
The NAMPT Inhibitor FK866 Increases Metformin Sensitivity in Pancreatic Cancer Cells

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Dose-Response & Growth Assays
2.3. NAD/NADH Quantification
2.4. DCFDA Measurements
2.5. Seahorse Analysis
2.6. Animal Experiments
2.7. Tissue Histology
2.8. Total RNA Isolation and Sequencing
2.9. RNA-Seq Data Analysis
2.10. qPCR
2.11. Immunoblots
3. Results
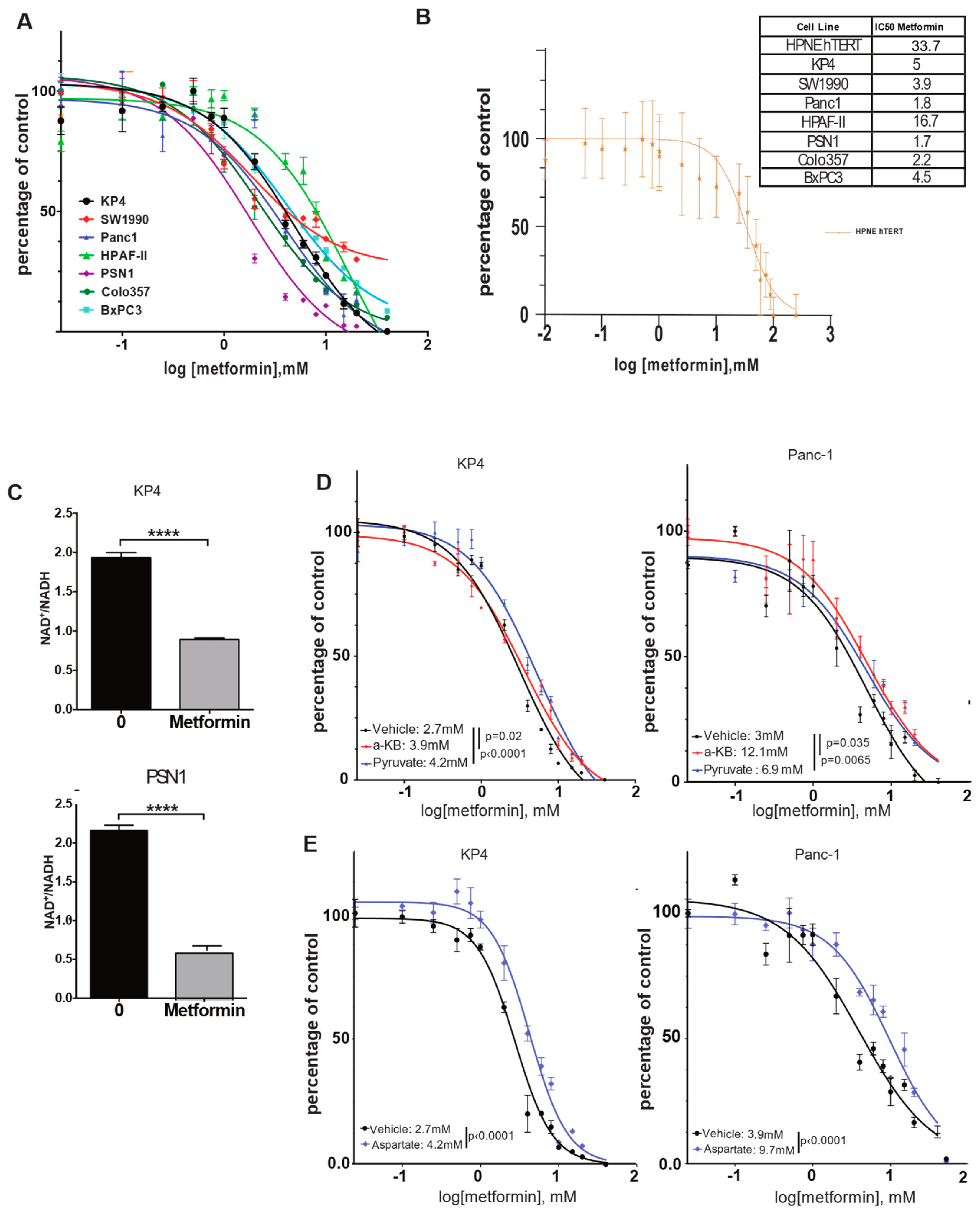
3.1. The NAD+/NADH Ratio Determines Metformin Sensitivity
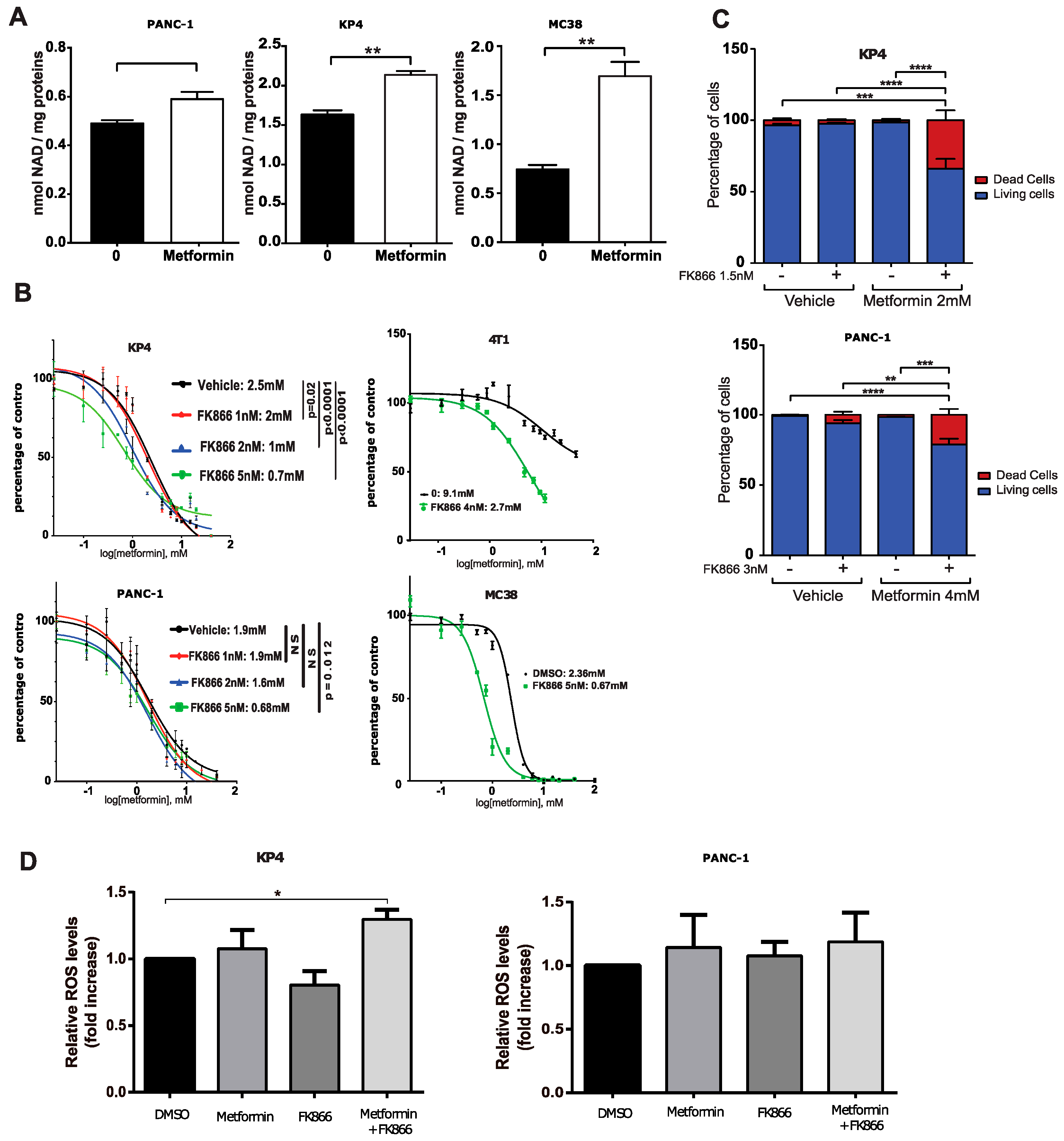
3.2. The NAMPT Inhibitor FK866 Increases Metformin Sensitivity
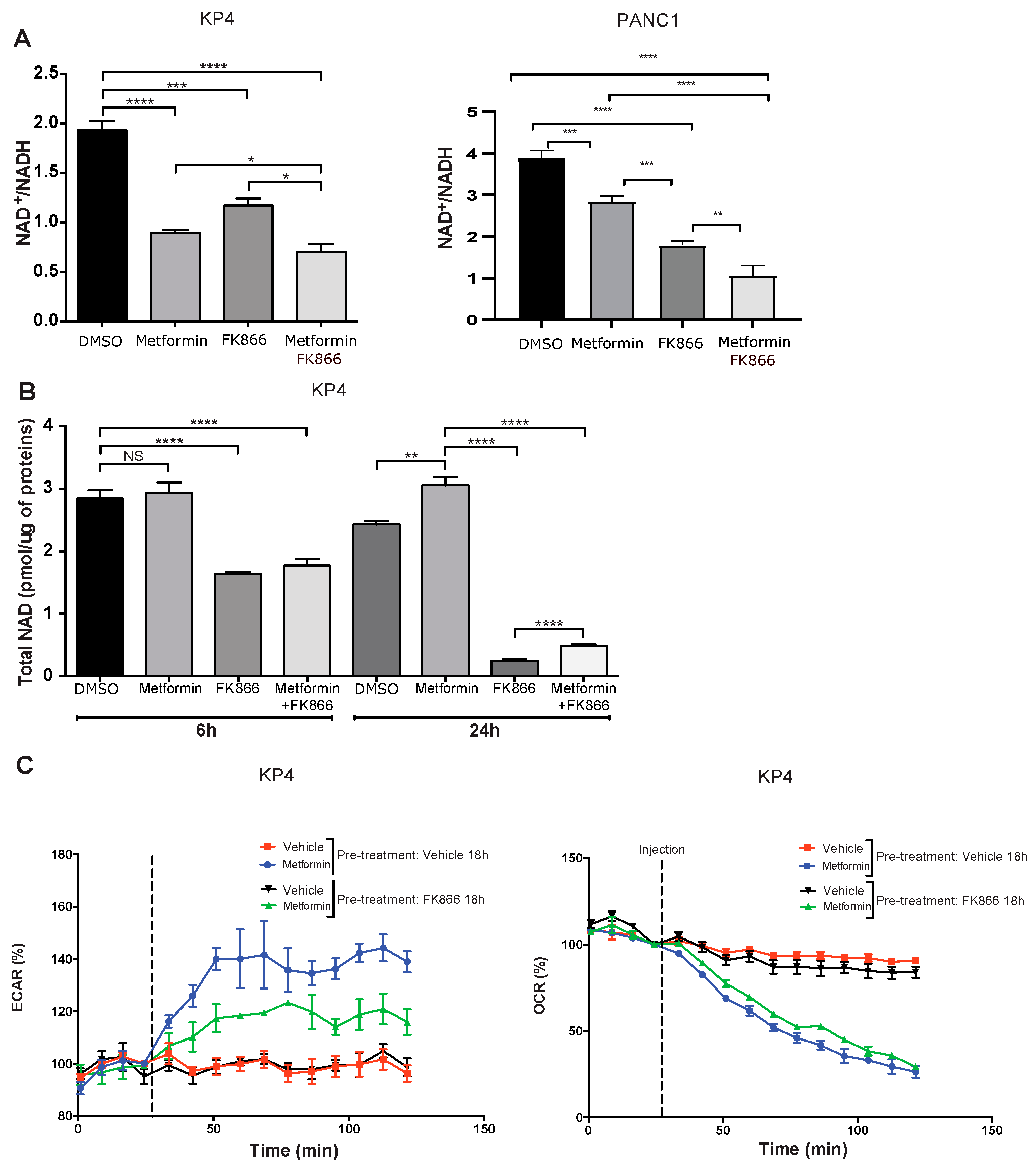
3.3. FK866 Inhibits Compensatory Glycolysis in Metformin-Treated Cells
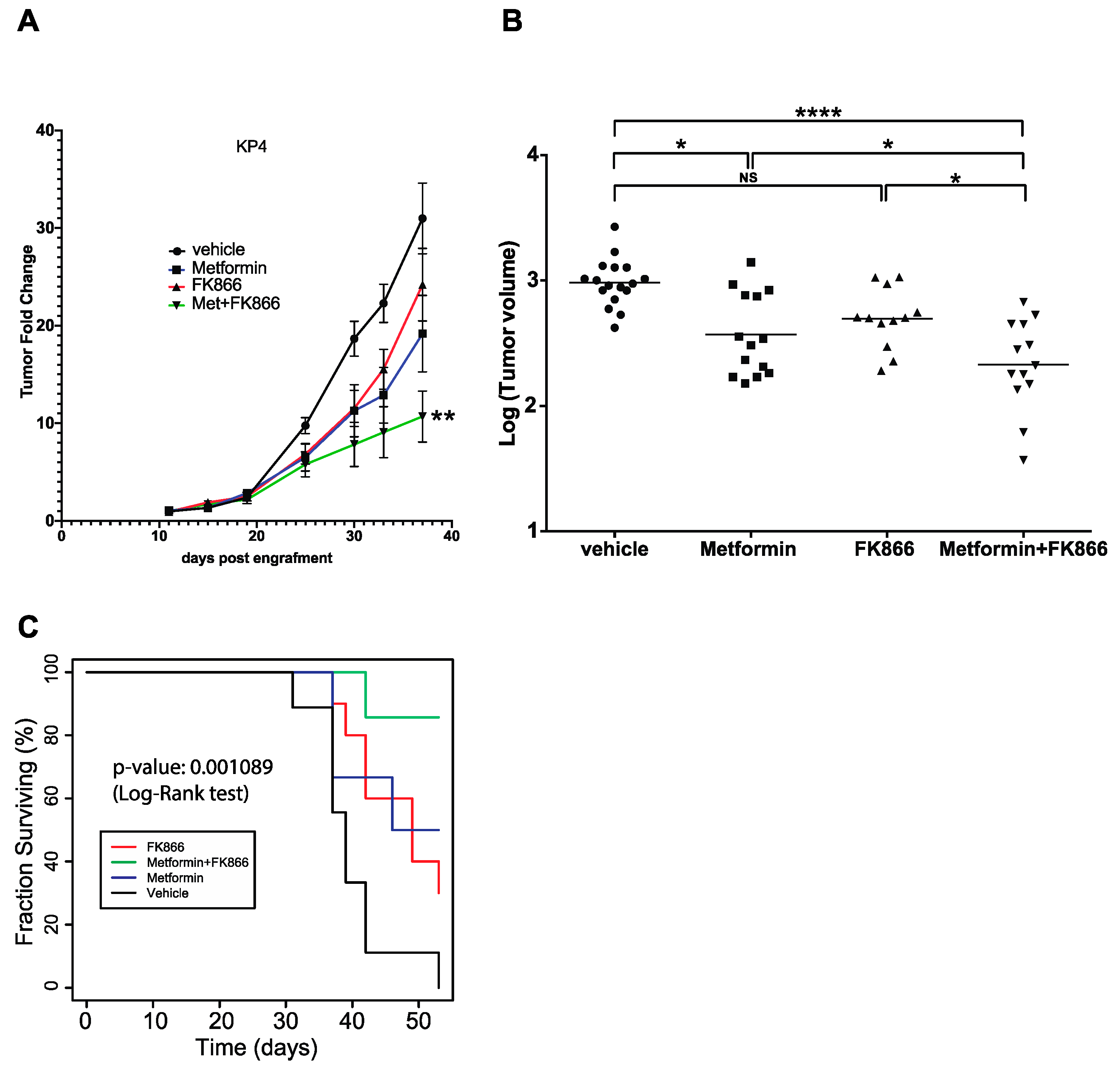
3.4. FK866 Improves Metformin Action on KP4 Xenografts in Nude Mice
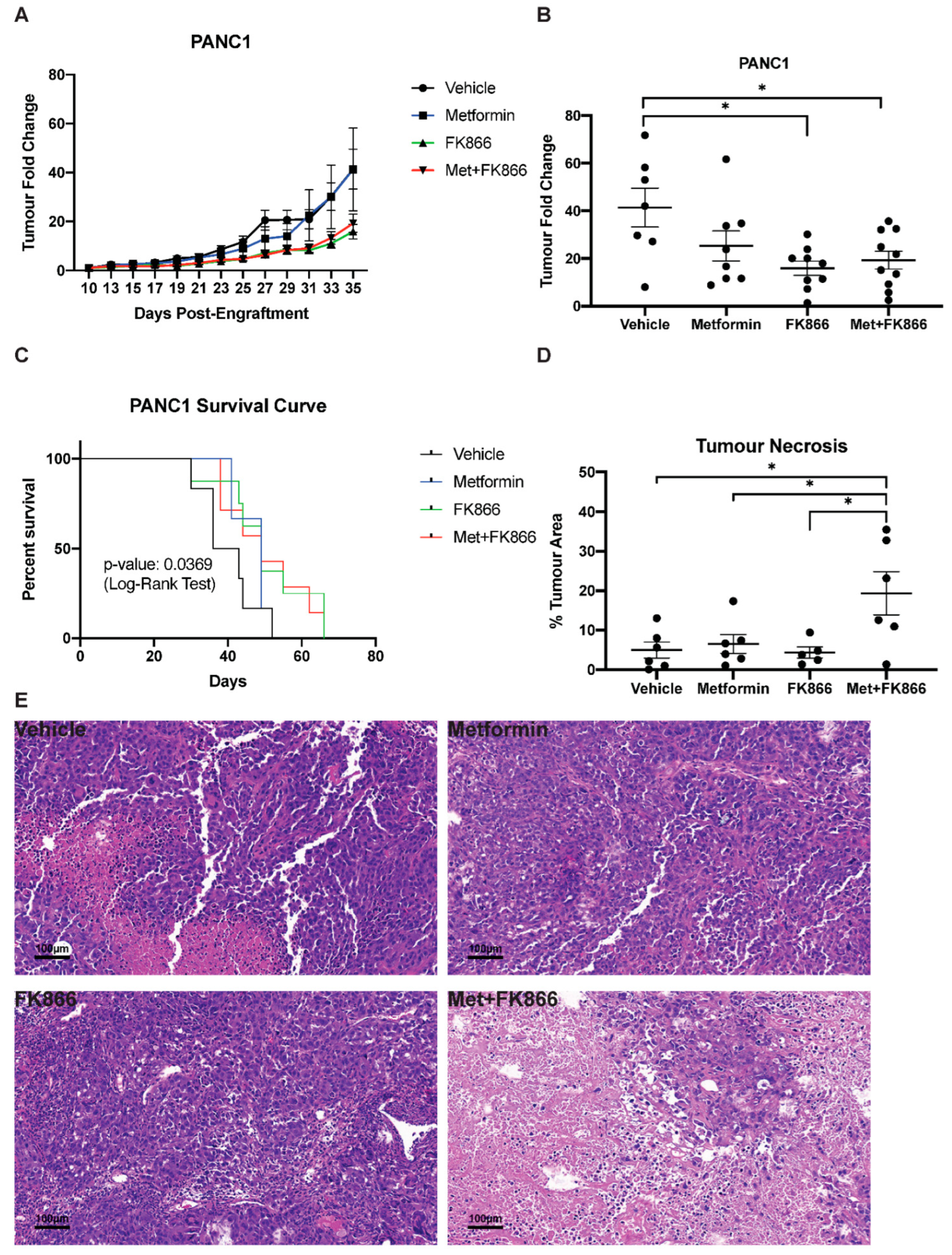
3.5. Metformin Does Not Cooperate with FK866 to Inhibit Growth of PANC-1 Xenografts in Nude Mice
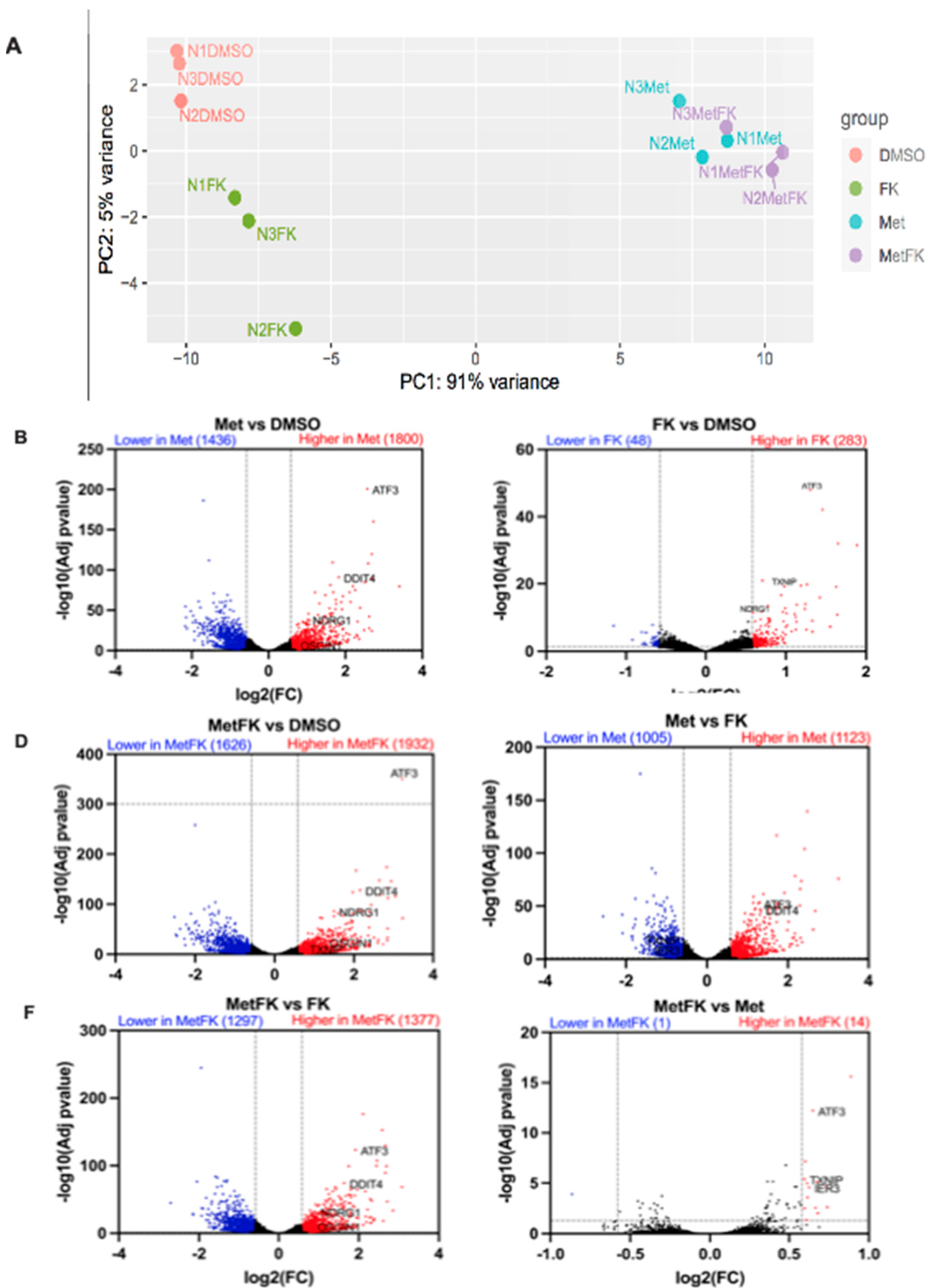
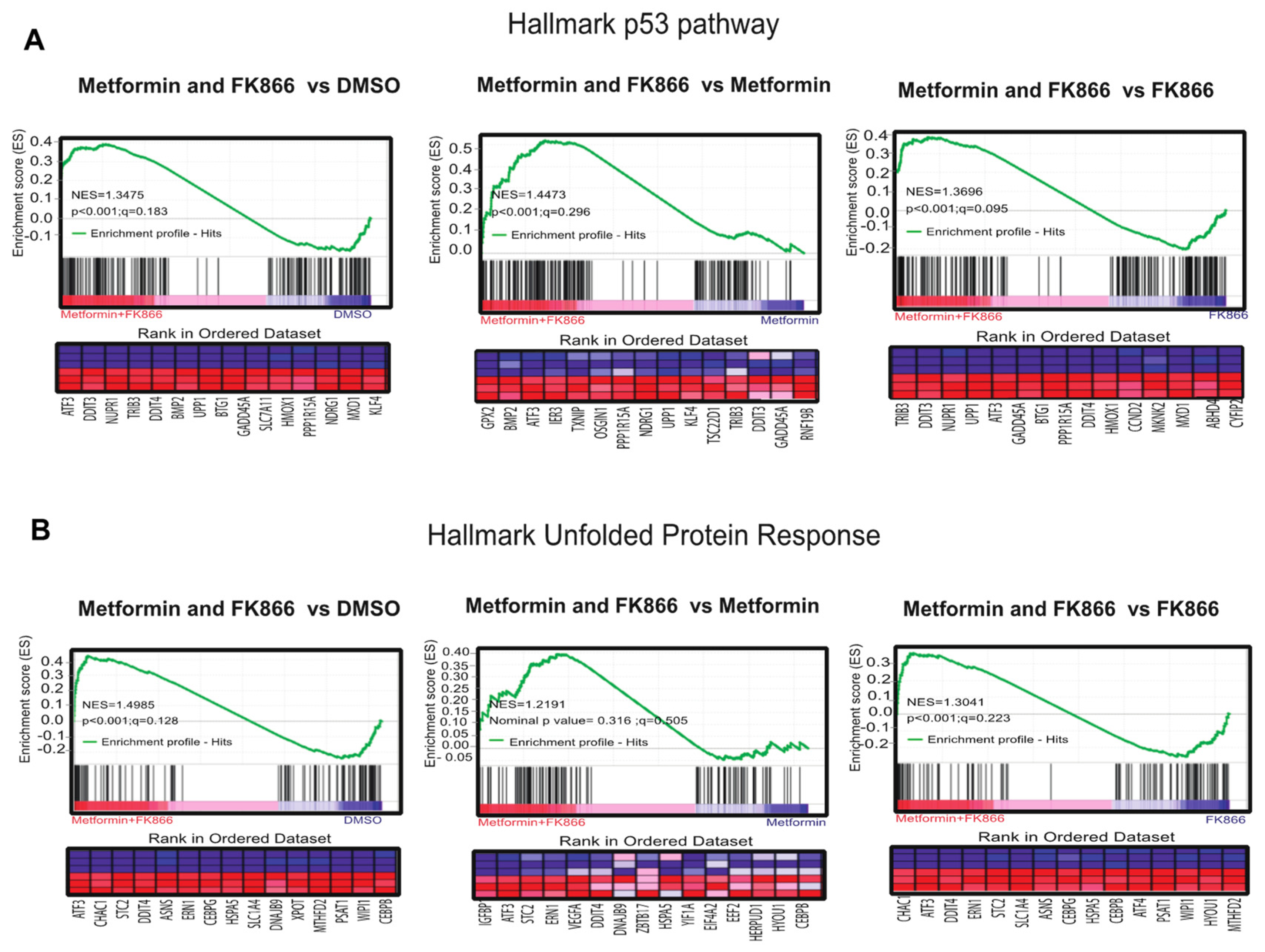
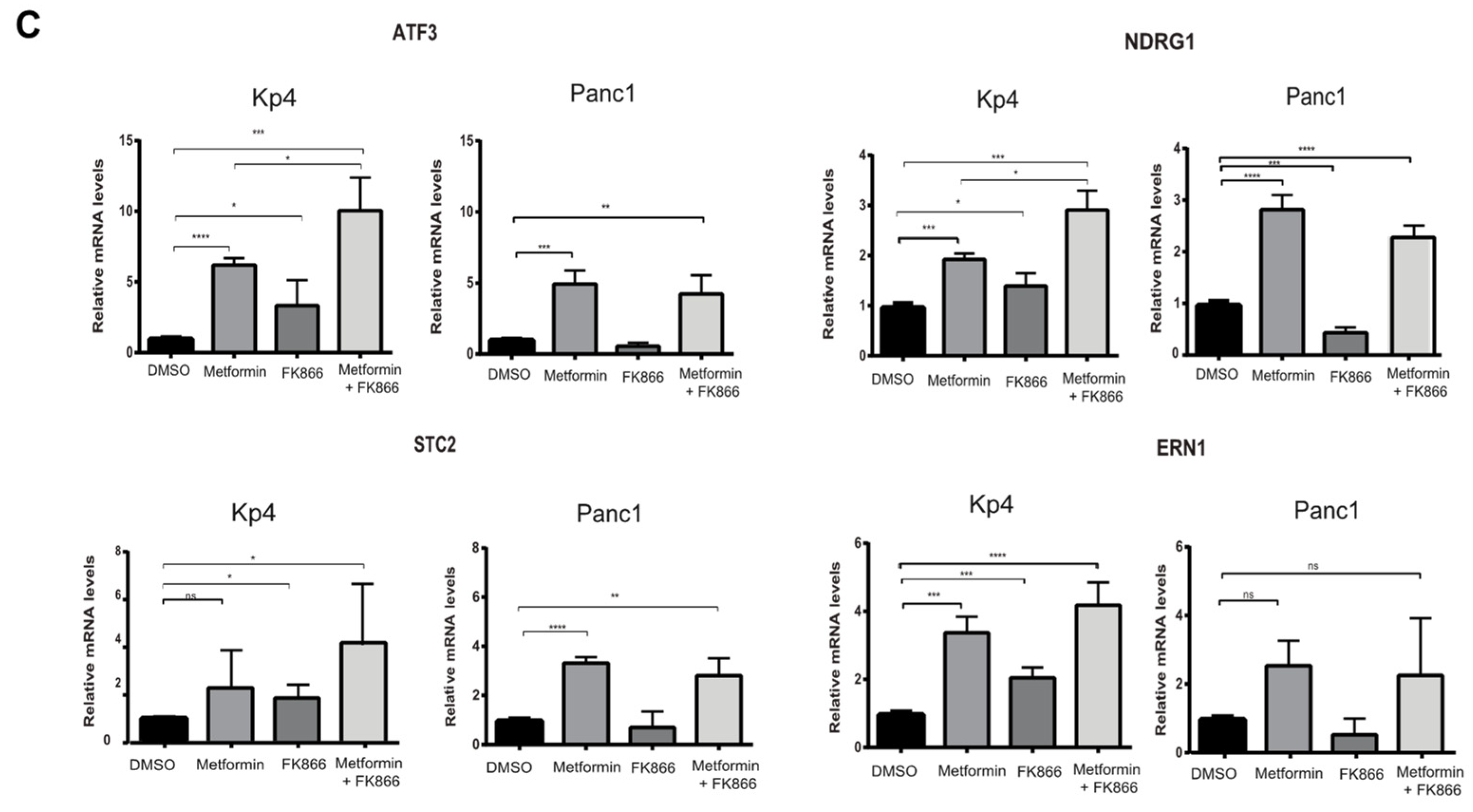
3.6. Combining FK866 with Metformin Restores p53 Signaling in Cancer Cells with Mutant p53
4. Discussion
Limitations of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Bardou, M.; Le Ray, I. Treatment of pancreatic cancer: A narrative review of cost-effectiveness studies. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Upadhyay, G.; Srivastava, R.K.; Shankar, S. Recent advances in pancreatic cancer: Biology, treatment, and prevention. Biochim. Biophys. Acta 2015, 1856, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Eibl, G.; Rozengurt, E. Metformin: Review of epidemiology and mechanisms of action in pancreatic cancer. Cancer Metastasis Rev. 2021, 40, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Dugnani, E.; Cereda, S.; Belli, C.; Balzano, G.; Nicoletti, R.; Liberati, D.; Pasquale, V.; Scavini, M.; Maggiora, P.; et al. (Ir)relevance of Metformin Treatment in Patients with Metastatic Pancreatic Cancer: An Open-Label, Randomized Phase II Trial. Clin. Cancer Res. 2016, 22, 1076–1085. [Google Scholar] [CrossRef]
