
Evaluation of the Synergistic Potential of Simultaneous Pan- or Isoform-Specific BET and SYK Inhibition in B-Cell Lymphoma: An In Vitro Approach

 , , ,
, , ,
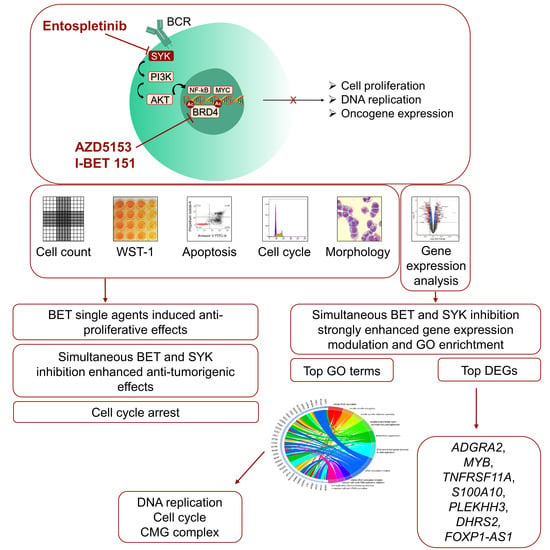
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Cell Lines, Reagents and Inhibitor Exposure
2.2. Cell Proliferation and WST-1 Proliferation Assay
2.3. Apoptosis Assay
2.4. Cell Cycle Analysis
2.5. May–Grunwald Giemsa Staining
2.6. RNA Extraction and Isolation
2.7. RNA Sequencing and Microarray Analyses
2.8. RNA Sequencing and Microarray Analysis Pipeline
2.9. Platform Comparison
2.10. Statistics, Reproducibility and Bliss Calculation
3. Results
3.1. Both the Isoform-Specific Bivalent- and Pan-BET Inhibitor Affect Proliferation and Metabolic Activity at Low Dosage in DLBCL and BL Cell Lines
3.2. Entospletinib Reduced Cell Viability of the DLBCL Cell Line SU-DHL-4 Selectively
3.3. Simultaneous BET and SYK Inhibition Revealed a Moderate Synergistic Effect Compared to Single Agent Response
3.4. Simultaneous BET and SYK Inhibition Modulates Cell Morphology Moderately
3.5. Simultaneous BET and SYK Inhibition-Induced Cell Cycle Blockade in DLBCL and BL Cell Lines, but Not Apoptosis
3.6. Combined BET and SYK Inhibition Boost the Changes in Gene Expression
3.7. RNAseq Data Validation by Microarray Analyses—Platform Comparison
3.8. Ento+AZD Combination Intensified the Gene Expression Changes Compared to the Single Agents and Induced a Specific Gene Set Modulation
3.9. Gene Ontology Enrichment Analyses Identified DNA Replication as the Biological Process Most Modulated by the Ento+AZD Combined Exposure
3.10. GO Term Clusters Revealed DNA Replication and Cell Cycle as the Most Affected Cell Biological Processes by the Ento+AZD Combination
4. Discussion
4.1. Both BET Inhibitors Efficiently Affected Cell Proliferation in B-Lymphoma Cell Lines
4.2. Simultaneous BET and SYK Inhibition Additionally Affected Cell’s Response
4.3. Simultaneous BET and SYK Inhibition Identified a Combination-Specific Gene Signature in DLBCL Cell Line SU-DHL-4
4.4. Overlapping DEGs between RNAseq and Microarray Platform in DLBCL Cell Line SU-DHL-4
4.5. Comparison of Identified DEGs Affected by Ento+AZD Combinaton with External Gene Lists
4.6. Gene Ontology (GO) Enrichment Analyses Identified Combination-Specific Biological Processes
4.7. Main GO Clusters in SU-DHL-4 after Combined Exposure
5. Conclusions and Future Perspective
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Panagis, F.; Sarah, P.; Maria, M.; Tracy, K.; Jean-Philippe, L.; Dalia, B.; Ildiko, F.; Rudolf, V.; Susanne, M.; Tony, P.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef]
- Cox, O.B.; Krojer, T.; Collins, P.; Monteiro, O.; Talon, R.; Bradley, A.; Fedorov, O.; Amin, J.; Marsden, B.D.; Spencer, J.; et al. A poised fragment library enables rapid synthetic expansion yielding the first reported inhibitors of PHIP(2), an atypical bromodomain. Chem. Sci. 2016, 7, 2322–2330. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.J.; Ornaghi, P.; Yang, J.; Lowe, N.; Evans, P.R.; Ballario, P.; Neuhaus, D.; Filetici, P.; Travers, A.A. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase Gcn5p. EMBO J. 2000, 19, 6141–6149. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and Characterization of Super-Enhancer-Associated Dependencies in Diffuse Large B Cell Lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.