
HIF-Dependent CKB Expression Promotes Breast Cancer Metastasis, Whereas Cyclocreatine Therapy Impairs Cellular Invasion and Improves Chemotherapy Efficacy

 , , ,
, , , 
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. PyMT Mammary Tumor Epithelial Cells (MTECs)
2.3. RNA Extraction and Gene Expression Microarray Analysis
2.4. Real-Time Quantitative PCR (qPCR)
2.5. Immunofluorescence of Cultured Cells
2.6. Protein Extraction and Western Blotting
2.7. Creatine Kinase Enzymatic Activity Assay
2.8. Patient Database Mining
2.9. Promoter Analysis and Chromatin Immunoprecipitation (ChIP) Assays
2.10. Generation of PyMT Ckb Knockdown Cells
2.11. PyMT Cell Proliferation Assay by WST-1
2.12. Wound Healing Assays
2.13. Human Breast Cancer Cell Lines, Mycoplasma Testing and Cell Line Authentication
2.14. Transient Ectopic Expression of CKB in MDA-MB-231 Cells
2.15. Invasion Assays
2.16. Intracellular ATP Levels
2.17. Seahorse Bioanalyzer Assays
2.18. Chemotaxis Assays
2.19. Cell Cycle Analysis
2.20. PyMT Primary Tumor Generation
2.21. Tissue Immunohistochemistry and Quantification
2.22. Tail Vein Assays in PyMT Mice
2.23. Invadopodia Assays Using Human TNBC Cells
2.24. Growth Inhibition during Chemotherapy Treatment
2.25. Statistical Analysis
3. Results
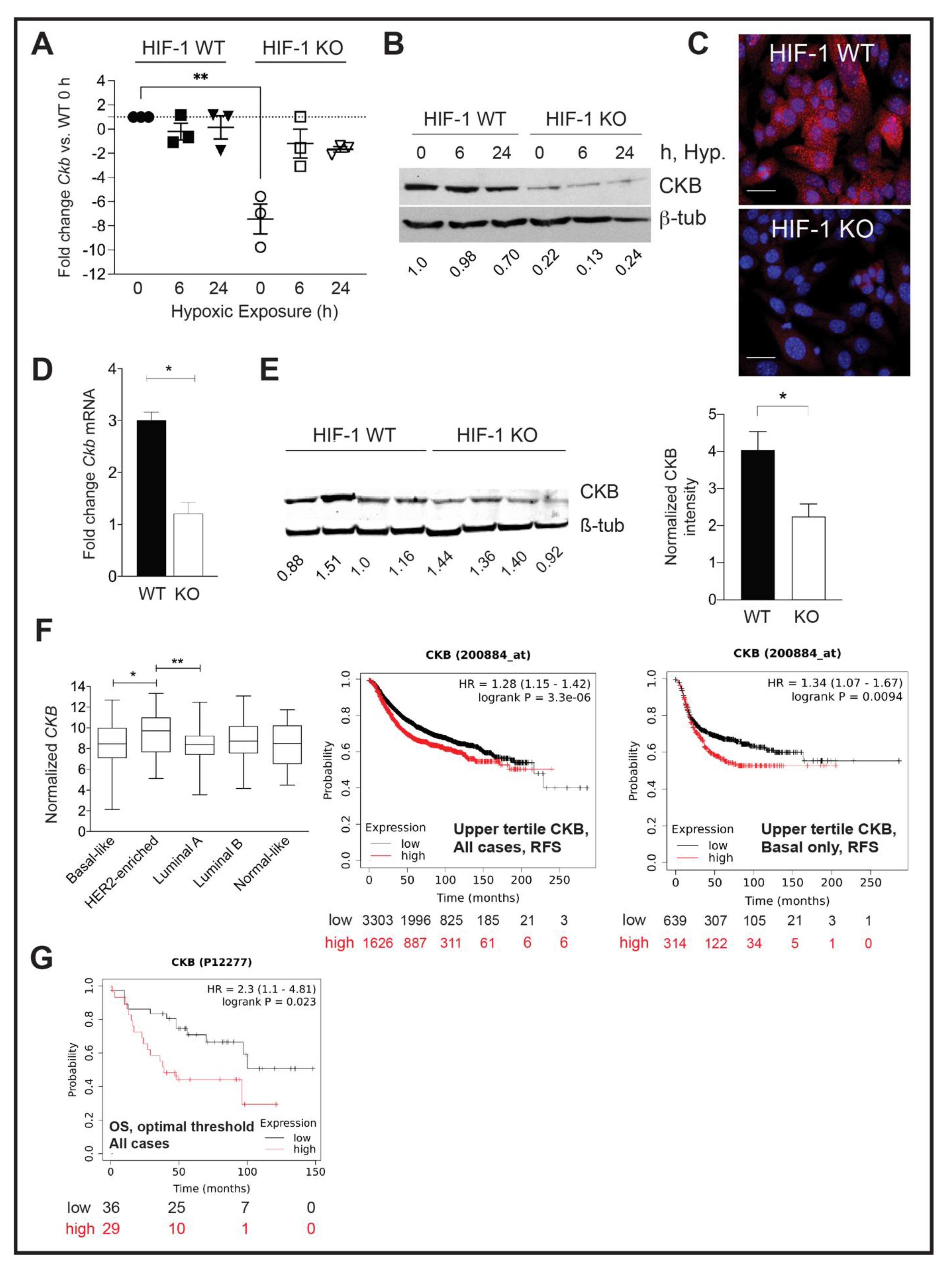
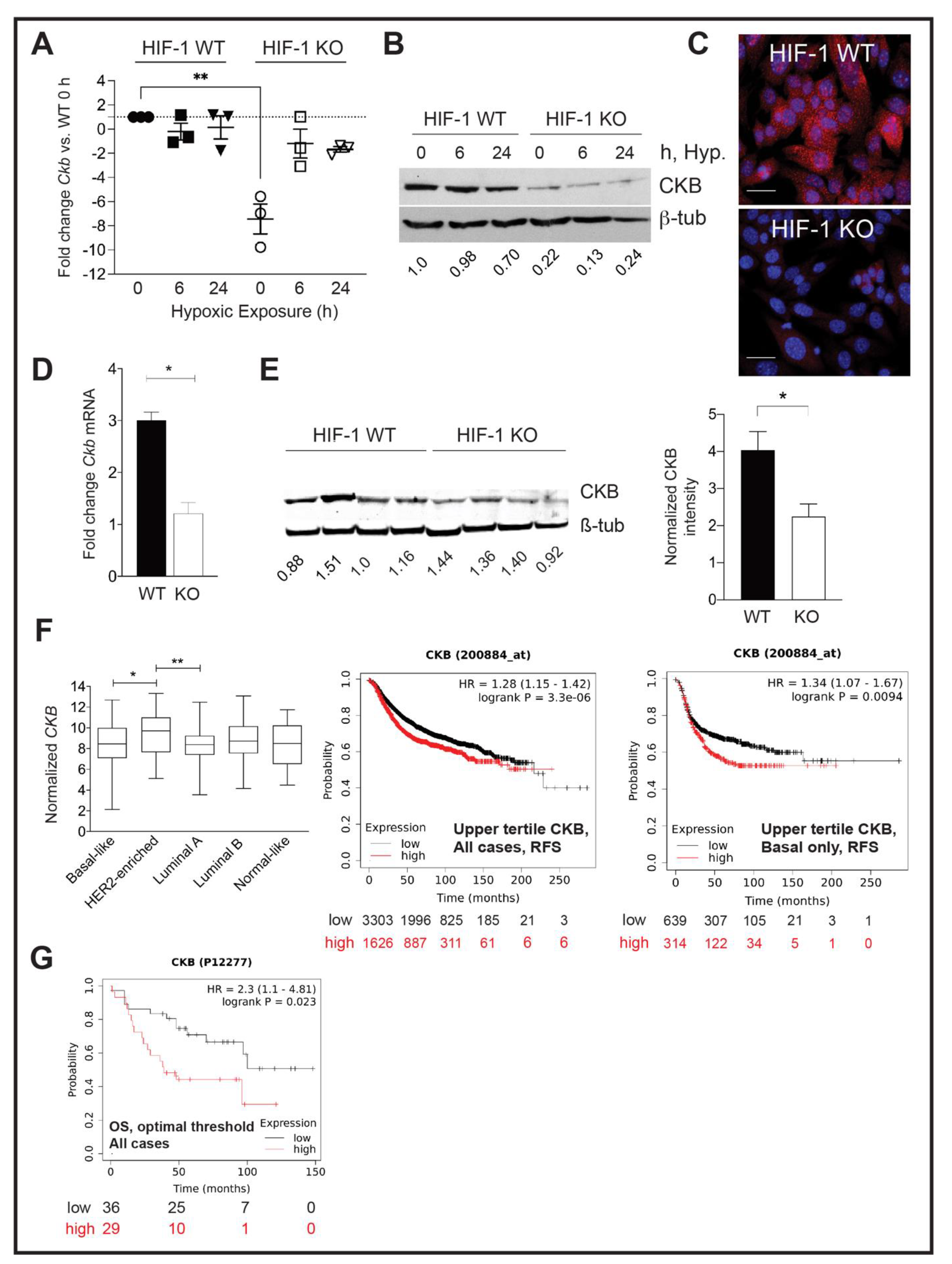
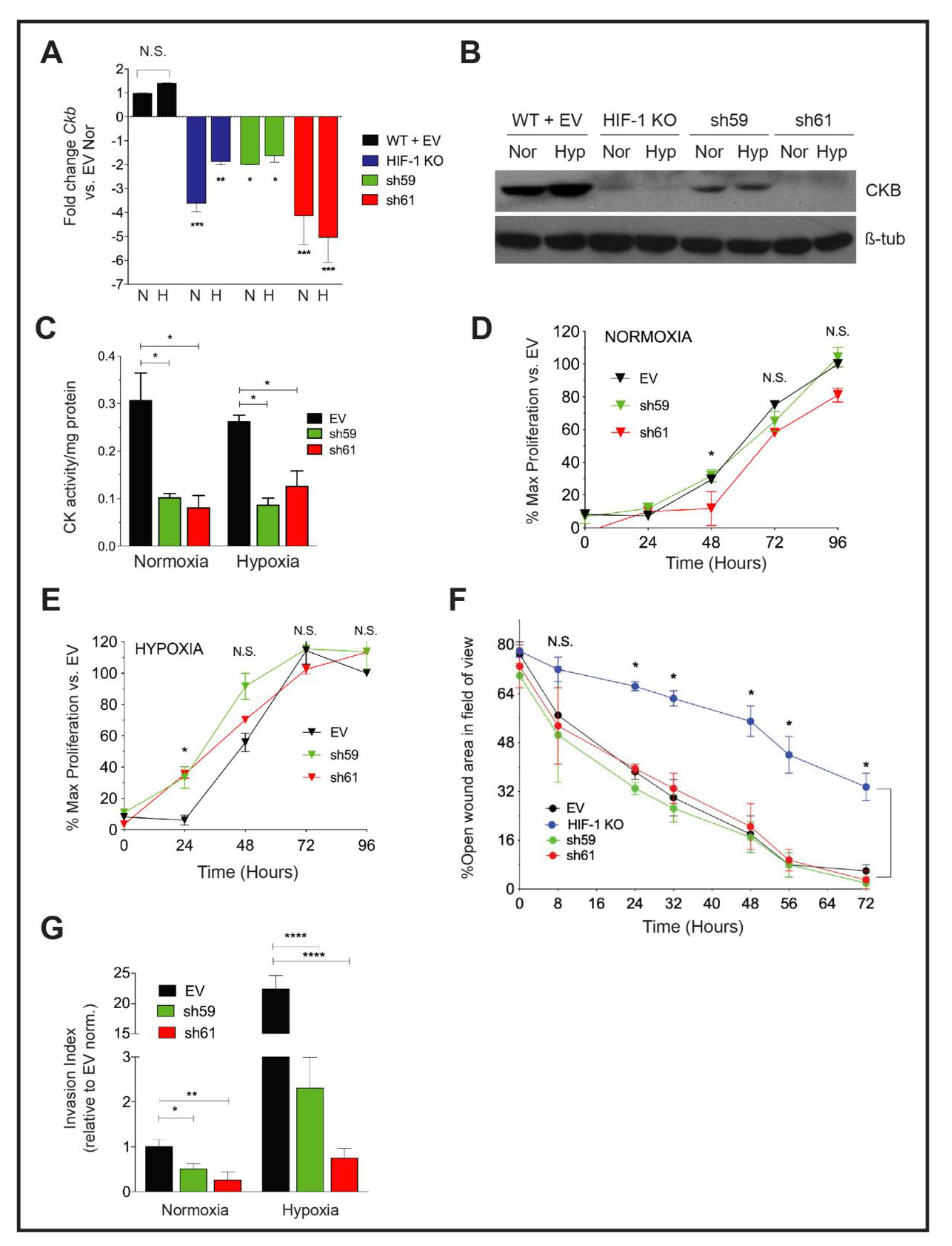
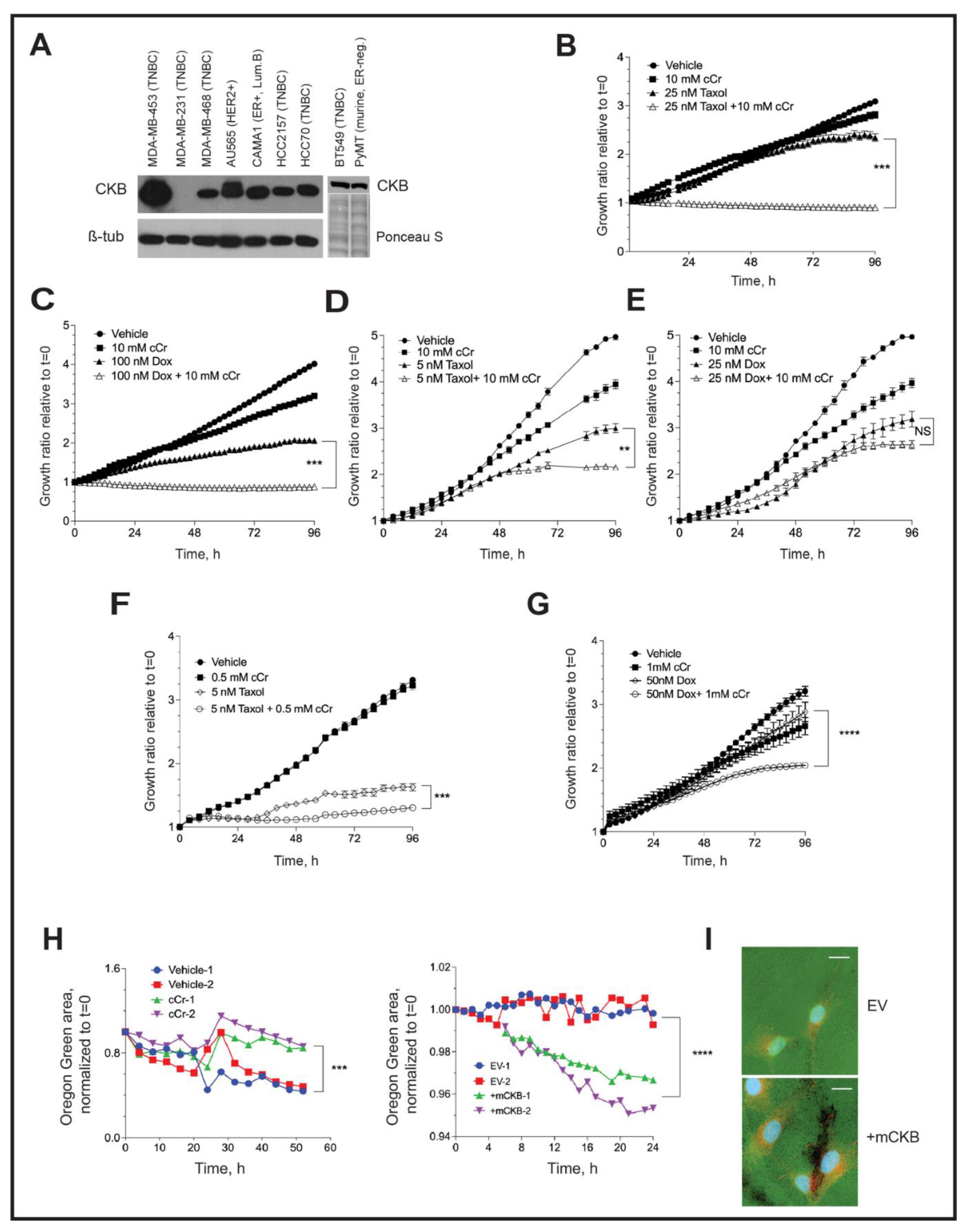
3.1. Ckb Is a HIF-1α Dependent Target in Breast Cancer
3.2. CKB Levels Are Correlated with Shortened Regression-Free Survival
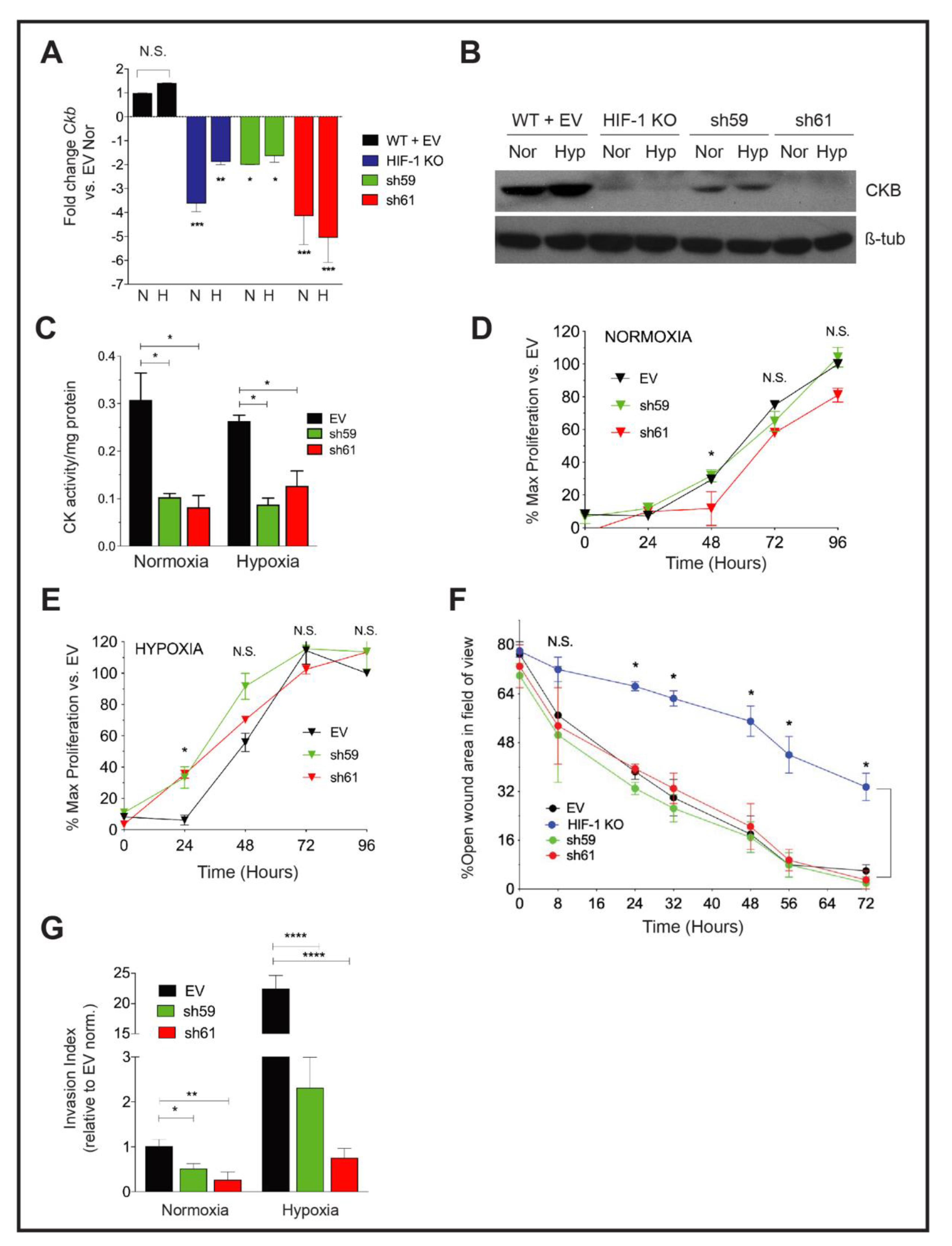
3.3. Knockdown of Ckb Impairs Cell Invasion
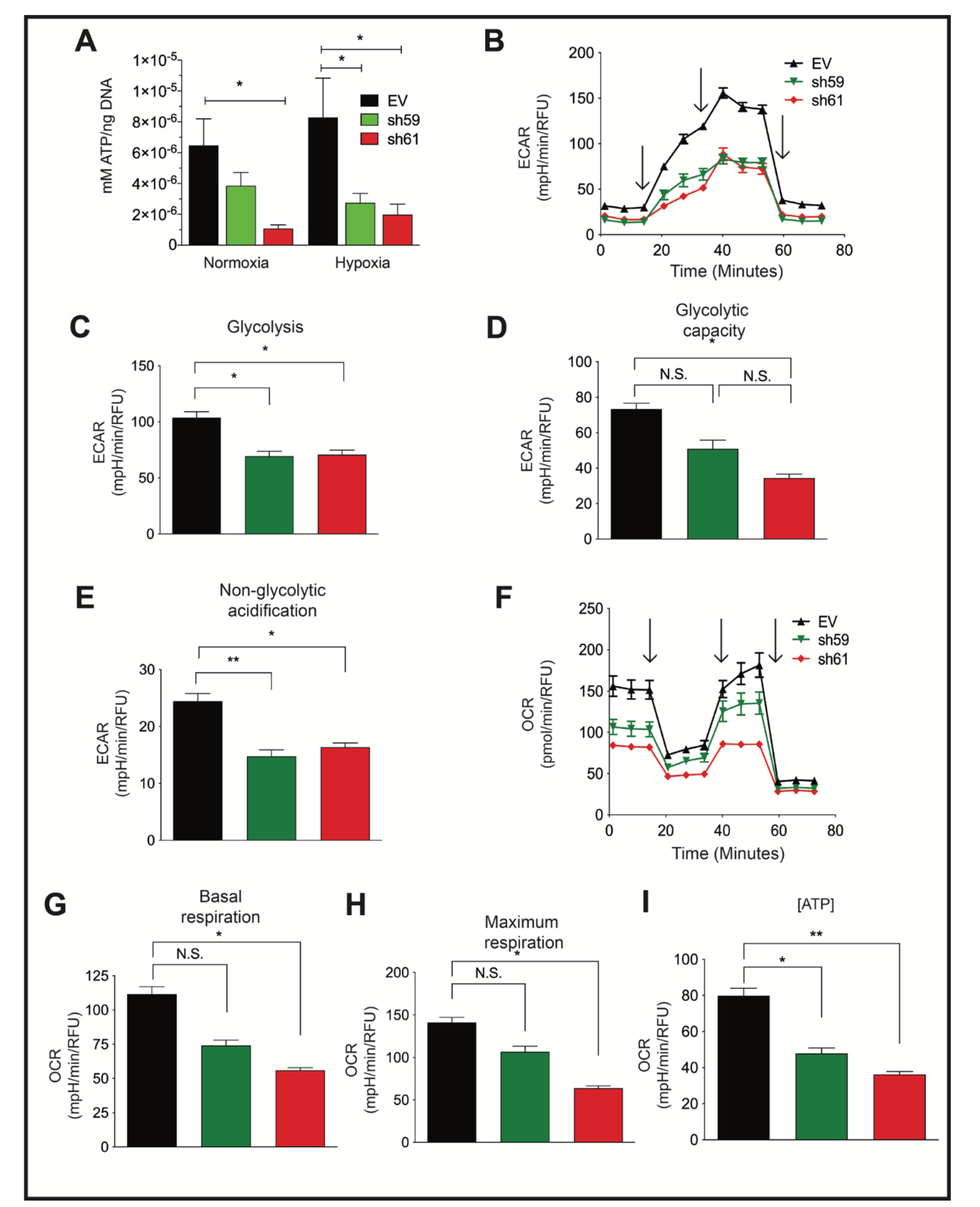
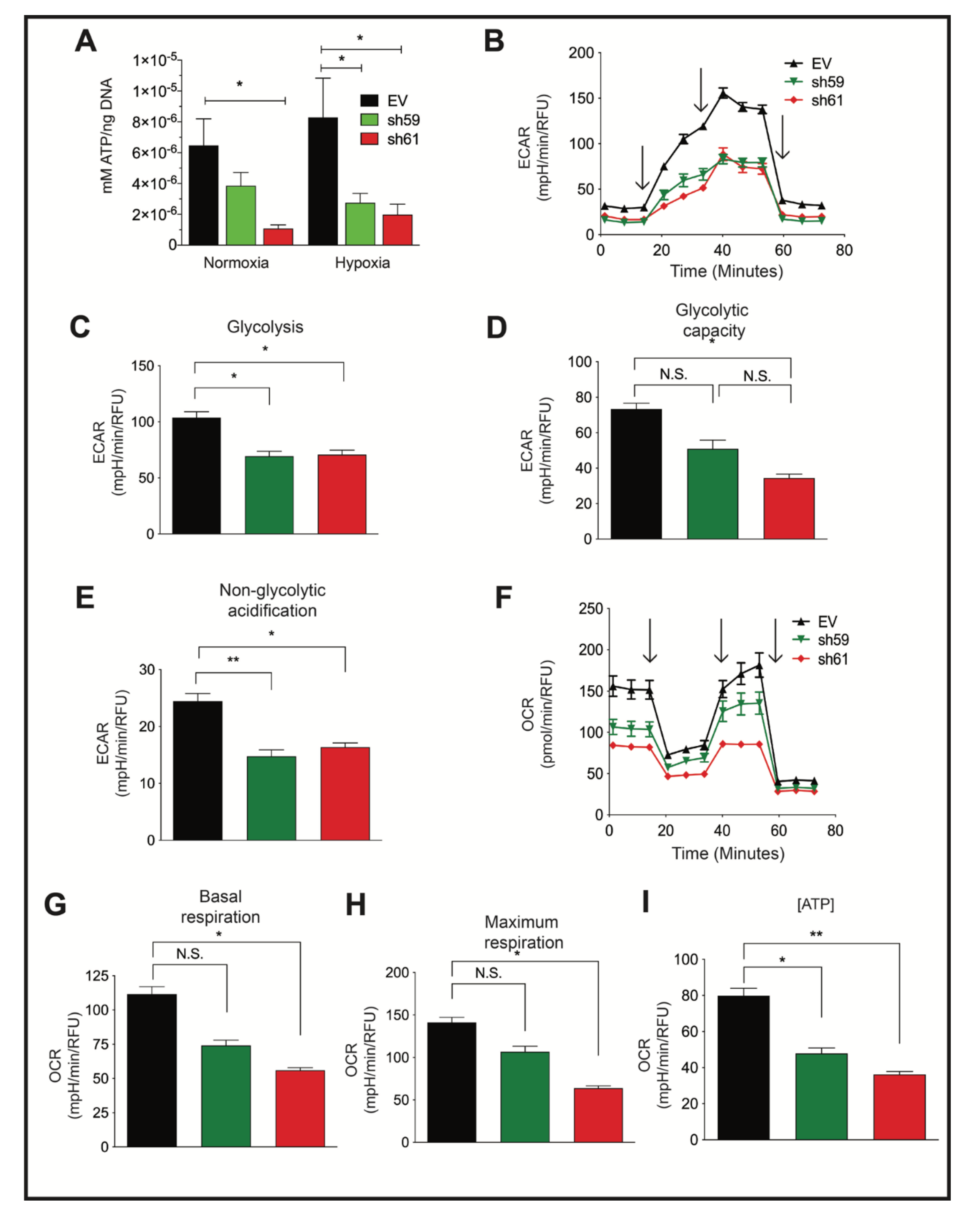
3.4. Loss of CKB Represses Glycolysis and Oxidative Phosphorylation
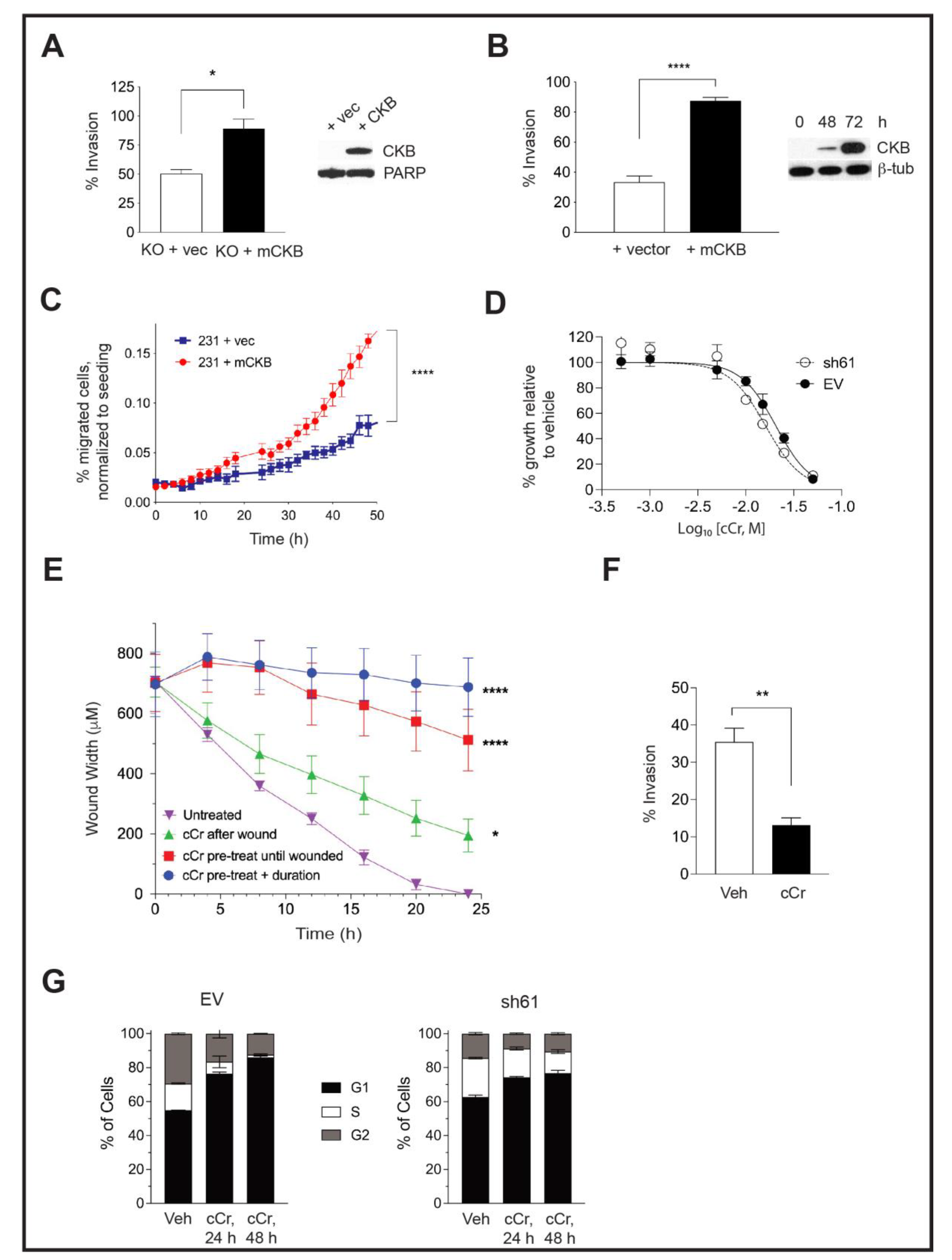
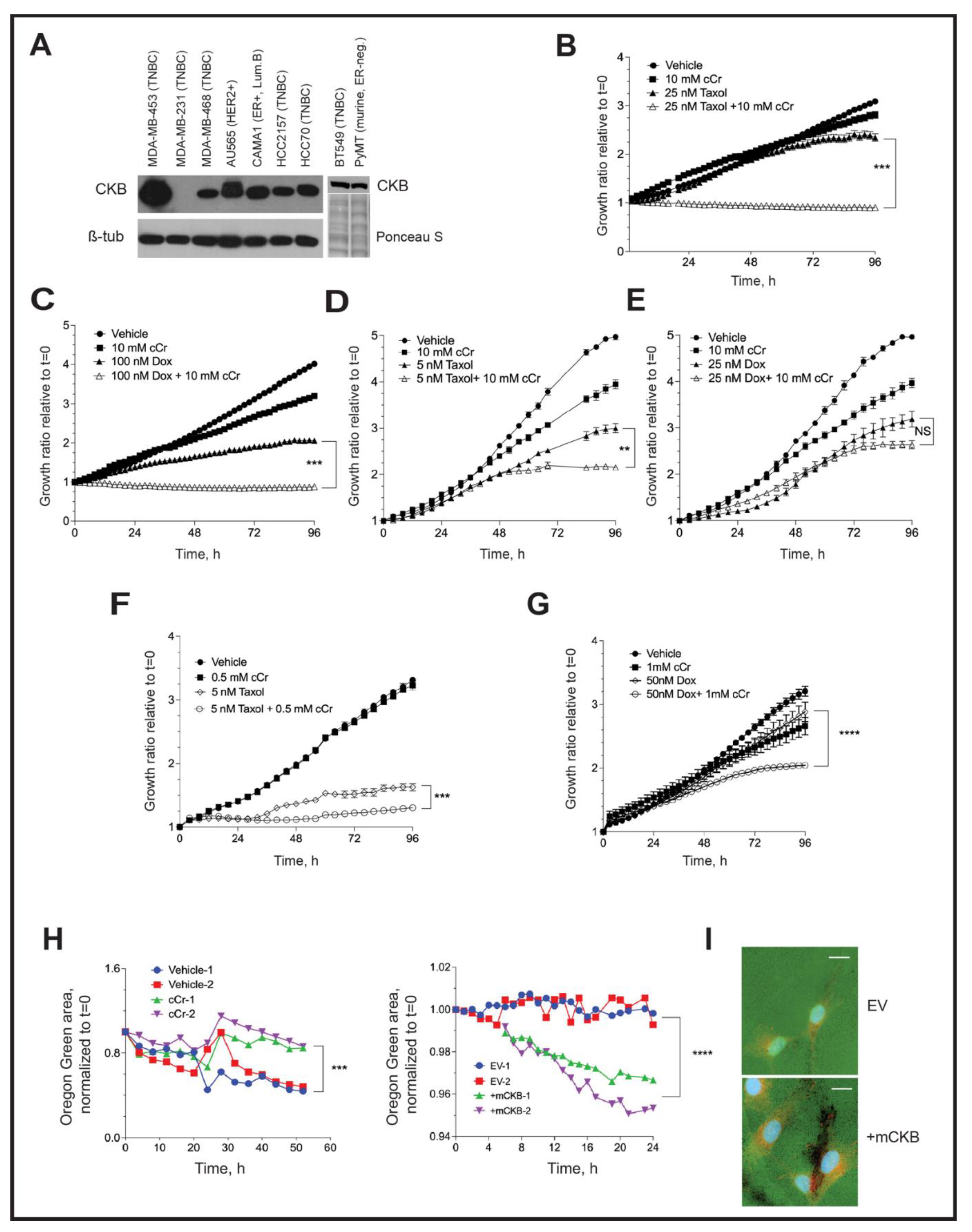
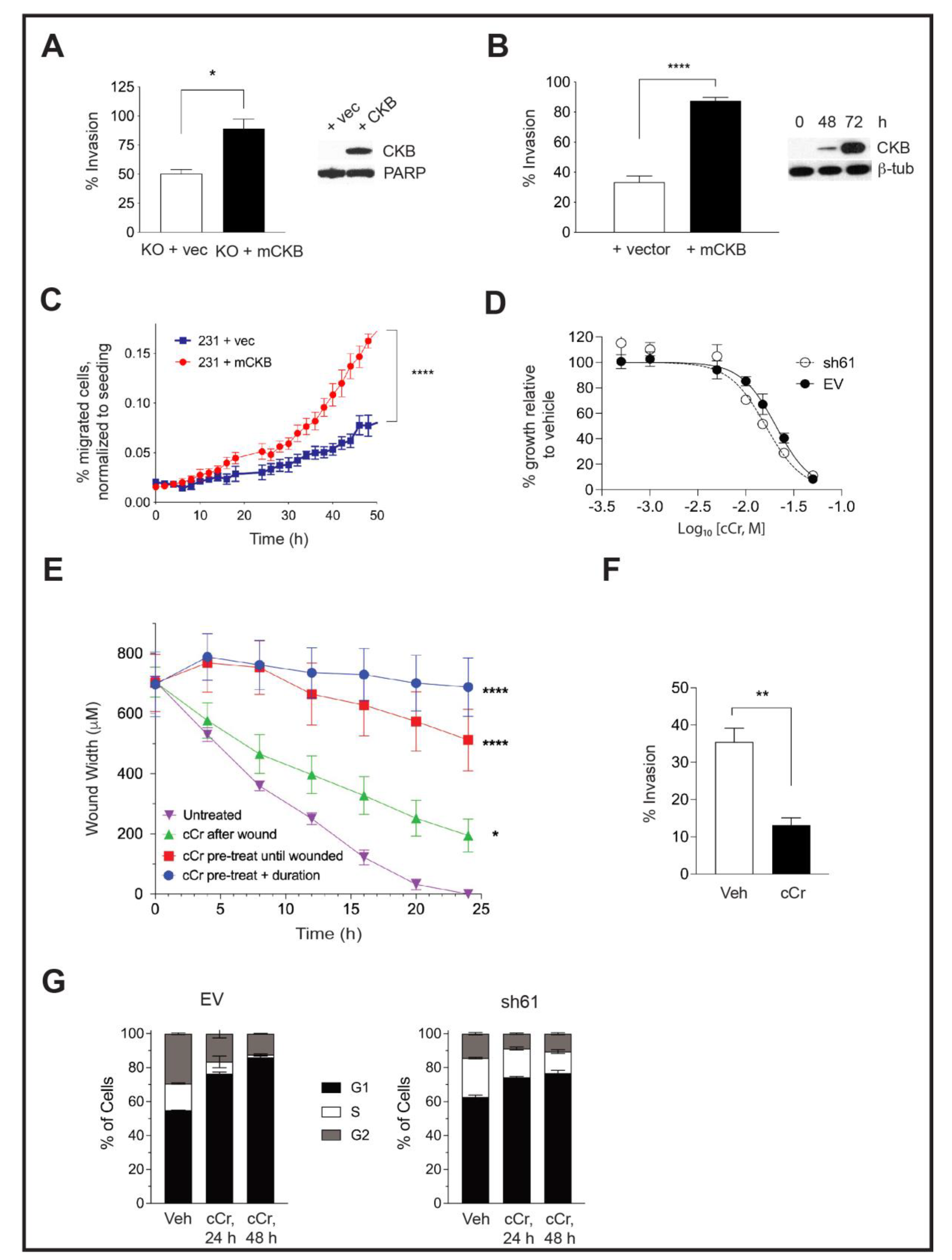
3.5. Ectopic CKB Expression Promotes, but Cyclocreatine Treatment Represses, Cell Migration and Invasion
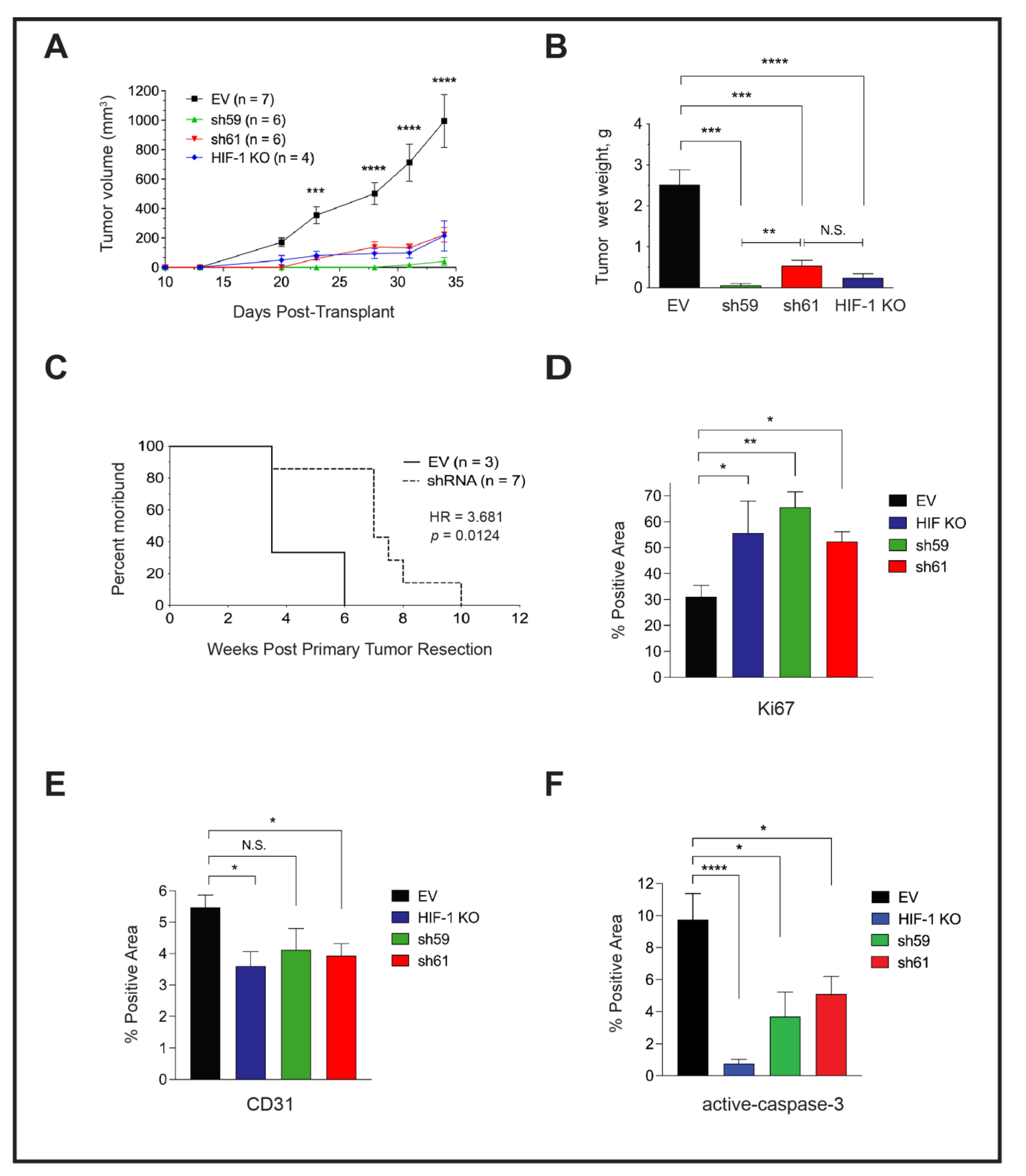
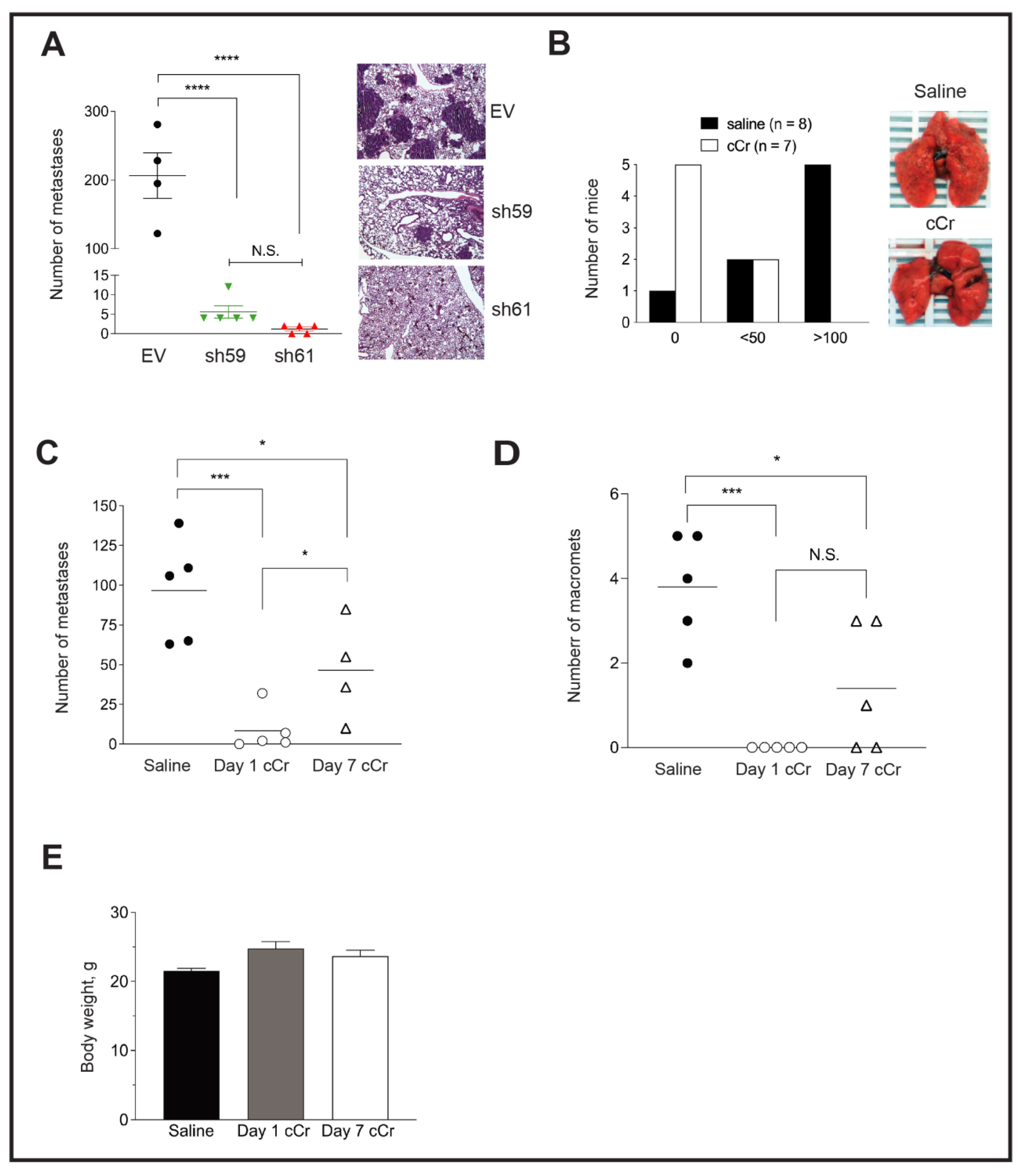
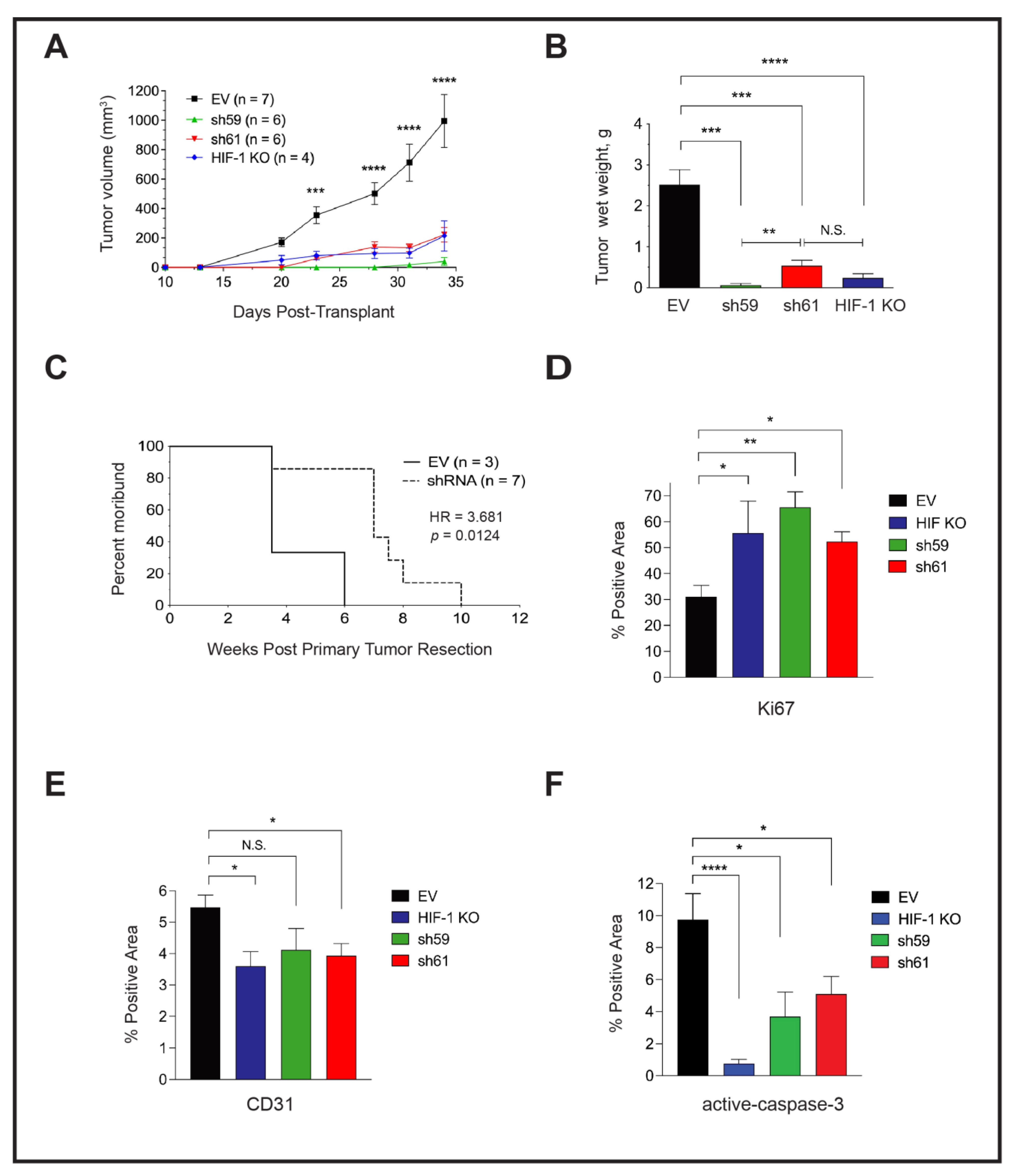
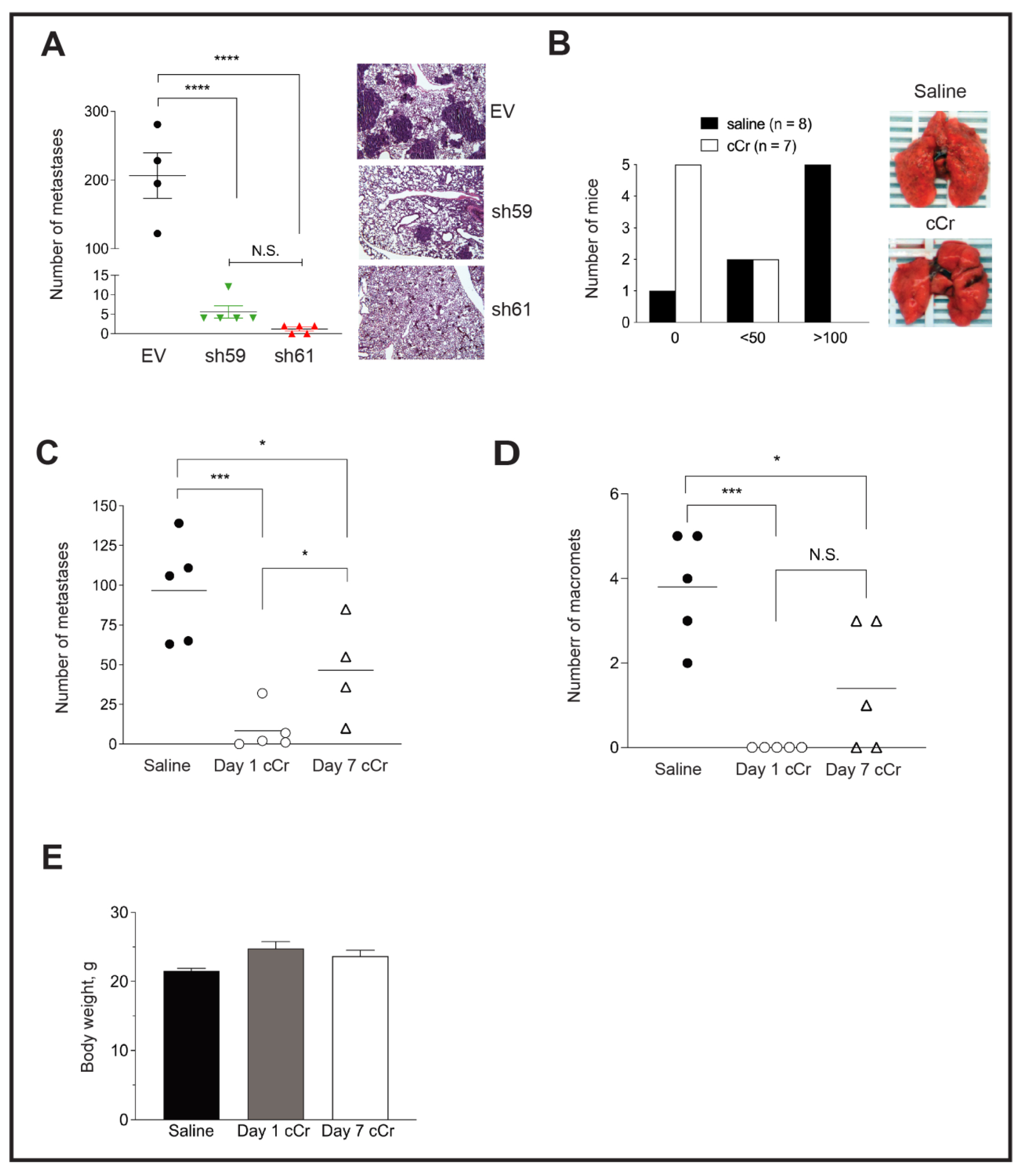
3.6. Loss of Ckb Suppresses Tumor Growth and Inhibits Lung Metastases
3.7. Either Ckb deletion or Systemic Treatment with Cyclocreatine Blocks Lung Metastasis
3.8. Addition of Cyclocreatine to Paclitaxel or Doxorubicin Enhances Growth Inhibition
3.9. Invadopodia Formation in TNBC Cells Depends on Creatine Kinase Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts: Female Breast Cancer. Available online: https://seer.cancer.gov/statfacts/html/breast.html (accessed on 16 August 2021).
