
Stromal Characteristics and Impact on New Therapies for Metastatic Triple-Negative Breast Cancer

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Immunotherapy
3. Tumor Microenvironment and Immune Modulation
4. Antibody–Drug Conjugates
5. Tumor Microenvironment and ADCs
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef] [Green Version]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological complete response and long-term clinical benefit in breast cancer: The CTNeoBC pooled analysis. Lancet 2014, 384, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Bagegni, N.A.; Tao, Y.; Ademuyiwa, F.O. Clinical outcomes with neoadjuvant versus adjuvant chemotherapy for triple negative breast cancer: A report from the National Cancer Database. PLoS ONE 2019, 14, e0222358. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13 Pt 1, 4429–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, N.U.; Claus, E.; Sohl, J.; Razzak, A.R.; Arnaout, A.; Winer, E.P. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: High incidence of central nervous system metastases. Cancer 2008, 113, 2638–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [Green Version]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 11, 620773. [Google Scholar] [CrossRef]
- Deligne, C.; Midwood, K.S. Macrophages and Extracellular Matrix in Breast Cancer: Partners in Crime or Protective Allies? Front. Oncol. 2021, 11, 620773. [Google Scholar] [CrossRef] [PubMed]
- Yue, B. Biology of the extracellular matrix: An overview. J. Glaucoma 2014, 23 (Suppl. 1), S20–S23. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef] [Green Version]
- Henke, E.; Nandigama, R.; Ergun, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2019, 6, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, T.; Packham, G.; Murphy, L.B.; Bateman, A.C.; Conti, J.A.; Fine, D.R.; Johnson, C.D.; Benyon, R.C.; Iredale, J.P. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004, 10, 7427–7437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conklin, M.W.; Eickhoff, J.C.; Riching, K.M.; Pehlke, C.A.; Eliceiri, K.W.; Provenzano, P.P.; Friedl, A.; Keely, P.J. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am. J. Pathol. 2011, 178, 1221–1232. [Google Scholar] [CrossRef]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunderlikova, B. Clinical significance of immunohistochemically detected extracellular matrix proteins and their spatial distribution in primary cancer. Crit. Rev. Oncol. Hematol. 2016, 105, 127–144. [Google Scholar] [CrossRef]
- Hanley, C.J.; Noble, F.; Ward, M.; Bullock, M.; Drifka, C.; Mellone, M.; Manousopoulou, A.; Johnston, H.E.; Hayden, A.; Thirdborough, S.; et al. A subset of myofibroblastic cancer-associated fibroblasts regulate collagen fiber elongation, which is prognostic in multiple cancers. Oncotarget 2016, 7, 6159–6174. [Google Scholar] [CrossRef] [Green Version]
- Esbona, K.; Yi, Y.; Saha, S.; Yu, M.; Van Doorn, R.R.; Conklin, M.W.; Graham, D.S.; Wisinski, K.B.; Ponik, S.M.; Eliceiri, K.W.; et al. The Presence of Cyclooxygenase 2, Tumor-Associated Macrophages, and Collagen Alignment as Prognostic Markers for Invasive Breast Carcinoma Patients. Am. J. Pathol. 2018, 188, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Tomko, L.A.; Hill, R.C.; Barrett, A.; Szulczewski, J.M.; Conklin, M.W.; Eliceiri, K.W.; Keely, P.J.; Hansen, K.C.; Ponik, S.M. Targeted matrisome analysis identifies thrombospondin-2 and tenascin-C in aligned collagen stroma from invasive breast carcinoma. Sci. Rep. 2018, 8, 12941. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Hastings, J.F.; Skhinas, J.N.; Fey, D.; Croucher, D.R.; Cox, T.R. The extracellular matrix as a key regulator of intracellular signalling networks. Br. J. Pharmacol. 2019, 176, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Jain, R.K. Normalizing tumor microenvironment to treat cancer: Bench to bedside to biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.X.; Chauhan, V.P.; Posada, J.; Ng, M.R.; Wu, M.W.; Adstamongkonkul, P.; Huang, P.; Lindeman, N.; Langer, R.; Jain, R.K. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 4558–4566. [Google Scholar] [CrossRef] [Green Version]
- Bates, A.M.; O’Leary, K.A.; Emma, S.; Nystuen, E.; Sumiec, E.G.; Schuler, L.A.; Morris, Z.S. Enhancing immunogenicity in immunologically cold ER+ breast cancer using estrogen receptor blockade and radiation therapy. In Proceedings of the AACR 2020 Virtual Meeting-II Proceedings, Philadelphia, PA, USA, 27–29 April and 22–24 June 2020. [Google Scholar]
- Primeau, A.J.; Rendon, A.; Hedley, D.; Lilge, L.; Tannock, I.F. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin. Cancer Res. 2005, 11 Pt 1, 8782–8788. [Google Scholar] [CrossRef] [Green Version]