-W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, E.; Gaudio, E.; Lejeune, P.; Tarantelli, C.; Cascione, L.; Kwee, I.; Spriano, F.; Rinaldi, A.; Mensah, A.A.; Chung, E.; et al. Preclinical evaluation of the BET bromodomain inhibitor BAY 1238097 for the treatment of lymphoma. Br. J. Haematol. 2017, 178, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Spriano, F.; Stathis, A.; Bertoni, F. Targeting BET bromodomain proteins in cancer: The example of lymphomas. Pharmacol. Ther. 2020, 2020, 107631. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Future Sci. OA 2019, 5. [Google Scholar] [CrossRef]
- McDaniel, K.F.; Wang, L.; Soltwedel, T.; Fidanze, S.D.; Hasvold, L.A.; Liu, D.; Mantei, R.A.; Pratt, J.K.; Sheppard, G.S.; Bui, M.H.; et al. Discovery of N-(4-(2,4-Difluorophenoxy)-3-(6-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-4-yl)phenyl)ethanesulfonamide (ABBV-075/Mivebresib), a Potent and Orally Available Bromodomain and Extraterminal Domain (BET) Family Bromodomain Inhibitor. J. Med. Chem. 2017, 60, 8369–8384. [Google Scholar] [CrossRef] [PubMed]
- Noel, J.K.; Iwata, K.; Ooike, S.; Sugahara, K.; Nakamura, H.; Daibata, M. Abstract C244: Development of the BET bromodomain inhibitor OTX015. Mol. Cancer Ther. 2013, 12, C244. [Google Scholar] [CrossRef]
- Albrecht, B.K.; Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Côté, A.; Leblanc, Y.; Nasveschuk, C.G.; Bellon, S.; Bergeron, L.; Campbell, R.; et al. Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J. Med. Chem. 2016, 59, 1330–1339. [Google Scholar] [CrossRef]
- Rhyasen, G.W.; Hattersley, M.M.; Yao, Y.; Dulak, A.; Wang, W.; Petteruti, P.; Dale, I.L.; Boiko, S.; Cheung, T.; Zhang, J.; et al. AZD5153: A novel bivalent BET bromodomain inhibitor highly active against hematologic malignancies. Mol. Cancer Ther. 2016, 15, 2563–2574. [Google Scholar] [CrossRef]
- Sheppard, G.S.; Wang, L.; Fidanze, S.D.; Hasvold, L.A.; Liu, D.; Pratt, J.K.; Park, C.H.; Longenecker, K.L.; Qiu, W.; Torrent, M.; et al. Discovery of N-Ethyl-4-[2-(4-fluoro-2,6-dimethyl-phenoxy)-5-(1-hydroxy-1-methyl-ethyl)phenyl]-6-methyl-7-oxo-1 H-pyrrolo[2,3-c]pyridine-2-carboxamide (ABBV-744), a BET Bromodomain Inhibitor with Selectivity for the Second Bromodomain. J. Med. Chem. 2020, 63, 5585–5623. [Google Scholar] [CrossRef]
- Hogg, S.J.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.A.; Darcy, P.K.; Martin, B.P.; Spencer, A.; et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 2017, 18, 2162–2174. [Google Scholar] [CrossRef]
- Zhao, X.; Lwin, T.; Zhang, X.; Huang, A.; Wang, J.; E Marquez, V.; Chen-Kiang, S.; Dalton, W.S.; Sotomayor, E.; Tao, J. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia 2013, 27, 2341–2350. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, S.V.; Bhadury, J.; Nilsson, L.M.; Green, L.C.; McLure, K.G.; Nilsson, J.A. BET bromodomain inhibitors synergize with ATR inhibitors to induce DNA damage, apoptosis, senescence-associated secretory pathway and ER stress in Myc-induced lymphoma cells. Oncogene 2016, 35, 4689–4697. [Google Scholar] [CrossRef] [PubMed]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef]
- Küppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Shah, B.A.; Fiskus, W.; Qi, J.; Rajapakshe, K.; Coarfa, C.; Li, L.; Devaraj, S.G.T.; Sharma, S.K.; Zhang, L.; et al. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood 2015, 126, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Hacken, E.T.; Sivina, M.; Clarke, A.; Thompson, P.A.; Jain, N.; Ferrajoli, A.; Estrov, Z.; Keating, M.J.; Wierda, W.G.; et al. The BET inhibitor GS-5829 targets chronic lymphocytic leukemia cells and their supportive microenvironment. Leukemia 2020, 34, 1588–1598. [Google Scholar] [CrossRef]
- Tinsley, S.; Meja, K.; Shepherd, C.; Khwaja, A. Synergistic induction of cell death in haematological malignancies by combined phosphoinositide-3-kinase and BET bromodomain inhibition. Br. J. Haematol. 2015, 170, 275–278. [Google Scholar] [CrossRef]
- Cummin, T.E.C.; Cox, K.L.; Murray, T.D.; Turaj, A.H.; Dunning, L.; English, V.L.; Fell, R.; Packham, G.; Ma, Y.; Powell, B.; et al. BET inhibitors synergize with venetoclax to induce apoptosis in MYC-driven lymphomas with high BCL-2 expression. Blood Adv. 2020, 4, 3316–3328. [Google Scholar] [CrossRef]
- Mócsai, A.; Ruland, J.; Tybulewicz, V.L.J. The SYK tyrosine kinase: A crucial player in diverse biological functions. Nat. Rev. Immunol. 2010, 10, 387–402. [Google Scholar] [CrossRef]
- Sender, S.; Sekora, A.; Perez, S.V.; Chabanovska, O.; Becker, A.; Ngezahayo, A.; Junghanss, C.; Escobar, H.M. Precursor B-ALL cell lines differentially respond to syk inhibition by entospletinib. Int. J. Mol. Sci. 2021, 22, 592. [Google Scholar] [CrossRef]
- Koczan, D.; Fitzner, B.; Zettl, U.K.; Hecker, M. Microarray data of transcriptome shifts in blood cell subsets during S1P receptor modulator therapy. Sci. Data 2018, 5, 180145. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2015, 44, W90–W97. [Google Scholar] [CrossRef]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Hassler, M.R.; Schiefer, A.I.; Egger, G. Combating the epigenome: Epigenetic drugs against non-Hodgkin’s lymphoma. Epigenomics 2013, 5, 397–415. [Google Scholar] [CrossRef]
- Davis, R.E.; Ngo, V.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef]
- Kong, W.; Sender, S.; Perez, S.V.; Sekora, A.; Ruetgen, B.; Junghanss, C.; Nolte, I.; Escobar, H.M. Pan-and isoform-specific inhibition of the bromodomain and extra-terminal proteins and evaluation of synergistic potential with entospletinib in canine lymphoma. Anticancer Res. 2020, 40, 3781–3792. [Google Scholar] [CrossRef]
- Takimoto-Shimomura, T.; Tsukamoto, T.; Maegawa, S.; Fujibayashi, Y.; Matsumura-Kimoto, Y.; Mizuno, Y.; Chinen, Y.; Shimura, Y.; Mizutani, S.; Horiike, S.; et al. Dual targeting of bromodomain-containing 4 by AZD5153 and BCL2 by AZD4320 against B-cell lymphomas concomitantly overexpressing c-MYC and BCL2. Investig. New Drugs 2019, 37, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, E.; Mondello, P.; Erazo, T.; Portelinha, A.; Liu, Y.; Scallion, M.; Asgari, Z.; Philip, J.; Hilden, P.; Valli, D.; et al. BET Inhibition-Induced GSK3β Feedback Enhances Lymphoma Vulnerability to PI3K Inhibitors. Cell Rep. 2018, 24, 2155–2166. [Google Scholar] [CrossRef]