- Bailey, K.M.; Wojtkowiak, J.W.; Hashim, A.I.; Gillies, R.J. Targeting the metabolic microenvironment of tumors. Adv. Pharmacol. 2012, 65, 63–107. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [Green Version]
- Dang, N.H.; Singla, A.K.; Mackay, E.M.; Jirik, F.R.; Weljie, A.M. Targeted Cancer Therapeutics: Biosynthetic and Energetic Pathways Characterized by Metabolomics and the Interplay with Key Cancer Regulatory Factors. Curr. Pharm. Des. 2013, 20, 2637–2647. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [Green Version]
- Chiche, J.; Brahimi-Horn, M.C.; Pouyssegur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohwer, N.; Cramer, T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist. Update 2011, 14, 191–201. [Google Scholar] [CrossRef]
- Brown, J.M. Exploiting the hypoxic cancer cell: Mechanisms and therapeutic strategies. Mol. Med. Today 2000, 6, 157–162. [Google Scholar] [CrossRef]
- Schwab, L.P.; Peacock, D.L.; Majumdar, D.; Ingels, J.F.; Jensen, L.C.; Smith, K.D.; Cushing, R.C.; Seagroves, T.N. Hypoxia-inducible factor 1alpha promotes primary tumor growth and tumor-initiating cell activity in breast cancer. Breast Cancer Res. 2012, 14, R6. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Choi, H.K.; Lee, K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur. J. Med. Chem. 2012, 49, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Doedens, A.; Johnson, R.S. Transgenic models to understand hypoxia-inducible factor function. Methods EnzyMol. 2007, 435, 87–105. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallimann, T.; Wyss, M.; Brdiczka, D.; Nicolay, K.; Eppenberger, H.M. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: The ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem. J. 1992, 281 Pt 1, 21–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallimann, T.; Tokarska-Schlattner, M.; Schlattner, U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and creatinine metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef]
- Glover, L.E.; Bowers, B.E.; Saeedi, B.; Ehrentraut, S.F.; Campbell, E.L.; Bayless, A.J.; Dobrinskikh, E.; Kendrick, A.A.; Kelly, C.J.; Burgess, A.; et al. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 19820–19825. [Google Scholar] [CrossRef] [Green Version]
- Streijger, F.; Oerlemans, F.; Ellenbroek, B.A.; Jost, C.R.; Wieringa, B.; Van der Zee, C.E. Structural and behavioural consequences of double deficiency for creatine kinases BCK and UbCKmit. Behav. Brain Res. 2005, 157, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Mooney, S.M.; Rajagopalan, K.; Williams, B.H.; Zeng, Y.; Christudass, C.S.; Li, Y.; Yin, B.; Kulkarni, P.; Getzenberg, R.H. Creatine kinase brain overexpression protects colorectal cells from various metabolic and non-metabolic stresses. J. Cell Biochem. 2011, 112, 1066–1075. [Google Scholar] [CrossRef] [Green Version]
