
Dissection of the MKK3 Functions in Human Cancer: A Double-Edged Sword?


Simple Summary
Abstract
1. Introduction
2. Oncogenic MKK3 Functions
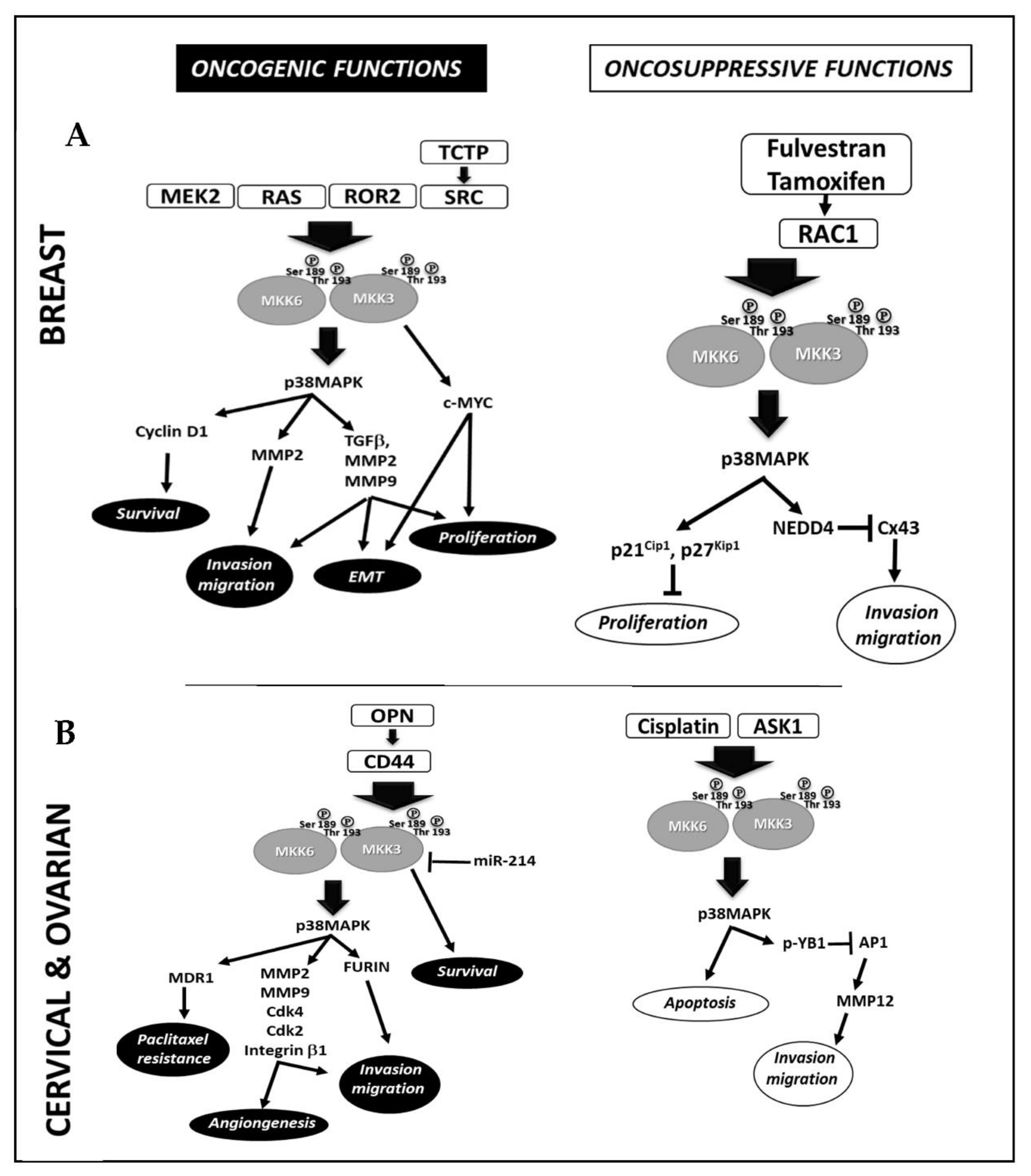
2.1. Breast Cancer
2.2. Cervical and Ovarian Cancer
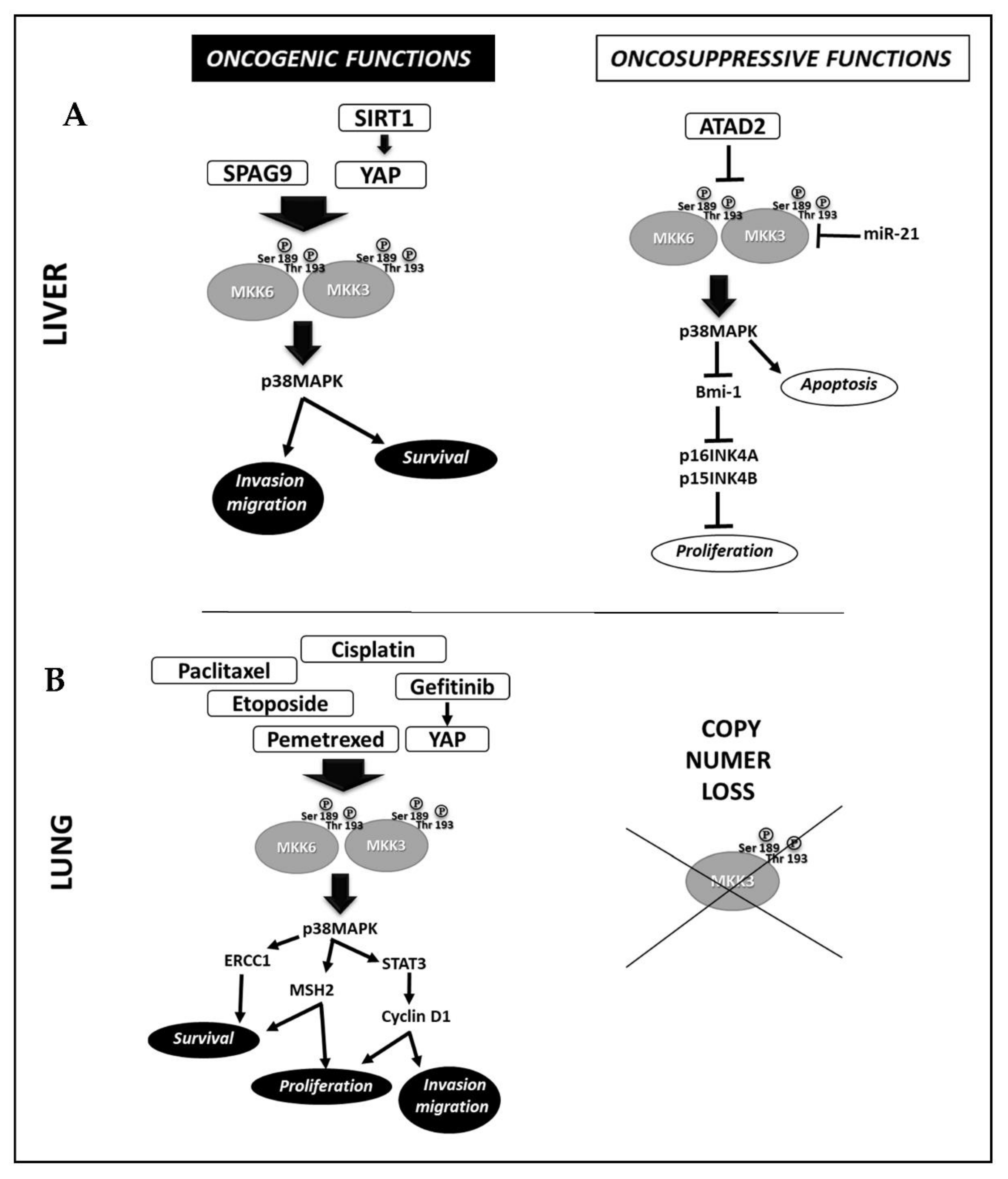
2.3. Liver Cancer
2.4. Melanoma
2.5. Prostate Cancer
2.6. Esophageal Cancer
2.7. Lung Cancer
2.8. Colorectal Cancer
3. Oncosuppressive MKK3 Functions
3.1. Glioblastoma
3.2. Breast Cancer
3.3. Lung Cancer
3.4. Gastric Cancer
3.5. Liver Cancer
3.6. Cervical and Ovarian Cancer
3.7. Esophageal Cancer
4. Conclusions

{kind=link}
{kind=link}
| Disease | Oncogenic Functions Regulatory Partners | References | Tumor-Suppressing Functions Regulatory Partners | References |
|---|---|---|---|---|
| breast cancer | MEK2/Cyclin D1 *; RAS/MMP2 *; RORO2/MMP2 *; ROR2/MMP9 *; ROR2/TGF-β, TCTP/SCR *, c-MYC | [2,3,7,8,9,10,11,12,13,14] | p21Cip1, p27Kip1; RAC1/Cx43 * | [39,40] |
| cervical and ovarian cancer | miR214; OPN/CD44/NF-KB/FURIN *; MDR1; MMP2; MMP9; CDK4; CDK2; Integrin β1 | [15,16,17,18,19] | ASK1; ASK1/p-YB1* | [46,47,48] |
| liver cancer | SIRT1/YAP *; SPAG9 | [20,21] | Bmi1-p16INK4A, -p15INK4B; ATAD2, miR-21 | [43,44,45] |
| melanoma | miR-21; L1CAM | [22,23] | ||
| prostate carcinoma | GRP78/α2M *; TRAIL/MCL1 *; SRC/SF * | [24,25,26] | ||
| esophageal cancer | MKK3/6-p38MAPK | [27] | GADD45/p53; miR19b-3p/STAT3 | [49,50] |
| lung cancer | p38MAPK; ERCC1; MSH2; STAT3 | [28,29,30,31,32,33,34] | MKK3 CNA | [41] |
| colorectal cancer | MAPK11-ERCC1 | [1,8,9,35] | ||
| glioblastoma | GMF-β | [37,38] | ||
| gastric cancer | DDIT3 | [42] |
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CRC | Colorectal cancer |
| p38MAPK | p38 mitogen-activated protein kinases |
| RAS | Rat sarcoma |
| RAF | Rapidly Accelerated Fibrosarcoma |
| BC | Breast Cancer |
| 5-FU | 5-fluorouracil |
| MMP-2 | Metalloproteinase-2 |
| MMP-9 | Metalloproteinase-9 |
| EGFR | Epidermis Growth Factor |
| ECM | Extracellular Matrix degradation |
| ER | Estrogen Receptor |
| Mir-214 | Micro-RNA 214 |
| ROR-2 | Receptor Tyrosine Kinase-like Orphan receptor 2 |
| OPN | Osteopontin |
| PBSA | 2-Phenylbenzimidazole 5-sulphonic acid |
| CDK4 | Cyclin-dependent Kinase 4 |
| CDK2 | Cyclin-dependent Kinase 2 |
| DUSP1 | Dual Specificity phosphatase 1 |
| HCC | Hepatocarcinoma |
| SIRT1 | Sirtuin-1 |