- Kordes, S.; Pollak, M.N.; Zwinderman, A.H.; Mathot, R.A.; Weterman, M.J.; Beeker, A.; Punt, C.J.; Richel, D.J.; Wilmink, J.W. Metformin in patients with advanced pancreatic cancer: A double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015, 16, 839–847. [Google Scholar] [CrossRef]
- Wan, G.; Sun, X.; Li, F.; Wang, X.; Li, C.; Li, H.; Yu, X.; Cao, F. Survival Benefit of Metformin Adjuvant Treatment For Pancreatic Cancer Patients: A Systematic Review and Meta-Analysis. Cell Physiol. Biochem. 2018, 49, 837–847. [Google Scholar] [CrossRef]
- Chen, K.; Qian, W.; Jiang, Z.; Cheng, L.; Li, J.; Sun, L.; Zhou, C.; Gao, L.; Lei, M.; Yan, B.; et al. Metformin suppresses cancer initiation and progression in genetic mouse models of pancreatic cancer. Mol. Cancer 2017, 16, 131. [Google Scholar] [CrossRef]
- Kisfalvi, K.; Moro, A.; Sinnett-Smith, J.; Eibl, G.; Rozengurt, E. Metformin inhibits the growth of human pancreatic cancer xenografts. Pancreas 2013, 42, 781–785. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Deschenes-Simard, X.; Parisotto, M.; Rowell, M.C.; Le Calve, B.; Igelmann, S.; Moineau-Vallee, K.; Saint-Germain, E.; Kalegari, P.; Bourdeau, V.; Kottakis, F.; et al. Circumventing senescence is associated with stem cell properties and metformin sensitivity. Aging Cell 2019, 18, e12889. [Google Scholar] [CrossRef]
- Liu, L.; Patnana, P.K.; Xie, X.; Frank, D.; Nimmagadda, S.C.; Rosemann, A.; Liebmann, M.; Klotz, L.; Opalka, B.; Khandanpour, C. High Metabolic Dependence on Oxidative Phosphorylation Drives Sensitivity to Metformin Treatment in MLL/AF9 Acute Myeloid Leukemia. Cancers 2022, 14, 486. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell Rep. 2018, 25, 3047–3058.e4. [Google Scholar] [CrossRef]
- Moiseeva, O.; Deschenes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Igelmann, S.; Lessard, F.; Uchenunu, O.; Bouchard, J.; Fernandez-Ruiz, A.; Rowell, M.C.; Lopes-Paciencia, S.; Papadopoli, D.; Fouillen, A.; Ponce, K.J.; et al. A hydride transfer complex reprograms NAD metabolism and bypasses senescence. Mol. Cell 2021, 81, 3848–3865.e19. [Google Scholar] [CrossRef]