- Zarghami, N.; Giai, M.; Yu, H.; Roagna, R.; Ponzone, R.; Katsaros, D.; Sismondi, P.; Diamandis, E.P. Creatine kinase BB isoenzyme levels in tumour cytosols and survival of breast cancer patients. Br. J. Cancer 1996, 73, 386–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.H.; Chen, X.J.; Ou, W.B.; Zhang, Q.; Lv, Z.R.; Zhan, Y.; Ma, L.; Huang, T.; Yan, Y.B.; Zhou, H.M. Knockdown of creatine kinase B inhibits ovarian cancer progression by decreasing glycolysis. Int. J. Biochem. Cell Biol. 2013, 45, 979–986. [Google Scholar] [CrossRef]
- Chen, H.; Pimienta, G.; Gu, Y.; Sun, X.; Hu, J.; Kim, M.S.; Chaerkady, R.; Gucek, M.; Cole, R.N.; Sukumar, S.; et al. Proteomic characterization of Her2/neu-overexpressing breast cancer cells. Proteomics 2010, 10, 3800–3810. [Google Scholar] [CrossRef]
- Chen, W.Z.; Pang, B.; Yang, B.; Zhou, J.G.; Sun, Y.H. Differential proteome analysis of conditioned medium of BPH-1 and LNCaP cells. Chin. Med. J. (Engl.) 2011, 124, 3806–3809. [Google Scholar]
- Xu, Y.; Cao, L.Q.; Jin, L.Y.; Chen, Z.C.; Zeng, G.Q.; Tang, C.E.; Li, G.Q.; Duan, C.J.; Peng, F.; Xiao, Z.Q.; et al. Quantitative proteomic study of human lung squamous carcinoma and normal bronchial epithelial acquired by laser capture microdissection. J. BioMed Biotechnol. 2012, 2012, 510418. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.Q.; Zhang, P.F.; Deng, X.; Yu, F.L.; Li, C.; Xu, Y.; Yi, H.; Li, M.Y.; Hu, R.; Zuo, J.H.; et al. Identification of candidate biomarkers for early detection of human lung squamous cell cancer by quantitative proteomics. Mol. Cell Proteomics 2012, 11, M111.013946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teicher, B.A.; Menon, K.; Northey, D.; Liu, J.; Kufe, D.W.; Kaddurah-Daouk, R. Cyclocreatine in cancer chemotherapy. Cancer Chemother. Pharmacol. 1995, 35, 411–416. [Google Scholar] [CrossRef]
- Mulvaney, P.T.; Stracke, M.L.; Nam, S.W.; Woodhouse, E.; O’Keefe, M.; Clair, T.; Liotta, L.A.; Khaddurah-Daouk, R.; Schiffmann, E. Cyclocreatine inhibits stimulated motility in tumor cells possessing creatine kinase. Int. J. Cancer 1998, 78, 46–52. [Google Scholar] [CrossRef]