| SPAG9 | Sperm-associated antigen 9 |
| L1CAM | L1 cell adhesion molecule |
| GRP-78 | Immunoglobulin Heavy chain-binding Protein |
| BiP | Binding Immunoglobulin Protein |
| EMT | Epithelium Mesenchymal Transition |
| MAPK | Mitogen-Activated Protein Kinase |
| MAPKKK | MAP kinase kinase kinase |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| TPL2 | Tumor progression locus 2 |
| MEKK3 | Mitogen-activated protein kinase kinase kinase 3 |
| MAPKK/MKK | Mitogen-activated protein kinase kinase |
| ara-c | Cytosine arabinoside |
| CPT-11 | Camptothecin-11 |
| SN-38 | 7-etil-10-idrossi-camptotecina |
| FOLFOX | Folinic acid, 5– Fluorouracil, Oxaliplatin |
| ATM | ataxia-telangiectasia mutated |
| MKP-1 | Mitogen-activated protein kinase 1 |
| TP53 | tumor protein p53 |
| HuR | Hu antigen R |
| MKNK1 | MAPK-interacting kinase 1 |
| PFS | Progression-free survival |
| OM | Oxymatrine |
| PAI-1 | Plasminogen activator inhibitor 1 |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| SMAD | Small mother against decapentaplegic |
| TGFβ-1 | Transforming growth factor beta 1 |
| PXR | Pregnane X receptor |
| PCNA | Proliferating Cell Nuclear Antigen |
| CPT11 | Irinotecan |
| AKT | Protein kinase B |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| TAK1 | TGFbeta Activated Kinase 1 |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| p70S6K | Ribosomal protein S6 kinase |
| ERK1/2 | Extracellular signal-regulated-1/2-ER2 |
| PP2AC | Serine/threonine-protein phosphatase 2A catalytic subunit alpha isoform |
| 4E-BP1 | eIF4E binding protein-1 |
| mTOR | Mammalian phosphorylation target of rapamycin |
| SAPK/JNK | c-Jun N-terminal kinases |
| RT | Radiotherapy |
| eIF4E | Eukaryotic translation initiation factor 4E |
References
- Stramucci, L.; Pranteda, A.; Stravato, A.; Amoreo, C.A.; Pennetti, A.; Diodoro, M.G.; Bartolazzi, A.; Milella, M.; Bossi, G. MKK3 sustains cell proliferation and survival through p38DELTA MAPK activation in colorectal cancer. Cell Death Dis. 2019, 10, 842. [Google Scholar] [CrossRef] [PubMed]