- Cheng, G.; Lanza-Jacoby, S. Metformin decreases growth of pancreatic cancer cells by decreasing reactive oxygen species: Role of NOX4. Biochem. Biophys. Res. Commun. 2015, 465, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Cardaci, S.; Zheng, L.; MacKay, G.; van den Broek, N.J.; MacKenzie, E.D.; Nixon, C.; Stevenson, D.; Tumanov, S.; Bulusu, V.; Kamphorst, J.J.; et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol. 2015, 17, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Lucas, S.; Soave, C.; Nabil, G.; Ahmed, Z.S.O.; Chen, G.; El-Banna, H.A.; Dou, Q.P.; Wang, J. Pharmacological Inhibitors of NAD Biosynthesis as Potential An ticancer Agents. Recent Pat. Anticancer Drug Discov. 2017, 12, 190–207. [Google Scholar] [CrossRef]
- Ju, H.Q.; Zhuang, Z.N.; Li, H.; Tian, T.; Lu, Y.X.; Fan, X.Q.; Zhou, H.J.; Mo, H.Y.; Sheng, H.; Chiao, P.J.; et al. Regulation of the Nampt-mediated NAD salvage pathway and its therapeutic implications in pancreatic cancer. Cancer Lett. 2016, 379, 1–11. [Google Scholar] [CrossRef]
- Davis, K.; Dunseth, C.D.; Mott, S.L.; Cramer-Morales, K.L.; Miller, A.M.; Ear, P.H.; Mezhir, J.J.; Bellizzi, A.M.; Chan, C.H.F. Nicotinamide phosphoribosyltransferase expression and clinical outcome of resected stage I/II pancreatic ductal adenocarcinoma. PLoS ONE 2019, 14, e0213576. [Google Scholar] [CrossRef]
- Hasmann, M.; Schemainda, I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef]
- Mayer, M.J.; Klotz, L.H.; Venkateswaran, V. Metformin and prostate cancer stem cells: A novel therapeutic target. Prostate Cancer Prostatic Dis. 2015, 18, 303–309. [Google Scholar] [CrossRef]
- Chini, C.C.; Guerrico, A.M.; Nin, V.; Camacho-Pereira, J.; Escande, C.; Barbosa, M.T.; Chini, E.N. Targeting of NAD metabolism in pancreatic cancer cells: Potential novel therapy for pancreatic tumors. Clin. Cancer Res. 2014, 20, 120–130. [Google Scholar] [CrossRef]
- Tan, B.; Dong, S.; Shepard, R.L.; Kays, L.; Roth, K.D.; Geeganage, S.; Kuo, M.S.; Zhao, G. Inhibition of Nicotinamide Phosphoribosyltransferase (NAMPT), an Enzyme Essential for NAD+ Biosynthesis, Leads to Altered Carbohydrate Metabolism in Cancer Cells. J. Biol. Chem. 2015, 290, 15812–15824. [Google Scholar] [CrossRef]