- Loo, J.M.; Scherl, A.; Nguyen, A.; Man, F.Y.; Weinberg, E.; Zeng, Z.; Saltz, L.; Paty, P.B.; Tavazoie, S.F. Extracellular metabolic energetics can promote cancer progression. Cell 2015, 160, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Papalazarou, V.; Zhang, T.; Paul, N.R.; Juin, A.; Cantini, M.; Maddocks, O.D.K.; Salmeron-Sanchez, M.; Machesky, L.M. The creatine-phosphagen system is mechanoresponsive in pancreatic adenocarcinoma and fuels invasion and metastasis. Nat. Metab. 2020, 2, 62–80. [Google Scholar] [CrossRef]
- Kuiper, J.W.; van Horssen, R.; Oerlemans, F.; Peters, W.; van Dommelen, M.M.; te Lindert, M.M.; ten Hagen, T.L.; Janssen, E.; Fransen, J.A.; Wieringa, B. Local ATP generation by brain-type creatine kinase (CK-B) facilitates cell motility. PLoS ONE 2009, 4, e5030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Horssen, R.; Janssen, E.; Peters, W.; van de Pasch, L.; Lindert, M.M.; van Dommelen, M.M.; Linssen, P.C.; Hagen, T.L.; Fransen, J.A.; Wieringa, B. Modulation of cell motility by spatial repositioning of enzymatic ATP/ADP exchange capacity. J. Biol. Chem. 2009, 284, 1620–1627. [Google Scholar] [CrossRef] [Green Version]
- Dent, R.; Hanna, W.M.; Trudeau, M.; Rawlinson, E.; Sun, P.; Narod, S.A. Pattern of metastatic spread in triple-negative breast cancer. Breast Cancer Res. Treat 2009, 115, 423–428. [Google Scholar] [CrossRef]
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Nagy, A.; Lanczky, A.; Menyhart, O.; Gyorffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 9227. [Google Scholar] [CrossRef]
- Brooks, D.L.; Schwab, L.P.; Krutilina, R.; Parke, D.N.; Sethuraman, A.; Hoogewijs, D.; Schorg, A.; Gotwald, L.; Fan, M.; Wenger, R.H.; et al. ITGA6 is directly regulated by hypoxia-inducible factors and enriches for cancer stem cell activity and invasion in metastatic breast cancer models. Mol. Cancer 2016, 15, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oosthuyse, B.; Moons, L.; Storkebaum, E.; Beck, H.; Nuyens, D.; Brusselmans, K.; Van Dorpe, J.; Hellings, P.; Gorselink, M.; Heymans, S.; et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 2001, 28, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Ha, Y.; Ahn, C.H.; Huang, X.; Kim, J.M.; Park, S.R.; Park, H.; Park, H.C.; Kim, S.W.; Lee, M. A hypoxia-inducible gene expression system using erythropoietin 3′ untranslated region for the gene therapy of rat spinal cord injury. Neurosci. Lett. 2007, 412, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.L.; Seagroves, T.N. Chromatin Immunoprecipitation of HIF-alpha in Breast Tumor Cells Using Wild Type and Loss of Function Models. Methods Mol. Biol. 2018, 1742, 67–79. [Google Scholar] [CrossRef]
- Martin, K.H.; Hayes, K.E.; Walk, E.L.; Ammer, A.G.; Markwell, S.M.; Weed, S.A. Quantitative measurement of invadopodia-mediated extracellular matrix proteolysis in single and multicellular contexts. J. Vis. Exp. 2012, 66, e4119. [Google Scholar] [CrossRef]