- Huth, H.W.; Albarnaz, J.D.; Torres, A.A.; Bonjardim, C.A.; Ropert, C. MEK2 controls the activation of MKK3/MKK6-p38 axis involved in the MDA-MB-231 breast cancer cell survival: Correlation with cyclin D1 expression. Cell. Signal. 2016, 28, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, E.S.; Noh, D.Y.; Hwang, K.T.; Moon, A. H-Ras-specific upregulation of granulocyte colony-stimulating factor promotes human breast cell invasion via matrix metalloproteinase-2. Cytokine 2011, 55, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Shi, Q.; Wang, W. Double agents: Genes with both oncogenic and tumor-suppressor functions. Oncogenesis 2018, 7, 25. [Google Scholar] [CrossRef]
- Aranko, A.S.; Oeemig, J.S.; Kajander, T.; Iwaï, H. Intermolecular domain swapping induces intein-mediated protein alternative splicing. Nat. Chem. Biol. 2013, 9, 616–622. [Google Scholar] [CrossRef]
- Raingeaud, J.; Whitmarsh, A.J.; Barrett, T.; Dérijard, B.; Davis, R.J. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 1996, 16, 1247–1255. [Google Scholar] [CrossRef]
- Ivanov, A.A.; Gonzalez-Pecchi, V.; Khuri, L.F.; Niu, Q.; Wang, Y.; Xu, Y.; Bai, Y.; Mo, X.; Prochownik, E.V.; Johns, M.A.; et al. OncoPPi-informed discovery of mitogen-activated protein kinase kinase 3 as a novel binding partner of c-Myc. Oncogene 2017, 36, 5852–5860. [Google Scholar] [CrossRef]
- Gurtner, A.; Starace, G.; Norelli, G.; Piaggio, G.; Sacchi, A.; Bossi, G. Mutant p53-induced up-regulation of mito-gen-activated protein kinase kinase 3 contributes to gain of function. J. Biol. Chem. 2010, 285, 14160–14169. [Google Scholar] [CrossRef]
- Baldari, S.; Ubertini, V.; Garufi, A.; D’Orazi, G.; Bossi, G. Targeting MKK3 as a novel anticancer strategy: Molecular mechanisms and therapeutical implications. Cell Death Dis. 2015, 6, e1621. [Google Scholar] [CrossRef]
- Shin, I.; Kim, S.; Song, H.; Kim, H.R.; Moon, A. H-Ras-specific activation of Rac-MKK3/6-p38 pathway: Its critical role in invasion and migration of breast epithelial cells. J. Biol. Chem. 2005, 280, 14675–14683. [Google Scholar] [CrossRef]
- Xu, J.; Shi, J.; Tang, W.; Jiang, P.; Guo, M.; Zhang, B.; Ma, G. ROR2 promotes the epithelial-mesenchymal transition by regulating MAPK/p38 signaling pathway in breast cancer. J. Cell. Biochem. 2020, 121, 4142–4153. [Google Scholar] [CrossRef]
- Jung, J.; Kim, H.Y.; Kim, M.; Sohn, K.; Lee, K. Translationally controlled tumor protein induces human breast epithelial cell transformation through the activation of Src. Oncogene 2011, 30, 2264–2274. [Google Scholar] [CrossRef]
- Tuynder, M.; Susini, L.; Prieur, S.; Besse, S.; Fiucci, G.; Amson, R.; Telerman, A. Biological models and genes of tumor reversion: Cellular reprogramming through tpt1/TCTP and SIAH-1. Proc. Natl. Acad. Sci. USA 2002, 99, 14976–14981. [Google Scholar] [CrossRef]