- Keshari, K.R.; Wilson, D.M.; Van Criekinge, M.; Sriram, R.; Koelsch, B.L.; Wang, Z.J.; VanBrocklin, H.F.; Peehl, D.M.; O’Brien, T.; Sampath, D.; et al. Metabolic response of prostate cancer to nicotinamide phophoribosyltransferase inhibition in a hyperpolarized MR/PET compatible bioreactor. Prostate 2015, 75, 1601–1609. [Google Scholar] [CrossRef][Green Version]
- Tan, B.; Young, D.A.; Lu, Z.H.; Wang, T.; Meier, T.I.; Shepard, R.L.; Roth, K.; Zhai, Y.; Huss, K.; Kuo, M.S.; et al. Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, in human cancer cells: Metabolic basis and potential clinical implications. J. Biol. Chem. 2013, 288, 3500–3511. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Goldinger, S.M.; Gobbi Bischof, S.; Fink-Puches, R.; Klemke, C.D.; Dreno, B.; Bagot, M.; Dummer, R. Efficacy and Safety of APO866 in Patients With Refractory or Relapsed Cutaneous T-Cell Lymphoma: A Phase 2 Clinical Trial. JAMA Dermatol. 2016, 152, 837–839. [Google Scholar] [CrossRef]
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nakamura, A.; Morioka, M.S.; Inoue, S.; Tamamori-Adachi, M.; Yamada, K.; Taketani, K.; Kawauchi, J.; Tanaka-Okamoto, M.; Miyoshi, J.; et al. Systems analysis of ATF3 in stress response and cancer reveals opposing effects on pro-apoptotic genes in p53 pathway. PLoS ONE 2011, 6, e26848. [Google Scholar] [CrossRef]
- Sharma, K.; Vu, T.T.; Cook, W.; Naseri, M.; Zhan, K.; Nakajima, W.; Harada, H. p53-independent Noxa induction by cisplatin is regulated by ATF3/ATF4 in head and neck squamous cell carcinoma cells. Mol. Oncol. 2018, 12, 788–798. [Google Scholar] [CrossRef]
- Schafer, H.; Trauzold, A.; Sebens, T.; Deppert, W.; Folsch, U.R.; Schmidt, W.E. The proliferation-associated early response gene p22/PRG1 is a novel p53 target gene. Oncogene 1998, 16, 2479–2487. [Google Scholar] [CrossRef][Green Version]