- Tallarida, R.J. An overview of drug combination analysis with isobolograms. J. Pharmacol. Exp. Ther. 2006, 319, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Bergnes, G.; Yuan, W.; Khandekar, V.S.; O’Keefe, M.M.; Martin, K.J.; Teicher, B.A.; Kaddurah-Daouk, R. Creatine and phosphocreatine analogs: Anticancer activity and enzymatic analysis. Oncol. Res. 1996, 8, 121–130. [Google Scholar]
- Hunter, K.W.; Broman, K.W.; Voyer, T.L.; Lukes, L.; Cozma, D.; Debies, M.T.; Rouse, J.; Welch, D.R. Predisposition to efficient mammary tumor metastatic progression is linked to the breast cancer metastasis suppressor gene Brms1. Cancer Res. 2001, 61, 8866–8872. [Google Scholar]
- Wang, F.; Samudio, I.; Safe, S. Transcriptional activation of rat creatine kinase B by 17beta-estradiol in MCF-7 cells involves an estrogen responsive element and GC-rich sites. J. Cell Biochem. 2001, 84, 156–172. [Google Scholar] [CrossRef]
- Stiehl, D.P.; Bordoli, M.R.; Abreu-Rodriguez, I.; Wollenick, K.; Schraml, P.; Gradin, K.; Poellinger, L.; Kristiansen, G.; Wenger, R.H. Non-canonical HIF-2alpha function drives autonomous breast cancer cell growth via an AREG-EGFR/ErbB4 autocrine loop. Oncogene 2012, 31, 2283–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramani, M.; Day, B.W.; Schoen, R.E.; Getzenberg, R.H. Altered expression and localization of creatine kinase B, heterogeneous nuclear ribonucleoprotein F, and high mobility group box 1 protein in the nuclear matrix associated with colon cancer. Cancer Res. 2006, 66, 763–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillie, J.W.; O’Keefe, M.; Valinski, H.; Hamlin, H.A., Jr.; Varban, M.L.; Kaddurah-Daouk, R. Cyclocreatine (1-carboxymethyl-2-iminoimidazolidine) inhibits growth of a broad spectrum of cancer cells derived from solid tumors. Cancer Res. 1993, 53, 3172–3178. [Google Scholar]
- Martin, K.; Winslow, E.; Okeefe, M.; Khandekar, V.; Hamlin, A.; Lillie, J.; Kaddurahdaouk, R. Specific targeting of tumor cells by the creatine analog cyclocreatine. Int. J. Oncol. 1996, 9, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.J.; Winslow, E.R.; Kaddurah-Daouk, R. Cell cycle studies of cyclocreatine, a new anticancer agent. Cancer Res. 1994, 54, 5160–5165. [Google Scholar]
- Hoosein, N.M.; Martin, K.J.; Abdul, M.; Logothetis, C.J.; Kaddurah-Daouk, R. Antiproliferative effects of cyclocreatine on human prostatic carcinoma cells. Anticancer Res. 1995, 15, 1339–1342. [Google Scholar]
- Miller, E.E.; Evans, A.E.; Cohn, M. Inhibition of rate of tumor growth by creatine and cyclocreatine. Proc. Natl. Acad. Sci. USA 1993, 90, 3304–3308. [Google Scholar] [CrossRef] [Green Version]