- Yang, X.; Amgad, M.; Cooper, L.A.D.; Du, Y.; Fu, H.; Ivanov, A.A. High expression of MKK3 is associated with worse clinical outcomes in African American breast cancer patients. J. Transl. Med. 2020, 18, 334. [Google Scholar] [CrossRef]
- Peng, R.; Cheng, X.; Zhang, Y.; Lu, X.; Hu, Z. miR-214 down-regulates MKK3 and suppresses malignant phenotypes of cervical cancer cells. Gene 2020, 724, 144146. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, S.; Luan, X.; Li, Y.; Liu, M.; Li, X.; Liu, T.; Tang, H. MicroRNA-214 is aberrantly expressed in cervical cancers and inhibits the growth of HeLa cells. IUBMB Life 2009, 61, 1075–1082. [Google Scholar] [CrossRef]
- Kumar, V.; Behera, R.; Lohite, K.; Karnik, S.; Kundu, G.C. p38 kinase is crucial for osteopontin-induced furin expression that supports cervical cancer progression. Cancer Res. 2010, 70, 10381–10391. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, J.H.; Ahn, E.K.; Cho, Y.R.; Han, S.; Lee, C.H.; Bae, G.U.; Oh, J.S.; Kim, K.B.; Seo, D.W. Novel functions for 2-phenylbenzimidazole-5-sulphonic acid: Inhibition of ovarian cancer cell responses and tumour angiogenesis. J. Cell. Mol. Med. 2020, 24, 2688–2700. [Google Scholar] [CrossRef]
- Kang, Y.S.; Seok, H.J.; Jeong, E.J.; Kim, Y.; Yun, S.J.; Min, J.K.; Kim, S.J.; Kim, J.S. DUSP1 induces paclitaxel resistance through the regulation of p-glycoprotein expression in human ovarian cancer cells. Biochem. Biophys. Res. Commun. 2016, 478, 403–409. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, R.; Zhang, X.; Qiao, Y.; Liu, X.; Chang, Y.; Yu, Y.; Sun, F.; Wang, J. SIRT1 increases YAP- and MKK3-dependent p38 phosphorylation in mouse liver and human hepatocellular carcinoma. Oncotarget 2016, 7, 11284–11298. [Google Scholar] [CrossRef]
- Luo, S.; Ren, B.; Zou, G.; Liu, J.; Chen, W.; Huang, Y.; Chen, X.; Fu, Y. SPAG9/MKK3/p38 axis is a novel therapeutic target for liver cancer. Oncol. Rep. 2019, 41, 2329–2336. [Google Scholar] [CrossRef]
- Zhou, M.; Yu, X.; Jing, Z.; Wu, W.; Lu, C. Overexpression of microRNA--21 inhibits the growth and metastasis of mela-noma cells by targeting MKK3. Mol. Med. Rep. 2019, 20, 1797–1807. [Google Scholar]