- Jin, H.; Suh, D.S.; Kim, T.H.; Yeom, J.H.; Lee, K.; Bae, J. IER3 is a crucial mediator of TAp73beta-induced apoptosis in cervical cancer and confers etoposide sensitivity. Sci. Rep. 2015, 5, 8367. [Google Scholar] [CrossRef]
- Yao, H.; Li, P.; Venters, B.J.; Zheng, S.; Thompson, P.R.; Pugh, B.F.; Wang, Y. Histone Arg modifications and p53 regulate the expression of OKL38, a mediator of apoptosis. J. Biol. Chem. 2008, 283, 20060–20068. [Google Scholar] [CrossRef]
- Stein, S.; Thomas, E.K.; Herzog, B.; Westfall, M.D.; Rocheleau, J.V.; Jackson, R.S., 2nd; Wang, M.; Liang, P. NDRG1 is necessary for p53-dependent apoptosis. J. Biol. Chem. 2004, 279, 48930–48940. [Google Scholar]
- Le, N.T.; Richardson, D.R. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: A link between iron metabolism and proliferation. Blood 2004, 104, 2967–2975. [Google Scholar] [CrossRef]
- Ellisen, L.W.; Ramsayer, K.D.; Johannessen, C.M.; Yang, A.; Beppu, H.; Minda, K.; Oliner, J.D.; McKeon, F.; Haber, D.A. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol. Cell 2002, 10, 995–1005. [Google Scholar] [CrossRef]
- Yin, H.; Wang, K.; Das, A.; Li, G.; Song, Y.; Luo, R.; Cheung, J.P.Y.; Zhang, T.; Li, S.; Yang, C. The REDD1/TXNIP Complex Accelerates Oxidative Stress-Induced Apoptosis of Nucleus Pulposus Cells through the Mitochondrial Pathway. Oxidative Med. Cell. Longev. 2021, 2021, 7397516. [Google Scholar] [CrossRef]
- Hartleben, G.; Schorpp, K.; Kwon, Y.; Betz, B.; Tsokanos, F.F.; Dantes, Z.; Schafer, A.; Rothenaigner, I.; Monroy Kuhn, J.M.; Morigny, P.; et al. Combination therapies induce cancer cell death through the integrated stress response and disturbed pyrimidine metabolism. EMBO Mol. Med. 2021, 13, e12461. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Zhan, Q.; Bae, I.; Fornace, A.J., Jr. Mammalian GADD34, an apoptosis- and DNA damage-inducible gene. J. Biol. Chem. 1997, 272, 13731–13737. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Lu, D.; Hai, T.; Harding, H.P.; Wang, X.; Ron, D.; Cavener, D.R.; Wek, R.C. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell Biol. 2004, 24, 1365–1377. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; Pollyea, D.A.; Culp-Hill, R.; Reisz, J.A.; Nemkov, T.; Gehrke, S.; Gamboni, F.; Krug, A.; Winters, A.; et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2020, 27, 748–764.e4. [Google Scholar] [CrossRef]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305. [Google Scholar] [CrossRef]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.Y.; Yoon, H. Chemotherapy Resistance Explained through Endoplasmic Reticulum Stress-Dependent Signaling. Cancers 2019, 11, 338. [Google Scholar] [CrossRef]
- Boudreau, M.W.; Duraki, D.; Wang, L.; Mao, C.; Kim, J.E.; Henn, M.A.; Tang, B.; Fanning, S.W.; Kiefer, J.; Tarasow, T.M.; et al. A small-molecule activator of the unfolded protein response eradicates human breast tumors in mice. Sci. Transl. Med. 2021, 13, eabf1383. [Google Scholar] [CrossRef]
- Mousa, S.A.; Gallati, C.; Simone, T.; Dier, E.; Yalcin, M.; Dyskin, E.; Thangirala, S.; Hanko, C.; Rebbaa, A. Dual targeting of the antagonistic pathways mediated by Sirt1 and TXNIP as a putative approach to enhance the efficacy of anti-aging interventions. Aging (Albany NY) 2009, 1, 412–424. [Google Scholar] [CrossRef]
- Qin, K.; Zhang, N.; Zhang, Z.; Nipper, M.; Zhu, Z.; Leighton, J.; Xu, K.; Musi, N.; Wang, P. SIRT6-mediated transcriptional suppression of Txnip is critical for pancreatic beta cell function and survival in mice. Diabetologia 2018, 61, 906–918. [Google Scholar] [CrossRef]
- Jia, Y.; Cui, R.; Wang, C.; Feng, Y.; Li, Z.; Tong, Y.; Qu, K.; Liu, C.; Zhang, J. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 2020, 32, 101534. [Google Scholar] [CrossRef]
- Xu, Y.; Nasri, M.; Dannenmann, B.; Mir, P.; Zahabi, A.; Welte, K.; Morishima, T.; Skokowa, J. NAMPT/SIRT2-mediated inhibition of the p53-p21 signaling pathway is indispensable for maintenance and hematopoietic differentiation of human iPS cells. Stem Cell Res. Ther. 2021, 12, 112. [Google Scholar] [CrossRef]
- Duncan, R.M.; Reyes, L.; Moats, K.; Robinson, R.M.; Murphy, S.A.; Kaur, B.; Stessman, H.A.F.; Dolloff, N.G. ATF3 Coordinates Antitumor Synergy between Epigenetic Drugs and Protein Disulfide Isomerase Inhibitors. Cancer Res. 2020, 80, 3279–3291. [Google Scholar] [CrossRef]