- Goetz, J.G.; Minguet, S.; Navarro-Lerida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibanez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riching, K.M.; Cox, B.L.; Salick, M.R.; Pehlke, C.; Riching, A.S.; Ponik, S.M.; Bass, B.R.; Crone, W.C.; Jiang, Y.; Weaver, A.M.; et al. 3D collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys. J. 2014, 107, 2546–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudin, M.J.; Jonas, O.; Kosciuk, T.; Broye, L.C.; Guido, B.C.; Wyckoff, J.; Riquelme, D.; Lamar, J.M.; Asokan, S.B.; Whittaker, C.; et al. Tumor Cell-Driven Extracellular Matrix Remodeling Drives Haptotaxis during Metastatic Progression. Cancer Discov. 2016, 6, 516–531. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Schwenzer, A.; Rupp, T.; Murdamoothoo, D.; Vegliante, R.; Lefebvre, O.; Klein, A.; Hussenet, T.; Orend, G. Tenascin-C Promotes Tumor Cell Migration and Metastasis through Integrin alpha9beta1-Mediated YAP Inhibition. Cancer Res. 2018, 78, 950–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, Y.K.; Choi, J.E.; Kang, S.H.; Lee, S.J. Epithelial-Mesenchymal Transition Phenotype Is Associated with Clinicopathological Factors That Indicate Aggressive Biological Behavior and Poor Clinical Outcomes in Invasive Breast Cancer. J. Breast Cancer 2015, 18, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, L.; Hong, D.; Chen, Z.; Zhang, J.; Fu, L.; Pan, D.; Zhang, Y.; Xu, Y.; Gan, S.; et al. Baicalein inhibits fibronectin-induced epithelial-mesenchymal transition by decreasing activation and upregulation of calpain-2. Cell Death Dis. 2019, 10, 341. [Google Scholar] [CrossRef]
- Wang, Z.; Xiong, S.; Mao, Y.; Chen, M.; Ma, X.; Zhou, X.; Ma, Z.; Liu, F.; Huang, Z.; Luo, Q.; et al. Periostin promotes immunosuppressive premetastatic niche formation to facilitate breast tumour metastasis. J. Pathol. 2016, 239, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Barcus, C.E.; O’Leary, K.A.; Brockman, J.L.; Rugowski, D.E.; Liu, Y.; Garcia, N.; Yu, M.; Keely, P.J.; Eliceiri, K.W.; Schuler, L.A. Elevated collagen-I augments tumor progressive signals, intravasation and metastasis of prolactin-induced estrogen receptor alpha positive mammary tumor cells. Breast Cancer Res. 2017, 19, 9. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Malik, G.; Knowles, L.M.; Dhir, R.; Xu, S.; Yang, S.; Ruoslahti, E.; Pilch, J. Plasma fibronectin promotes lung metastasis by contributions to fibrin clots and tumor cell invasion. Cancer Res. 2010, 70, 4327–4334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Corsa, C.A.; Ponik, S.M.; Prior, J.L.; Piwnica-Worms, D.; Eliceiri, K.W.; Keely, P.J.; Longmore, G.D. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat. Cell Biol. 2013, 15, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, S.; Inman, D.R.; Li, W.J.; Ponik, S.M.; Keely, P.J. Mechano-Signal Transduction in Mesenchymal Stem Cells Induces Prosaposin Secretion to Drive the Proliferation of Breast Cancer Cells. Cancer Res. 2017, 77, 6179–6189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, S.V.; Grither, W.R.; Brenot, A.; Hwang, P.Y.; Barcus, C.E.; Ernst, M.; Pence, P.; Walter, C.; Pathak, A.; Longmore, G.D. DDR2 controls breast tumor stiffness and metastasis by regulating integrin mediated mechanotransduction in CAFs. eLife 2019, 8, e45508. [Google Scholar] [CrossRef]
- Liu, J.; Liao, S.; Diop-Frimpong, B.; Chen, W.; Goel, S.; Naxerova, K.; Ancukiewicz, M.; Boucher, Y.; Jain, R.K.; Xu, L. TGF-beta blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2012, 109, 16618–16623. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.D.; Cabral, H.; Stylianopoulos, T.; Jain, R.K. Improving cancer immunotherapy using nanomedicines: Progress, opportunities and challenges. Nat. Rev. Clin. Oncol. 2020, 17, 251–266. [Google Scholar] [CrossRef]
- Sangaletti, S.; Chiodoni, C.; Tripodo, C.; Colombo, M.P. The good and bad of targeting cancer-associated extracellular matrix. Curr. Opin. Pharmacol. 2017, 35, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Lampi, M.C.; Reinhart-King, C.A. Targeting extracellular matrix stiffness to attenuate disease: From molecular mechanisms to clinical trials. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Mushtaq, M.U.; Papadas, A.; Pagenkopf, A.; Flietner, E.; Morrow, Z.; Chaudhary, S.G.; Asimakopoulos, F. Tumor matrix remodeling and novel immunotherapies: The promise of matrix-derived immune biomarkers. J. Immunother. Cancer 2018, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Senkus, E.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rutgers, E.; Zackrisson, S.; Cardoso, F.; Committee, E.G. Primary breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26 (Suppl. 5), v8–v30. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology—Breast Cancer. Page BINV-10. Available online: https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf (accessed on 18 January 2022).