- Fenouille, N.; Bassil, C.F.; Ben-Sahra, I.; Benajiba, L.; Alexe, G.; Ramos, A.; Pikman, Y.; Conway, A.S.; Burgess, M.R.; Li, Q.; et al. The creatine kinase pathway is a metabolic vulnerability in EVI1-positive acute myeloid leukemia. Nat. Med. 2017, 23, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Kurmi, K.; Hitosugi, S.; Yu, J.; Boakye-Agyeman, F.; Wiese, E.K.; Larson, T.R.; Dai, Q.; Machida, Y.J.; Lou, Z.; Wang, L.; et al. Tyrosine Phosphorylation of Mitochondrial Creatine Kinase 1 Enhances a Druggable Tumor Energy Shuttle Pathway. Cell Metab. 2018, 28, 833–847.e838. [Google Scholar] [CrossRef] [Green Version]
- Vranic, S.; Gatalica, Z.; Wang, Z.Y. Update on the molecular profile of the MDA-MB-453 cell line as a model for apocrine breast carcinoma studies. Oncol. Lett. 2011, 2, 1131–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, C.; Cannet, A.; Descamps, S.; Sirvent, A.; Simon, V.; Roche, S.; Benistant, C. ABL tyrosine kinase inhibition variable effects on the invasive properties of different triple negative breast cancer cell lines. PLoS ONE 2015, 10, e0118854. [Google Scholar] [CrossRef]
- Martin, K.J.; Vassallo, C.D.; Teicher, B.A.; Kaddurah-Daouk, R. Microtubule stabilization and potentiation of taxol activity by the creatine analog cyclocreatine. Anticancer Drugs 1995, 6, 419–426. [Google Scholar] [CrossRef]
- de Heer, E.C.; Jalving, M.; Harris, A.L. HIFs, angiogenesis, and metabolism: Elusive enemies in breast cancer. J. Clin. Investig. 2020, 130, 5074–5087. [Google Scholar] [CrossRef] [PubMed]
- Gatza, M.L.; Kung, H.N.; Blackwell, K.L.; Dewhirst, M.W.; Marks, J.R.; Chi, J.T. Analysis of tumor environmental response and oncogenic pathway activation identifies distinct basal and luminal features in HER2-related breast tumor subtypes. Breast Cancer Res. 2011, 13, R62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)alpha: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Decking, U.K.; Alves, C.; Wallimann, T.; Wyss, M.; Schrader, J. Functional aspects of creatine kinase isoenzymes in endothelial cells. Am. J. Physiol. Cell Physiol. 2001, 281, C320–C328. [Google Scholar] [CrossRef]
- Loike, J.D.; Kozler, V.F.; Silverstein, S.C. Creatine kinase expression and creatine phosphate accumulation are developmentally regulated during differentiation of mouse and human monocytes. J. Exp. Med. 1984, 159, 746–757. [Google Scholar] [CrossRef] [Green Version]
- Rahbani, J.F.; Roesler, A.; Hussain, M.F.; Samborska, B.; Dykstra, C.B.; Tsai, L.; Jedrychowski, M.P.; Vergnes, L.; Reue, K.; Spiegelman, B.M.; et al. Creatine kinase B controls futile creatine cycling in thermogenic fat. Nature 2021, 590, 480–485. [Google Scholar] [CrossRef]