- Subedi, A.; Liu, Q.; Ayyathan, D.M.; Sharon, D.; Cathelin, S.; Hosseini, M.; Xu, C.; Voisin, V.; Bader, G.D.; D’Alessandro, A.; et al. Nicotinamide phosphoribosyltransferase inhibitors selectively induce apoptosis of AML stem cells by disrupting lipid homeostasis. Cell Stem Cell 2021, 28, 1851–1867.e8. [Google Scholar] [CrossRef] [PubMed]
- Del Nagro, C.; Xiao, Y.; Rangell, L.; Reichelt, M.; O’Brien, T. Depletion of the central metabolite NAD leads to oncosis-mediated cell death. J. Biol. Chem. 2014, 289, 35182–35192. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.; Parisotto, M.; Rowell, M.C.; Dore, A.; Fernandez Ruiz, A.; Lefrancois, G.; Kalegari, P.; Ferbeyre, G.; Schmitzer, A.R. Phenylethynylbenzyl-modified biguanides inhibit pancreatic cancer tumor growth. Sci. Rep. 2021, 11, 9854. [Google Scholar] [CrossRef] [PubMed]
- De Santi, M.; Baldelli, G.; Diotallevi, A.; Galluzzi, L.; Schiavano, G.F.; Brandi, G. Metformin prevents cell tumorigenesis through autophagy-related cell death. Sci. Rep. 2019, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Sica, V.; Bravo-San Pedro, J.M.; Izzo, V.; Pol, J.; Pierredon, S.; Enot, D.; Durand, S.; Bossut, N.; Chery, A.; Souquere, S.; et al. Lethal Poisoning of Cancer Cells by Respiratory Chain Inhibition plus Dimethyl alpha-Ketoglutarate. Cell Rep. 2019, 27, 820–834.e9. [Google Scholar] [CrossRef]
- Fan, J.; Hitosugi, T.; Chung, T.W.; Xie, J.; Ge, Q.; Gu, T.L.; Polakiewicz, R.D.; Chen, G.Z.; Boggon, T.J.; Lonial, S.; et al. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol. Cell Biol. 2011, 31, 4938–4950. [Google Scholar] [CrossRef]
- Hanse, E.A.; Ruan, C.; Kachman, M.; Wang, D.; Lowman, X.H.; Kelekar, A. Cytosolic malate dehydrogenase activity helps support glycolysis in actively proliferating cells and cancer. Oncogene 2017, 36, 3915–3924. [Google Scholar] [CrossRef]
- Hulea, L.; Gravel, S.P.; Morita, M.; Cargnello, M.; Uchenunu, O.; Im, Y.K.; Lehuede, C.; Ma, E.H.; Leibovitch, M.; McLaughlan, S.; et al. Translational and HIF-1alpha-Dependent Metabolic Reprogramming Underpin Metabolic Plasticity and Responses to Kinase Inhibitors and Biguanides. Cell metabolism 2018, 28, 817–832.e8. [Google Scholar] [CrossRef]