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; Curigliano, G.; Aapro, M.S.; André, F.; Barrios, C.H.; Bergh, J.; Bhattacharyya, G.S.; Biganzoli, L.; et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann. Oncol. 2020, 31, 1623–1649. [Google Scholar] [CrossRef]
- Savas, P.; Salgado, R.; Denkert, C.; Sotiriou, C.; Darcy, P.K.; Smyth, M.J.; Loi, S. Clinical relevance of host immunity in breast cancer: From TILs to the clinic. Nat. Rev. Clin. Oncol. 2016, 13, 228–241. [Google Scholar] [CrossRef]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Sabatier, R.; Finetti, P.; Mamessier, E.; Adelaide, J.; Chaffanet, M.; Ali, H.R.; Viens, P.; Caldas, C.; Birnbaum, D.; Bertucci, F. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget 2015, 6, 5449–5464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Diamond, J.R.; Hamilton, E.; Pohlmann, P.R.; Tolaney, S.M.; Chang, C.W.; Zhang, W.; Iizuka, K.; Foster, P.G.; Molinero, L.; et al. Atezolizumab Plus nab-Paclitaxel in the Treatment of Metastatic Triple-Negative Breast Cancer With 2-Year Survival Follow-up: A Phase 1b Clinical Trial. JAMA Oncol. 2019, 5, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.; Loi, S.; Toppmeyer, D.; Cescon, D.W.; De Laurentiis, M.; Nanda, R.; Winer, E.P.; Mukai, H.; Tamura, K.; Armstrong, A.; et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: Cohort B of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 405–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Soliman, H.H. nab-Paclitaxel as a potential partner with checkpoint inhibitors in solid tumors. Onco Targets Ther. 2017, 10, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emens, L.A.; Adams, S.; Barrios, C.H.; Diéras, V.; Iwata, H.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Winer, E.P.; Patel, S.; et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. 2021, 32, 983–993. [Google Scholar] [CrossRef]
- Miles, D.; Gligorov, J.; Andre, F.; Cameron, D.; Schneeweiss, A.; Barrios, C.; Xu, B.; Wardley, A.; Kaen, D.; Andrade, L.; et al. Primary results from IMpassion131, a double-blind placebo-controlled randomised phase III trial of first-line paclitaxel (PAC) ± atezolizumab (atezo) for unresectable locally advanced/metastatic triple-negative breast cancer (mTNBC). Ann. Oncol. 2020, 31, S1147. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Kulangara, K.; Zhang, N.; Corigliano, E.; Guerrero, L.; Waldroup, S.; Jaiswal, D.; Ms, M.J.; Shah, S.; Hanks, D.; Wang, J.; et al. Clinical Utility of the Combined Positive Score for Programmed Death Ligand-1 Expression and the Approval of Pembrolizumab for Treatment of Gastric Cancer. Arch. Pathol. Lab. Med. 2019, 143, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Rugo, H.S. LBA16-KEYNOTE-355: Final results from a randomized, double-blind phase III study of first-line pembrolizumab + chemotherapy vs placebo + chemotherapy for metastatic TNBC. Ann. Oncol. 2021, 32 (Suppl. 5), S1283–S1346. [Google Scholar]
- Vennapusa, B.; Baker, B.; Kowanetz, M.; Boone, J.; Menzl, I.; Bruey, J.M.; Fine, G.; Mariathasan, S.; McCaffery, I.; Mocci, S.; et al. Development of a PD-L1 Complementary Diagnostic Immunohistochemistry Assay (SP142) for Atezolizumab. Appl. Immunohistochem. Mol. Morphol. 2019, 27, 92–100. [Google Scholar] [CrossRef]