- Attanasio, F.; Caldieri, G.; Giacchetti, G.; van Horssen, R.; Wieringa, B.; Buccione, R. Novel invadopodia components revealed by differential proteomic analysis. Eur. J. Cell Biol. 2011, 90, 115–127. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, Z.; Yan, H.; Wang, W.; Wu, Z.; Zhang, F.; Zhang, Q.; Shi, G.; Du, J.; Cai, H.; et al. Creatine promotes cancer metastasis through activation of Smad2/3. Cell Metab. 2021, 33, 1111–1123.e1114. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, N.; Gray, R.S.; Li, H.; Ewald, A.J.; Zahnow, C.A.; Pan, D. A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev. 2014, 28, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Yang, X.; Zhang, W.; Wang, S.; Cui, X.; Du, T.; Li, T. Mechanical loading induces HIF-1alpha expression in chondrocytes via YAP. Biotechnol. Lett. 2020, 42, 1645–1654. [Google Scholar] [CrossRef]
- Jia, Y.; Li, H.Y.; Wang, J.; Wang, Y.; Zhang, P.; Ma, N.; Mo, S.J. Phosphorylation of 14-3-3zeta links YAP transcriptional activation to hypoxic glycolysis for tumorigenesis. Oncogenesis 2019, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, S.W.; Sunga, P. The effect of doxorubicin (adriamycin) on cytoplasmic microtubule system in cardiac cells. J. Mol. Cell Cardiol. 1987, 19, 1073–1083. [Google Scholar] [CrossRef]
- Cacciante, F.; Gennaro, M.; Sagona, G.; Mazziotti, R.; Lupori, L.; Cerri, E.; Putignano, E.; Butt, M.; Do, M.T.; McKew, J.C.; et al. Cyclocreatine treatment ameliorates the cognitive, autistic and epileptic phenotype in a mouse model of Creatine Transporter Deficiency. Sci. Rep. 2020, 10, 18361. [Google Scholar] [CrossRef]
- Uemura, T.; Ito, S.; Masuda, T.; Shimbo, H.; Goto, T.; Osaka, H.; Wada, T.; Couraud, P.O.; Ohtsuki, S. Cyclocreatine Transport by SLC6A8, the Creatine Transporter, in HEK293 Cells, a Human Blood-Brain Barrier Model Cell, and CCDSs Patient-Derived Fibroblasts. Pharm. Res. 2020, 37, 61. [Google Scholar] [CrossRef]
- Kurosawa, Y.; Degrauw, T.J.; Lindquist, D.M.; Blanco, V.M.; Pyne-Geithman, G.J.; Daikoku, T.; Chambers, J.B.; Benoit, S.C.; Clark, J.F. Cyclocreatine treatment improves cognition in mice with creatine transporter deficiency. J. Clin. Investig. 2012, 122, 2837–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.J.; Ha, J.; Oerlemans, F.; Lee, Y.J.; Lee, S.W.; Ryu, J.; Kim, H.J.; Lee, Y.; Kim, H.M.; Choi, J.Y.; et al. Brain-type creatine kinase has a crucial role in osteoclast-mediated bone resorption. Nat. Med. 2008, 14, 966–972. [Google Scholar] [CrossRef]