- Yi, Y.S.; Baek, K.S.; Cho, J.Y. L1 cell adhesion molecule induces melanoma cell motility by activation of mitogen-activated protein kinase pathways. Pharmazie 2014, 69, 461–467. [Google Scholar]
- Misra, U.K.; Pizzo, S.V. Ligation of cell surface GRP78 with antibody directed against the COOH-terminal domain of GRP78 suppresses Ras/MAPK and PI 3-kinase/AKT signaling while promoting caspase activation in human prostate cancer cells. Cancer Biol. Ther. 2010, 9, 142–152. [Google Scholar] [CrossRef]
- Son, J.K.; Varadarajan, S.; Bratton, S.B. TRAIL-activated stress kinases suppress apoptosis through transcriptional up-regulation of MCL-1. Cell Death Differ. 2010, 17, 1288–1301. [Google Scholar] [CrossRef]
- Fan, S.; Meng, Q.; Laterra, J.J.; Rosen, E.M. Role of Src signal transduction pathways in scatter factor-mediated cellular protection. J. Biol. Chem. 2009, 284, 7561–7577. [Google Scholar] [CrossRef]
- Xie, X.; Liu, K.; Liu, F.; Chen, H.; Wang, X.; Zu, X.; Ma, X.; Wang, T.; Wu, Q.; Zheng, Y.; et al. Gossypetin is a novel MKK3 and MKK6 inhibitor that suppresses esophageal cancer growth in vitro and in vivo. Cancer Lett. 2019, 442, 126–136. [Google Scholar] [CrossRef]
- Yeung, Y.T.; Yin, S.; Lu, B.; Fan, S.; Yang, R.; Bai, R.; Zhang, C.; Bode, A.M.; Liu, K.; Dong, Z. Losmapimod Overcomes Gefitinib Resistance in Non-small Cell Lung Cancer by Preventing Tetraploidization. EBioMedicine 2018, 28, 51–61. [Google Scholar] [CrossRef]
- Ko, J.C.; Chiu, H.C.; Wo, T.Y.; Huang, Y.J.; Tseng, S.C.; Huang, Y.C.; Chen, H.J.; Syu, J.J.; Chen, C.Y.; Jian, Y.T.; et al. Inhibition of p38 MAPK-dependent MutS homologue-2 (MSH2) expression by metformin enhances gefitinib-induced cytotoxicity in human squamous lung cancer cells. Lung Cancer 2013, 82, 397–406. [Google Scholar] [CrossRef]
- Tung, C.L.; Chiu, H.C.; Jian, Y.J.; Jian, Y.T.; Chen, C.Y.; Syu, J.J.; Wo, T.Y.; Huang, Y.J.; Tseng, S.C.; Lin, Y.W. Down-regulation of MSH2 expression by an Hsp90 inhibitor enhances pemetrexed-induced cytotoxicity in human non-small-cell lung cancer cells. Exp. Cell Res. 2014, 322, 345–354. [Google Scholar] [CrossRef]