- Torlakovic, E.; Lim, H.J.; Adam, J.; Barnes, P.; Bigras, G.; Chan, A.W.H.; Cheung, C.C.; Chung, J.H.; Couture, C.; Fiset, P.O.; et al. “Interchangeability” of PD-L1 immunohistochemistry assays: A meta-analysis of diagnostic accuracy. Mod. Pathol. 2020, 33, 4–17. [Google Scholar] [CrossRef]
- Dill, E.A.; Gru, A.A.; Atkins, K.A.; Friedman, L.A.; Moore, M.E.; Bullock, T.N.; Cross, J.V.; Dillon, P.M.; Mills, A.M. PD-L1 Expression and Intratumoral Heterogeneity Across Breast Cancer Subtypes and Stages: An Assessment of 245 Primary and 40 Metastatic Tumors. Am. J. Surg. Pathol. 2017, 41, 334–342. [Google Scholar] [CrossRef]
- Boman, C.; Zerdes, I.; Martensson, K.; Bergh, J.; Foukakis, T.; Valachis, A.; Matikas, A. Discordance of PD-L1 status between primary and metastatic breast cancer: A systematic review and meta-analysis. Cancer Treat. Rev. 2021, 99, 102257. [Google Scholar] [CrossRef]
- Schmid, P.; Dent, R.; O’Shaughnessy, J. Pembrolizumab for Early Triple-Negative Breast Cancer. Reply. N. Engl. J. Med. 2020, 382, e108. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P. KEYNOTE-522: Phase III study of neoadjuvant pembrolizumab + chemotherapy vs. placebo + chemotherapy, followed by adjuvant pembrolizumab vs. placebo for early-stage TNBC. In Proceedings of the ESMO Virtual Plenaries 2021, London, UK, 15 July 2021. [Google Scholar]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.V.; Blohmer, J.U.; Grischke, E.M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: Clinical results and biomarker analysis of GeparNuevo study. Ann. Oncol. 2019, 30, 1279–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianni, L.; Huang, C.S.; Egle, D.; Bermejo, B.; Zamagni, C.; Thill, M.; Anton, A.; Zambelli, S.; Bianchini, G.; Russo, S.; et al. Pathologic complete response (pCR) to neoadjuvant treatment with or without atezolizumab in triple negative, early high-risk and locally advanced breast cancer. NeoTRIPaPDL1 Michelangelo randomized study. In Proceedings of the San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 10–14 December 2019. [Google Scholar]
- Bai, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Triple-negative breast cancer therapeutic resistance: Where is the Achilles’ heel? Cancer Lett. 2021, 497, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Hammerl, D.; Martens, J.W.M.; Timmermans, M.; Smid, M.; Trapman-Jansen, A.M.; Foekens, R.; Isaeva, O.I.; Voorwerk, L.; Balcioglu, H.E.; Wijers, R.; et al. Spatial immunophenotypes predict response to anti-PD1 treatment and capture distinct paths of T cell evasion in triple negative breast cancer. Nat. Commun. 2021, 12, 5668. [Google Scholar] [CrossRef]
- Janse van Rensburg, H.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef] [Green Version]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Zhao, M.; Khaldoyanidi, S.K.; Discipio, R.G. The chemokine CCL18 causes maturation of cultured monocytes to macrophages in the M2 spectrum. Immunology 2012, 135, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Pollard, J.W. Therapeutic potential of chemokine signal inhibition for metastatic breast cancer. Pharmacol. Res. 2015, 100, 266–270. [Google Scholar] [CrossRef] [Green Version]
