park, H.S.; et al. Prognostic value of neutrophil-to-lymphocyte ratio in human epidermal growth factor receptor 2-negative breast cancer patients who received neoadjuvant chemotherapy. Sci. Rep. 2020, 10, 13078. [Google Scholar] [CrossRef]
- Pistelli, M.; De Lisa, M.; Ballatore, Z.; Caramanti, M.; Pagliacci, A.; Battelli, N.; Ridolfi, F.; Santoni, M.; Maccaroni, E.; Bracci, R.; et al. Pre-treatment neutrophil to lymphocyte ratio may be a useful tool in predicting survival in early triple negative breast cancer patients. BMC Cancer 2015, 15, 195. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.A.; Xi, J.; Luo, J.; Hassan, B.; Thomas, S.; Ma, C.X.; Campian, J.L. Neutrophil-to-lymphocyte ratio as a predictor of survival in patients with triple-negative breast cancer. Breast Cancer Res. Treat. 2019, 174, 443–452. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guerin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef]
- Guestini, F.; Ono, K.; Miyashita, M.; Ishida, T.; Ohuchi, N.; Nakagawa, S.; Hirakawa, H.; Tamaki, K.; Ohi, Y.; Rai, Y.; et al. Impact of Topoisomerase IIalpha, PTEN, ABCC1/MRP1, and KI67 on triple-negative breast cancer patients treated with neoadjuvant chemotherapy. Breast Cancer Res. Treat. 2019, 173, 275–288. [Google Scholar] [CrossRef]
- Britton, K.M.; Eyre, R.; Harvey, I.J.; Stemke-Hale, K.; Browell, D.; Lennard, T.W.J.; Meeson, A.P. Breast cancer, side population cells and ABCG2 expression. Cancer Lett. 2012, 323, 97–105. [Google Scholar] [CrossRef]
- Lee, H.E.; Kim, J.H.; Kim, Y.J.; Choi, S.Y.; Kim, S.W.; Kang, E.; Chung, I.Y.; Kim, I.A.; Kim, E.J.; Choi, Y.; et al. An increase in cancer stem cell population after primary systemic therapy is a poor prognostic factor in breast cancer. Br. J. Cancer 2011, 104, 1730–1738. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef]
- Merikhian, P.; Eisavand, M.R.; Farahmand, L. Triple-negative breast cancer: Understanding Wnt signaling in drug resistance. Cancer Cell Int. 2021, 21, 419. [Google Scholar] [CrossRef]
- Martin-Orozco, E.; Sanchez-Fernandez, A.; Ortiz-Parra, I.; Ayala-San Nicolas, M. WNT Signaling in Tumors: The Way to Evade Drugs and Immunity. Front. Immunol. 2019, 10, 2854. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Liu, M.; Kuo, F.; Capistrano, K.J.; Kang, D.; Nixon, B.G.; Shi, W.; Chou, C.; Do, M.H.; Stamatiades, E.G.; Gao, S.; et al. TGF-beta suppresses type 2 immunity to cancer. Nature 2020, 587, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Yu, X.; Li, S.; Tian, Y.; Liu, C. New Perspectives for Resistance to PARP Inhibitors in Triple-Negative Breast Cancer. Front. Oncol. 2020, 10, 578095. [Google Scholar] [CrossRef] [PubMed]
- Sengodan, S.K.; Hemalatha, S.K.; Nadhan, R.; Somanathan, T.; Mathew, A.P.; Chil, A.; Kopczynski, J.; Nair, R.S.; Kumar, J.M.; Srinivas, P. beta-hCG-induced mutant BRCA1 ignites drug resistance in susceptible breast tissue. Carcinogenesis 2019, 40, 1415–1426. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug Type | Drug | Total Trials | Completed | Active |
|---|---|---|---|---|
| Immune checkpoint inhibitors | pembrolizumab | 90 | 11 | 79 |
| atezolizumab | 55 | 3 | 52 | |
| Antibody-drug conjugates | sacitizumab-govitecan | 7 | 1 | 6 |
| trastuzumab-deruxtecam | 2 | 0 | 2 | |
| RTK pathway inhibitors | selumetinib | 2 | 0 | 2 |
| ipatasertib | 9 | 2 | 7 | |
| bemcentinib | 1 | 1 | 0 | |
| bevacizumab | 26 | 13 | 13 | |
| EMT pathway inhibitors | panobinostat | 3 | 1 | 2 |
| Androgen receptor inhibitors | bicalutamide | 4 | 0 | 4 |
| enzalutamide | 8 | 1 | 7 | |
| taselisib and enzalutamide | 1 | 1 | 0 | |
| DNA damage repair inhibitors | iniparib | 7 | 6 | 1 |
| olaparib | 26 | 7 | 19 | |
| talazoparib | 13 | 0 | 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newton, E.E.; Mueller, L.E.; Treadwell, S.M.; Morris, C.A.; Machado, H.L. Molecular Targets of Triple-Negative Breast Cancer: Where Do We Stand? Cancers 2022, 14, 482. https://doi.org/10.3390/cancers14030482
Newton EE, Mueller LE, Treadwell SM, Morris CA, Machado HL. Molecular Targets of Triple-Negative Breast Cancer: Where Do We Stand? Cancers. 2022; 14(3):482. https://doi.org/10.3390/cancers14030482
Chicago/Turabian StyleNewton, Emma E., Lauren E. Mueller, Scout M. Treadwell, Cindy A. Morris, and Heather L. Machado. 2022. "Molecular Targets of Triple-Negative Breast Cancer: Where Do We Stand?" Cancers 14, no. 3: 482. https://doi.org/10.3390/cancers14030482
APA StyleNewton, E. E., Mueller, L. E., Treadwell, S. M., Morris, C. A., & Machado, H. L. (2022). Molecular Targets of Triple-Negative Breast Cancer: Where Do We Stand? Cancers, 14(3), 482. https://doi.org/10.3390/cancers14030482