- Tsai, M.S.; Weng, S.H.; Chen, H.J.; Chiu, Y.F.; Huang, Y.C.; Tseng, S.C.; Kuo, Y.H.; Lin, Y.W. Inhibition of p38 MAPK-dependent excision repair cross-complementing 1 expression decreases the DNA repair capacity to sensitize lung cancer cells to etoposide. Mol. Cancer Ther. 2012, 11, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.C.; Huang, Y.C.; Chen, H.J.; Chiu, H.C.; Huang, Y.J.; Wo, T.Y.; Weng, S.H.; Lin, Y.W. Metformin-mediated downregulation of p38 mitogen-activated protein kinase-dependent excision repair cross-complementing 1 decreases DNA repair capacity and sensitizes human lung cancer cells to paclitaxel. Biochem. Pharmacol. 2013, 85, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Galan-Moya, E.M.; de la Cruz-Morcillo, M.A.; Valero, M.L.; Callejas-Valera, J.L.; Melgar-Rojas, P.; Losa, J.H.; Salcedo, M.; Fernández-Aramburo, A.; Cajal, S.R.Y.; Sánchez-Prieto, R. Balance between MKK6 and MKK3 mediates p38 MAPK associated resistance to cisplatin in NSCLC. PLoS ONE 2011, 6, e28406. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, C.; Mace, G.; Galban, S.; Fritsch, C.; Vintersten, K.; Black, E.; Gorospe, M.; Nebreda, A.R. Negative feedback regulation of MKK6 mRNA stability by p38alpha mitogen-activated protein kinase. Mol. Cell. Biol. 2003, 23, 370–381. [Google Scholar] [CrossRef]
- Stramucci, L.; Bossi, G. Approaching the challenges of MKK3/p38delta MAPK targeting for therapeutic purpose in col-orectal cancer. J. Exp. Clin. Cancer Res. 2019, 38, 504. [Google Scholar] [CrossRef]
- Bossi, G. MKK3 as oncotarget. Aging 2016, 8, 1–2. [Google Scholar] [CrossRef]
- Zhu, T.; Zhao, Y.; Zhang, J.; Li, L.; Zou, L.; Yao, Y.; Xu, Y. ß-Elemene inhibits proliferation of human glioblastoma cells and causes cell-cycle G0/G1 arrest via mutually compensatory activation of MKK3 and MKK6. Int. J. Oncol. 2011, 38, 419–426. [Google Scholar]
- Zhu, T.; Xu, Y.; Dong, B.; Zhang, J.; Wei, Z.; Yao, Y. β-elemene inhibits proliferation of human glioblastoma cells through the activation of glia maturation factor β and induces sensitization to cisplatin. Oncol. Rep. 2011, 26, 405–413. [Google Scholar]
- MacNeil, A.J.; Jiao, S.C.; McEachern, L.A.; Yang, Y.J.; Dennis, A.; Yu, H.; Xu, Z.; Marshall, J.S.; Lin, T.J. MAPK kinase 3 is a tumor suppressor with reduced copy number in breast cancer. Cancer Res. 2014, 74, 162–172. [Google Scholar] [CrossRef][Green Version]
- Tsai, C.F.; Cheng, Y.K.; Lu, D.Y.; Wang, S.L.; Chang, C.N.; Chang, P.C.; Yeh, W.L. Inhibition of estrogen receptor reduces connexin 43 expression in breast cancers. Toxicol. Appl. Pharmacol. 2018, 338, 182–190. [Google Scholar] [CrossRef]