- Esbona, K.; Inman, D.; Saha, S.; Jeffery, J.; Schedin, P.; Wilke, L.; Keely, P. COX-2 modulates mammary tumor progression in response to collagen density. Breast Cancer Res. 2016, 18, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Wuest, M.; Kuchar, M.; Sharma, S.K.; Richter, S.; Hamann, I.; Wang, M.; Vos, L.; Mackey, J.R.; Wuest, F.; Loser, R. Targeting lysyl oxidase for molecular imaging in breast cancer. Breast Cancer Res. 2015, 17, 107. [Google Scholar] [CrossRef] [Green Version]
- Takai, K.; Le, A.; Weaver, V.M.; Werb, Z. Targeting the cancer associated fibroblasts as a treatment in triple negative breast cancer. Oncotarget 2016, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, S.; Chen, I.X.; Liu, Y.; Ramjiawan, R.R.; Leung, C.H.; Gerweck, L.E.; Fukumura, D.; Loeffler, J.S.; Jain, R.K.; et al. Combining losartan with radiotherapy increases tumor control and inhibits lung metastases from a HER2/neu-positive orthotopic breast cancer model. Radiat. Oncol. 2021, 16, 48. [Google Scholar] [CrossRef]
- Kuroda, H.; Jamiyan, T.; Yamaguchi, R.; Kakumoto, A.; Abe, A.; Harada, O.; Masunaga, A. Tumor microenvironment in triple-negative breast cancer: The correlation of tumor-associated macrophages and tumor-infiltrating lymphocytes. Clin. Transl. Oncol. 2021, 23, 2513–2525. [Google Scholar] [CrossRef]
- Yu, T.; Di, G. Role of tumor microenvironment in triple-negative breast cancer and its prognostic significance. Chin. J. Cancer Res. 2017, 29, 237–252. [Google Scholar] [CrossRef]
- Bae, Y.K.; Kim, A.; Kim, M.K.; Choi, J.E.; Kang, S.H.; Lee, S.J. Fibronectin expression in carcinoma cells correlates with tumor aggressiveness and poor clinical outcome in patients with invasive breast cancer. Hum. Pathol. 2013, 44, 2028–2037. [Google Scholar] [CrossRef]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arwert, E.N.; Harney, A.S.; Entenberg, D.; Wang, Y.; Sahai, E.; Pollard, J.W.; Condeelis, J.S. A Unidirectional Transition from Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Rep. 2018, 23, 1239–1248. [Google Scholar] [CrossRef] [Green Version]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Reyes Torres, I.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harney, A.S.; Karagiannis, G.S.; Pignatelli, J.; Smith, B.D.; Kadioglu, E.; Wise, S.C.; Hood, M.M.; Kaufman, M.D.; Leary, C.B.; Lu, W.P.; et al. The Selective Tie2 Inhibitor Rebastinib Blocks Recruitment and Function of Tie2(Hi) Macrophages in Breast Cancer and Pancreatic Neuroendocrine Tumors. Mol. Cancer Ther. 2017, 16, 2486–2501. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wang, X.H.; Zhao, Y.X.; Chen, C.; Xu, X.Y.; Sun, Q.; Wu, H.Y.; Chen, M.; Sang, J.F.; Su, L.; et al. Cancer-Associated Fibroblasts Correlate with Tumor-Associated Macrophages Infiltration and Lymphatic Metastasis in Triple Negative Breast Cancer Patients. J. Cancer 2018, 9, 4635–4641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyrol, S.; Raccurt, M.; Gerard, F.; Gleyzal, C.; Grimaud, J.A.; Sommer, P. Lysyl oxidase gene expression in the stromal reaction to in situ and invasive ductal breast carcinoma. Am. J. Pathol. 