- Kurth, I.; Yamaguchi, N.; Andreu-Agullo, C.; Tian, H.S.; Sridhar, S.; Takeda, S.; Gonsalves, F.C.; Loo, J.M.; Barlas, A.; Manova-Todorova, K.; et al. Therapeutic targeting of SLC6A8 creatine transporter suppresses colon cancer progression and modulates human creatine levels. Sci. Adv. 2021, 7, eabi7511. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIF-1 WT | HIF-1 KO | EV | sh61 CKB KD |
|---|---|---|---|
| 16.76 mM ± 4.47 | 15.44 mM ± 3.95 | 13.73 mM ± 4.33 | 16.43 mM ± 2.98 |
| Cell Line | cCr C50, mM | Taxol C50, nM | DOX C50, nM |
|---|---|---|---|
| MDA-MB-468 | 19.15 ± 3.06 | 11.16 nM ± 2.94 | 343.6 ±126.6 |
| BT549 | 22.38 ±1.47 | 3.82 ± 0.44 | 28.28 ± 3.68 |
| MDA-MB-453 | 6.10 ± 1.48 | 6.03 ± 1.75 | 60.39 ± 11.53 |
| TNBC Models | Taxol @ IC50 +cCr @ IC30 Combination Indices (CI) | DOX @ IC50 +cCr @ IC30 Combination Indices (CI) | ||
|---|---|---|---|---|
| Cell Line | IC50, TAXOL CI | IC70, TAXOL CI | IC50, DOX CI | IC70, DOX CI |
| MDA-MB-468 | 1.06 | 0.52 | 0.61 | 0.39 |
| BT549 | 0.86 | 0.58 | 1.50 | 1.28 |
| MDA-MB-453 | 0.95 | 2.18 | 0.77 | 1.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krutilina, R.I.; Playa, H.; Brooks, D.L.; Schwab, L.P.; Parke, D.N.; Oluwalana, D.; Layman, D.R.; Fan, M.; Johnson, D.L.; Yue, J.; et al. HIF-Dependent CKB Expression Promotes Breast Cancer Metastasis, Whereas Cyclocreatine Therapy Impairs Cellular Invasion and Improves Chemotherapy Efficacy. Cancers 2022, 14, 27. https://doi.org/10.3390/cancers14010027
Krutilina RI, Playa H, Brooks DL, Schwab LP, Parke DN, Oluwalana D, Layman DR, Fan M, Johnson DL, Yue J, et al. HIF-Dependent CKB Expression Promotes Breast Cancer Metastasis, Whereas Cyclocreatine Therapy Impairs Cellular Invasion and Improves Chemotherapy Efficacy. Cancers. 2022; 14(1):27. https://doi.org/10.3390/cancers14010027
Chicago/Turabian StyleKrutilina, Raisa I., Hilaire Playa, Danielle L. Brooks, Luciana P. Schwab, Deanna N. Parke, Damilola Oluwalana, Douglas R. Layman, Meiyun Fan, Daniel L. Johnson, Junming Yue, and et al. 2022. "HIF-Dependent CKB Expression Promotes Breast Cancer Metastasis, Whereas Cyclocreatine Therapy Impairs Cellular Invasion and Improves Chemotherapy Efficacy" Cancers 14, no. 1: 27. https://doi.org/10.3390/cancers14010027
APA StyleKrutilina, R. I., Playa, H., Brooks, D. L., Schwab, L. P., Parke, D. N., Oluwalana, D., Layman, D. R., Fan, M., Johnson, D. L., Yue, J., Smallwood, H., & Seagroves, T. N. (2022). HIF-Dependent CKB Expression Promotes Breast Cancer Metastasis, Whereas Cyclocreatine Therapy Impairs Cellular Invasion and Improves Chemotherapy Efficacy. Cancers, 14(1), 27. https://doi.org/10.3390/cancers14010027