- Erdem, J.S.; Skaug, V.; Haugen, A.; Zienolddiny, S. Loss of MKK3 and MK2 Copy Numbers in Non-Small Cell Lung Cancer. J. Cancer 2016, 7, 512–515. [Google Scholar] [CrossRef]
- Zhang, B.; Han, H.; Fu, S.; Yang, P.; Gu, Z.; Zhou, Q.; Cao, Z. Dehydroeffusol inhibits gastric cancer cell growth and tumorigenicity by selectively inducing tumor-suppressive endoplasmic reticulum stress and a moderate apoptosis. Biochem. Pharmacol. 2016, 104, 8–18. [Google Scholar] [CrossRef]
- Wang, L.; Chen, C.; Feng, S.; Lei, P.; Tian, J. Mitogen-activated protein kinase kinase 3 induces cell cycle arrest via p38 activation mediated Bmi-1 downregulation in hepatocellular carcinoma. Mol. Med. Rep. 2016, 13, 243–248. [Google Scholar] [CrossRef]
- Lu, W.J.; Chua, M.S.; So, S.K. Suppression of ATAD2 inhibits hepatocellular carcinoma progression through activation of p53- and p38-mediated apoptotic signaling. Oncotarget 2015, 6, 41722–41735. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, Y.; Wei, J.; Jia, W.; Ge, Z.; Zhang, Z.; Liu, X. MicroRNA-21 promotes hepatocellular carcinoma HepG2 cell pro-liferation through repression of mitogen-activated protein kinase-kinase 3. BMC Cancer 2013, 13, 469. [Google Scholar] [CrossRef]
- Lee, C.H.; Ying, T.H.; Chiou, H.L.; Hsieh, S.C.; Wen, S.H.; Chou, R.H.; Hsieh, Y.H. Alpha-mangostin induces apoptosis through activation of reactive oxygen species and ASK1/p38 signaling pathway in cervical cancer cells. Oncotarget 2017, 8, 47425–47439. [Google Scholar] [CrossRef]
- Lin, C.L.; Ying, T.H.; Yang, S.F.; Chiou, H.L.; Chen, Y.S.; Kao, S.H.; Hsieh, Y.H. MTA2 silencing attenuates the metastatic potential of cervical cancer cells by in-hibiting AP1-mediated MMP12 expression via the ASK1/MEK3/p38/YB1 axis. Cell Death Dis. 2021, 12, 451. [Google Scholar] [CrossRef]
- Mansouri, A.; Ridgway, L.D.; Korapati, A.L.; Zhang, Q.; Tian, L.; Wang, Y.; Siddik, Z.H.; Mills, G.B.; Claret, F.X. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J. Biol. Chem. 2003, 278, 19245–19256. [Google Scholar] [CrossRef]
- Han, S.; Wang, Y.; Ma, J.; Wang, Z.; Wang, H.D.; Yuan, Q. Sulforaphene inhibits esophageal cancer progression via suppressing SCD and CDH3 expression, and activating the GADD45B-MAP2K3-p38-p53 feedback loop. Cell Death Dis. 2020, 11, 713. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, W.; Chen, Y.; Lin, Y.; Yang, X.; Wang, H.; Liu, Z. The miR-19b-3p-MAP2K3-STAT3 feedback loop regulates cell proliferation and invasion in esophageal squamous cell carcinoma. Mol. Oncol. 2021, 15, 1566–1583. [Google Scholar] [CrossRef]