1997, 150, 497–507. [Google Scholar]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disis, M.L.; Stanton, S.E. Triple-Negative Breats Cancer: Immune Modulation as the New Treatment Paradigm; American Society of Clinical Oncology: Alexandria, VA, USA, 2015; p. 6. [Google Scholar]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benner, B.; Scarberry, L.; Suarez-Kelly, L.P.; Duggan, M.C.; Campbell, A.R.; Smith, E.; Lapurga, G.; Jiang, K.; Butchar, J.P.; Tridandapani, S.; et al. Generation of monocyte-derived tumor-associated macrophages using tumor-conditioned media provides a novel method to study tumor-associated macrophages in vitro. J. Immunother. Cancer 2019, 7, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dollin, Y.; Rubin, J.; Carvajal, R.D.; Rached, H.; Nitzkorski, J.R. Pembrolizumab and tavokinogene telseplasmid electroporation in metastatic melanoma. Int. J. Surg. Case Rep. 2020, 77, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Kim, T.M.; Vicente, D.; Felip, E.; Lee, D.H.; Lee, K.H.; Lin, C.C.; Flor, M.J.; Di Nicola, M.; Alvarez, R.M.; et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-beta and PD-L1, in Second-Line Treatment of Patients With NSCLC: Results From an Expansion Cohort of a Phase 1 Trial. J. Thorac. Oncol. 2020, 15, 1210–1222. [Google Scholar] [CrossRef] [PubMed]
- Greiner, J.W.; Morillon, Y.M., 2nd; Schlom, J. NHS-IL12, a Tumor-Targeting Immunocytokine. Immunotargets Ther. 2021, 10, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M.; Cardillo, T.M.; Govindan, S.V.; Rossi, E.A.; Sharkey, R.M. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget 2015, 6, 22496–22512. [Google Scholar] [CrossRef] [Green Version]
- Lenárt, S.; Lenárt, P.; Šmarda, J.; Remšík, J.; Souček, K.; Beneš, P. Trop2: Jack of All Trades, Master of None. Cancers 2020, 12, 3328. [Google Scholar] [CrossRef]
- Zaman, S.; Jadid, H.; Denson, A.C.; Gray, J.E. Targeting Trop-2 in solid tumors: Future prospects. Onco Targets Ther. 2019, 12, 1781–1790. [Google Scholar] [CrossRef] [Green Version]
- Ambrogi, F.; Fornili, M.; Boracchi, P.; Trerotola, M.; Relli, V.; Simeone, P.; La Sorda, R.; Lattanzio, R.; Querzoli, P.; Pedriali, M.; et al. Trop-2 is a determinant of breast cancer survival. PLoS ONE 2014, 9, e96993. [Google Scholar] [CrossRef] [Green Version]
- Mathijssen, R.H.; van Alphen, R.J.; Verweij, J.; Loos, W.J.; Nooter, K.; Stoter, G.; Sparreboom, A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin. Cancer Res. 2001, 7, 2182–2194. [Google Scholar] [PubMed]
- Moon, S.J.; Govindan, S.V.; Cardillo, T.M.; D’Souza, C.A.; Hansen, H.J.; Goldenberg, D.M. Antibody conjugates of 7-ethyl-10-hydroxycamptothecin (SN-38) for targeted cancer chemotherapy. J. Med. Chem. 2008, 51, 6916–6926. [Google Scholar] [CrossRef] [Green Version]
- Criscitiello, C.; Morganti, S.; Curigliano, G. Antibody-drug conjugates in solid tumors: A look into novel targets. J. Hematol. Oncol. 2021, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M.; Stein, R.; Sharkey, R.M. The emergence of trophoblast cell-surface antigen 2 (TROP-2) as a novel cancer target. Oncotarget 2018, 9, 28989–29006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starodub, A.N.; Ocean, A.J.; Shah, M.A.; Guarino, M.J.; Picozzi, V.J.; Vahdat, L.T.; Thomas, S.S.; Govindan, S.V.; Maliakal, P.P.; Wegener, W.A.; et al. First-in-Human Trial of a Novel Anti-Trop-2 Antibody-SN-38 Conjugate, Sacituzumab Govitecan, for the Treatment of Diverse Metastatic Solid Tumors. Clin. Cancer Res. 2015, 21, 3870–3878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardia, A.; Mayer, I.A.; Diamond, J.R.; Moroose, R.L.; Isakoff, S.J.; Starodub, A.N.; Shah, N.C.; O’Shaughnessy, J.; Kalinsky, K.; Guarino, M.; et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients With Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2017, 35, 2141–2148. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- Rose, A.A.; Grosset, A.A.; Dong, Z.; Russo, C.; Macdonald, P.A.; Bertos, N.R.; St-Pierre, Y.; Simantov, R.; Hallett, M.; Park, M.; et al. Glycoprotein nonmetastatic B is an independent prognostic indicator of recurrence and a novel therapeutic target in breast cancer. Clin. Cancer Res. 2010, 16, 2147–2156. [Google Scholar] [CrossRef] [Green Version]