- Billington, R.A.; Travelli, C.; Ercolano, E.; Galli, U.; Roman, C.B.; Grolla, A.A.; Canonico, P.L.; Condorelli, F.; Genazzani, A.A. Characterization of NAD uptake in mammalian cells. J. Biol. Chem. 2008, 283, 6367–6374. [Google Scholar] [CrossRef]
- Pereira, F.V.; Melo, A.C.L.; Low, J.S.; de Castro, I.A.; Braga, T.T.; Almeida, D.C.; Batista de Lima, A.G.U.; Hiyane, M.I.; Correa-Costa, M.; Andrade-Oliveira, V.; et al. Metformin exerts antitumor activity via induction of multiple death pathways in tumor cells and activation of a protective immune response. Oncotarget 2018, 9, 25808–25825. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parisotto, M.; Vuong-Robillard, N.; Kalegari, P.; Meharwade, T.; Joumier, L.; Igelmann, S.; Bourdeau, V.; Rowell, M.-C.; Pollak, M.; Malleshaiah, M.; et al. The NAMPT Inhibitor FK866 Increases Metformin Sensitivity in Pancreatic Cancer Cells. Cancers 2022, 14, 5597. https://doi.org/10.3390/cancers14225597
Parisotto M, Vuong-Robillard N, Kalegari P, Meharwade T, Joumier L, Igelmann S, Bourdeau V, Rowell M-C, Pollak M, Malleshaiah M, et al. The NAMPT Inhibitor FK866 Increases Metformin Sensitivity in Pancreatic Cancer Cells. Cancers. 2022; 14(22):5597. https://doi.org/10.3390/cancers14225597
Chicago/Turabian StyleParisotto, Maxime, Nhung Vuong-Robillard, Paloma Kalegari, Thulaj Meharwade, Loick Joumier, Sebastian Igelmann, Véronique Bourdeau, Marie-Camille Rowell, Michael Pollak, Mohan Malleshaiah, and et al. 2022. "The NAMPT Inhibitor FK866 Increases Metformin Sensitivity in Pancreatic Cancer Cells" Cancers 14, no. 22: 5597. https://doi.org/10.3390/cancers14225597
APA StyleParisotto, M., Vuong-Robillard, N., Kalegari, P., Meharwade, T., Joumier, L., Igelmann, S., Bourdeau, V., Rowell, M.-C., Pollak, M., Malleshaiah, M., Schmitzer, A., & Ferbeyre, G. (2022). The NAMPT Inhibitor FK866 Increases Metformin Sensitivity in Pancreatic Cancer Cells. Cancers, 14(22), 5597. https://doi.org/10.3390/cancers14225597