- Lu, H.T.; Yang, D.D.; Wysk, M.; Gatti, E.; Mellman, I.; Davis, R.J.; Flavell, R.A. Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO J. 1999, 18, 1845–1857. [Google Scholar] [CrossRef]
- Sun, Y.; Di Zhang, D.; Guo, X.; Li, W.; Li, C.; Luo, J.; Zhou, M.; Xue, L. MKK3 modulates JNK-dependent cell migration and inva-sion. Cell Death Dis. 2019, 10, 149. [Google Scholar] [CrossRef]
- Behren, A.; Muhlen, S.; Sanhueza, G.A.A.; Schwager, C.; Plinkert, P.K.; Huber, P.E.; Abdollahi, A.; Simon, C. Pheno-type-assisted transcriptome analysis identified FOXM1 downstream from RAS-MKK3-p38 to regulate in vitro cellular invasion. Oncogene 2010, 29, 1519–1530. [Google Scholar] [CrossRef]
- Meng, L.; Zhang, Q.; Huang, X. Comprehensive Analysis of 5-Methylcytosine Profiles of Messenger RNA in Human High-Grade Serous Ovarian Cancer by MeRIP Sequencing. Cancer Manag. Res. 2021, 13, 6005. [Google Scholar] [CrossRef]
- Conceição, A.L.; Babeto, E.; Candido, N.M.; Franco, F.C.; de Zuccari, D.A.C.; Bonilha, J.L.; Cordeiro, J.A.; Calmon, M.F.; Rahal, P. Differential Expression of ADAM23, CDKN2A (P16), MMP14 and VIM Associated with Giant Cell Tumor of Bone. J. Cancer 2015, 6, 593–603. [Google Scholar] [CrossRef]
- Yang, X.; Fan, D.; Troha, A.H.; Ahn, H.M.; Qian, K.; Liang, B.; Du, Y.; Fu, H.; Ivanov, A.A. Discovery of the first chemical tools to regulate MKK3-mediated MYC activation in cancer. Bioorgan. Med. Chem. 2021, 45, 116324. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piastra, V.; Pranteda, A.; Bossi, G. Dissection of the MKK3 Functions in Human Cancer: A Double-Edged Sword? Cancers 2022, 14, 483. https://doi.org/10.3390/cancers14030483
Piastra V, Pranteda A, Bossi G. Dissection of the MKK3 Functions in Human Cancer: A Double-Edged Sword? Cancers. 2022; 14(3):483. https://doi.org/10.3390/cancers14030483
Chicago/Turabian StylePiastra, Valentina, Angelina Pranteda, and Gianluca Bossi. 2022. "Dissection of the MKK3 Functions in Human Cancer: A Double-Edged Sword?" Cancers 14, no. 3: 483. https://doi.org/10.3390/cancers14030483
APA StylePiastra, V., Pranteda, A., & Bossi, G. (2022). Dissection of the MKK3 Functions in Human Cancer: A Double-Edged Sword? Cancers, 14(3), 483. https://doi.org/10.3390/cancers14030483