- Yardley, D.A.; Weaver, R.; Melisko, M.E.; Saleh, M.N.; Arena, F.P.; Forero, A.; Cigler, T.; Stopeck, A.; Citrin, D.; Oliff, I.; et al. EMERGE: A Randomized Phase II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Advanced Glycoprotein NMB-Expressing Breast Cancer. J. Clin. Oncol. 2015, 33, 1609–1619. [Google Scholar] [CrossRef]
- Diéras, V.; Deluche, E.; Lusque, A. Trastuzumab deruxtecan (T-DXd) for advanced breast cancer patients (ABC), regardless HER2 status: A phase II study with biomarkers analysis (DAISY). In Proceedings of the 2021 San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 7–10 December 2021. [Google Scholar]
- Epenetos, A.A.; Snook, D.; Durbin, H.; Johnson, P.M.; Taylor-Papadimitriou, J. Limitations of radiolabeled monoclonal antibodies for localization of human neoplasms. Cancer Res. 1986, 46, 3183–3191. [Google Scholar]
- Bartelink, I.H.; Jones, E.F.; Shahidi-Latham, S.K.; Lee, P.R.E.; Zheng, Y.; Vicini, P.; van ’t Veer, L.; Wolf, D.; Iagaru, A.; Kroetz, D.L.; et al. Tumor Drug Penetration Measurements Could Be the Neglected Piece of the Personalized Cancer Treatment Puzzle. Clin. Pharmacol. Ther. 2019, 106, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Kagan, H.M.; Trackman, P.C. Properties and function of lysyl oxidase. Am. J. Respir. Cell Mol. Biol. 1991, 5, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Rossow, L.; Veitl, S.; Vorlova, S.; Wax, J.K.; Kuhn, A.E.; Maltzahn, V.; Upcin, B.; Karl, F.; Hoffmann, H.; Gatzner, S.; et al. LOX-catalyzed collagen stabilization is a proximal cause for intrinsic resistance to chemotherapy. Oncogene 2018, 37, 4921–4940. [Google Scholar] [CrossRef] [Green Version]
- Deonarain, M.P.; Yahioglu, G.; Stamati, I.; Marklew, J. Emerging formats for next-generation antibody drug conjugates. Expert Opin. Drug Discov. 2015, 10, 463–481. [Google Scholar] [CrossRef]
- Lucas, A.T.; Price, L.S.L.; Schorzman, A.N.; Storrie, M.; Piscitelli, J.A.; Razo, J.; Zamboni, W.C. Factors Affecting the Pharmacology of Antibody-Drug Conjugates. Antibodies 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xenaki, K.T.; Dorrestijn, B.; Muns, J.A.; Adamzek, K.; Doulkeridou, S.; Houthoff, H.; Oliveira, S.; van Bergen En Henegouwen, P.M. Homogeneous tumor targeting with a single dose of HER2-targeted albumin-binding domain-fused nanobody-drug conjugates results in long-lasting tumor remission in mice. Theranostics 2021, 11, 5525–5538. [Google Scholar] [CrossRef]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer-drug conjugate therapeutics: Advances, insights and prospects. Nat. Rev. Drug Discov. 2019, 18, 273–294. [Google Scholar] [CrossRef] [PubMed]
- Dal Corso, A.; Gebleux, R.; Murer, P.; Soltermann, A.; Neri, D. A non-internalizing antibody-drug conjugate based on an anthracycline payload displays potent therapeutic activity in vivo. J. Control. Release 2017, 264, 211–218. [Google Scholar] [CrossRef] [PubMed]
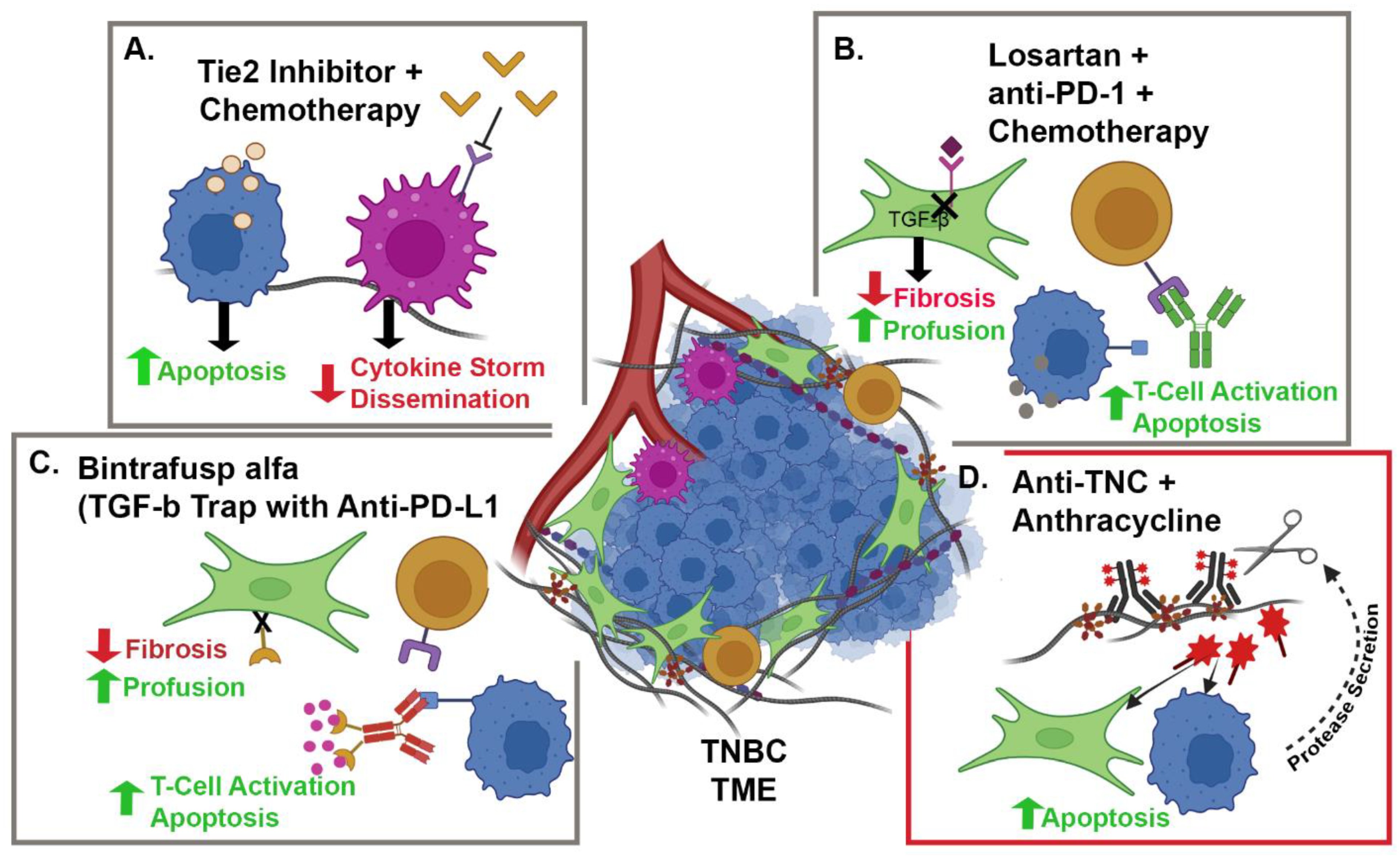

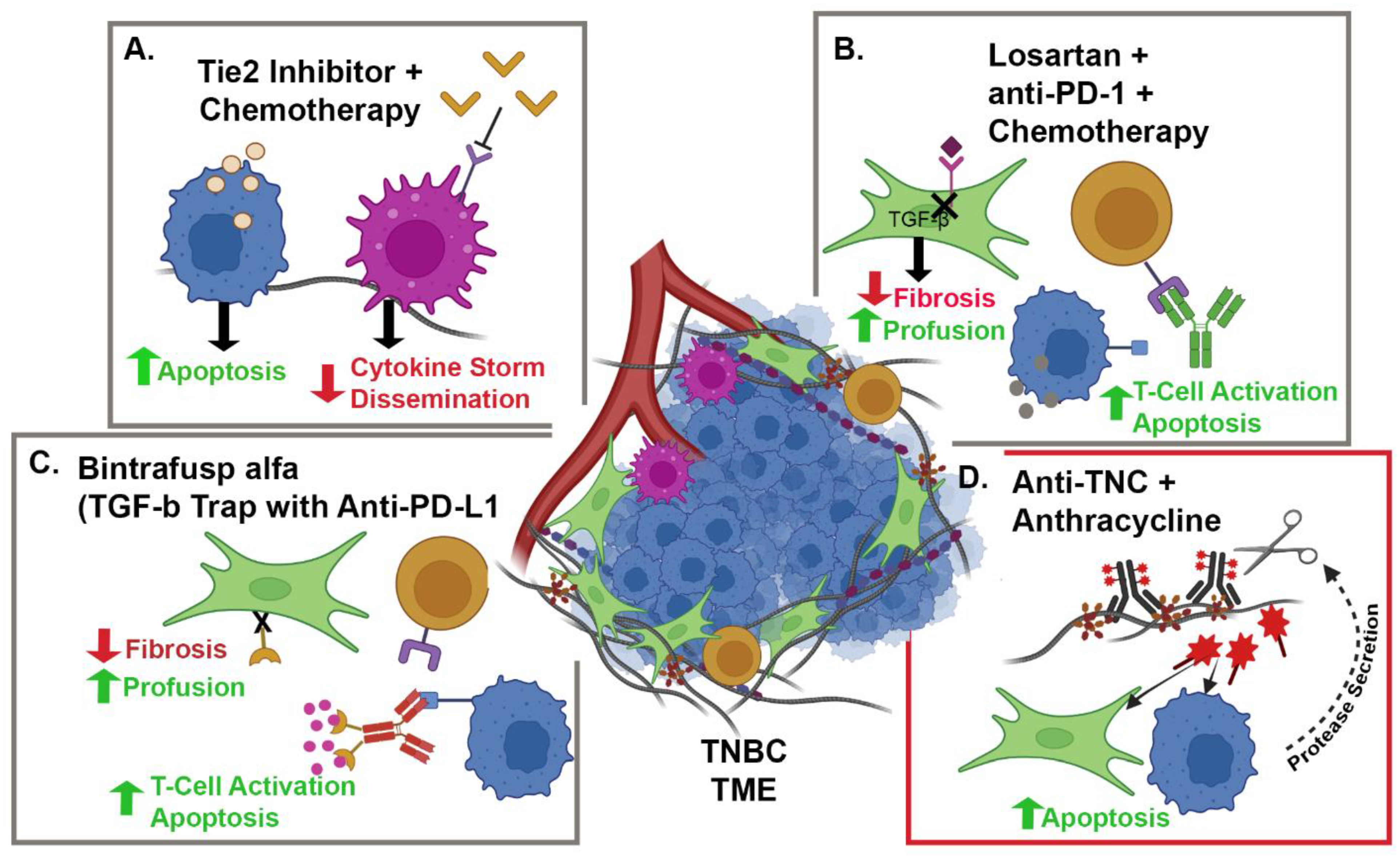{kind=link}
| Study | Study Groups | Line of Therapy | Total Number of Patients | Study Design | Progression-Free Survival | Overall Survival | Response Rate |
|---|---|---|---|---|---|---|---|
| Immunotherapy | |||||||
| IMPassion130 % | Atezolizumab + nab-paclitaxel vs. placebo + nab-paclitaxel | 1st | 902 | Phase III, randomized, double-blind, placebo-controlled trial | 7.2 vs. 5.5 months (p = 0.002) | 21.3 vs. 17.6 months (p = 0.08) | 56.0% vs. 45.9% |
| IMPassion131 ^ | Atezolizumab + paclitaxel vs. placebo + paclitaxel | 1st | 651 | Phase III randomized, double-blind, placebo-controlled trial | 5.7 vs. 5.6 months | 19.2 vs. 22.8 months | |
| KEYNOTE-355 & | Pembrolizumab + chemotherapy $ vs. placebo vs. chemotherapy | 1st | 847 | Phase III randomized, double-blind, placebo-controlled trial | 9.7 vs. 5.6 * months | 23.0 vs. 16.1 * months (p = 0.009) | 52.7% * |
| Sacituzumab govitecan | |||||||
| ASCENT + | Sacituzumab govitecan vs. chemotherapy # | ≥2 prior | 468 | Phase III, Randomized | 5.6 vs. 1.7 months (p < 0.001) | 12.1 vs. 6.7 months (p < 0.001) | 35% vs. 5% |
| Clinical Trial Identifier | Study Groups | Cancer | Stromal Target | Study Design | Pre-Clinical Reference |
|---|---|---|---|---|---|
| Immunotherapy | |||||
| NCT05097248 | Camrelizumab + Liposomal Doxorubicin + Losartan | TNBC | CAFs and PD-1 | Phase II, single-arm, open-label, prospective clinical trial | 88 |
| NCT02824575 | Paclitaxel + Rebastinib vs. Eribulin + Rebastinib | BC | TIE-2 Expressing Macrophages | Phase I non-randomized, open-label clinical trial | 95 |
| NCT03567720 | Pembrolizumab + Tavo + EP vs. Pembrolizumab + Tavo + EP + Nab-Paclitaxel | TNBC | IL-12 and PD-L1 | Phase II non-randomized, open-label, multicohort clinical trial | 97 |
| NCT04756505 | Bintrafusp alfa + NHS-IL-12 + Radiation | HR+, HER2 − BC | IL-12, PD-L1 and TGFβ | Phase I, open-label clinical trial | 98 and 99 |
| NCT03620201 | Bintrafusp alfa + chemotherapy | HER2+ BC | PD-L1 and TGFβ | Phase I, open-label clinical trial | 98 and 99 |
| NCT04489940 | Bintrafusp alfa | TNBC | PD-L1 and TGFβ | Phase II, open-label clinical trial | 98 and 99 |
| Antibody–Drug Conjugates | |||||
| NCT04969835 | AVA6000 | BC and Solid Tumors | FAP | Phase I, open-label, 3 + 3 clinical trial | Avacta Life Science Ltd. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fertal, S.A.; Poterala, J.E.; Ponik, S.M.; Wisinski, K.B. Stromal Characteristics and Impact on New Therapies for Metastatic Triple-Negative Breast Cancer. Cancers 2022, 14, 1238. https://doi.org/10.3390/cancers14051238
Fertal SA, Poterala JE, Ponik SM, Wisinski KB. Stromal Characteristics and Impact on New Therapies for Metastatic Triple-Negative Breast Cancer. Cancers. 2022; 14(5):1238. https://doi.org/10.3390/cancers14051238
Chicago/Turabian StyleFertal, Shelby A., Johanna E. Poterala, Suzanne M. Ponik, and Kari B. Wisinski. 2022. "Stromal Characteristics and Impact on New Therapies for Metastatic Triple-Negative Breast Cancer" Cancers 14, no. 5: 1238. https://doi.org/10.3390/cancers14051238
APA StyleFertal, S. A., Poterala, J. E., Ponik, S. M., & Wisinski, K. B. (2022). Stromal Characteristics and Impact on New Therapies for Metastatic Triple-Negative Breast Cancer. Cancers, 14(5), 1238. https://doi.org/10.3390/cancers14051238