- Cheng, S.; Coffey, G.; Zhang, X.H.; Shaknovich, R.; Song, Z.; Lu, P.; Pandey, A.; Melnick, A.M.; Sinha, U.; Wang, Y.L. SYK inhibition and response prediction in diffuse large B-cell lymphoma. Blood 2011, 118, 6342–6352. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, F.; Mancuso, M.R.; Shamloo, A.; Wang, H.-T.; Choksi, V.; Florek, M.; Su, H.; Fruttiger, M.; Young, W.L.; Heilshorn, S.C.; et al. Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science 2010, 330, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Nathans, J. Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical Wnt signaling. Dev. Cell 2014, 31, 248. [Google Scholar] [CrossRef]
- Du, J.; Qiu, X.; Lu, C.; Zhu, Y.; Kong, W.; Xu, M.; Zhang, X.; Tang, M.; Chen, J.; Li, Q.; et al. Molecular Landscape and Prognostic Biomarker Analysis of Advanced Pancreatic Cancer and Predictors of Treatment Efficacy of AG Chemotherapy. Front. Oncol. 2022, 12, 844527. [Google Scholar] [CrossRef]
- Pietrzyk, Ł.; Wdowiak, P. Serum TEM5 and TEM7 concentrations correlate with clinicopathologic features and poor prognosis of colorectal cancer patients. Adv. Med. Sci. 2019, 64, 402–408. [Google Scholar] [CrossRef]
- Cherry, A.E.; Vicente, J.J.; Xu, C.; Morrison, R.S.; Ong, S.; Wordeman, L.; Stella, N. GPR124 regulates microtubule assembly, mitotic progression, and glioblastoma cell proliferation. Glia 2019, 67, 1558. [Google Scholar] [CrossRef]
- Wang, Y.; Cho, S.-G.; Wu, X.; Siwko, S.; Liu, M. G-protein coupled receptor 124 (GPR124) in endothelial cells regulates vascular endothelial growth factor (VEGF)-induced tumor angiogenesis. Curr. Mol. Med. 2014, 14, 543–554. [Google Scholar] [CrossRef]
- DepMap, Broad: DepMap 22Q2 Public. Figshare. Dataset. 2022. Available online: https://figshare.com/articles/dataset/DepMap_22Q2_Public/19700056/2 (accessed on 8 June 2022).
- Dempsey, P.W.; Doyle, S.E.; He, J.Q.; Cheng, G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003, 14, 193–209. [Google Scholar] [CrossRef]
- González-Suárez, E.; Sanz-Moreno, A. RANK as a therapeutic target in cancer. FEBS J. 2016, 283, 2018–2033. [Google Scholar] [CrossRef] [PubMed]
- Knesebeck, A.V.D.; Felsberg, J.; Waha, A.; Hartmann, W.; Scheffler, B.; Glas, M.; Hammes, J.; Mikeska, T.; Yan, P.S.; Endl, E.; et al. RANK (TNFRSF11A) Is Epigenetically Inactivated and Induces Apoptosis in Gliomas. Neoplasia 2012, 14, 526. [Google Scholar] [CrossRef] [PubMed]
- Bonifaci, N.; Palafox, M.; Pellegrini, P.; Osorio, A.; Benítez, J.; Peterlongo, P.; Manoukian, S.; Peissel, B.; Zaffaroni, D.; Roversi, G.; et al. Evidence for a link between TNFRSF11A and risk of breast cancer. Breast Cancer Res. Treat. 2011, 129, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Chang, L.-Y.; Zhao, S.; Zhao, J.-J.; Xiong, Y.-J.; Cao, F.-Y.; Fu-Yuan, C.; Zhang, Q.; Wang, X.-Y.; Geng, M.-L.; et al. KLF5 promotes cervical cancer proliferation, migration and invasion in a manner partly dependent on TNFRSF11a expression. Sci. Rep. 2017, 7, 15683. [Google Scholar] [CrossRef]
- Liang, Q.; Wang, Y.; Lu, Y.; Zhu, Q.; Xie, W.; Tang, N.; Huang, L.; An, T.; Zhang, D.; Yan, A.; et al. RANK promotes colorectal cancer migration and invasion by activating the Ca2+-calcineurin/NFATC1-ACP5 axis. Cell Death Dis. 2021, 12, 336. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, Y.; Gao, J.; Zhong, X.; Wu, X.; Li, H. A nine-gene signature to improve prognosis prediction of colon carcinoma. Cell Cycle 2021, 20, 1021. [Google Scholar] [CrossRef]
- Ieranò, C.; Righelli, D.; D’Alterio, C.; Napolitano, M.; Portella, L.; Rea, G.; Auletta, F.; Santagata, S.; Trotta, A.M.; Guardascione, G.; et al. Original research: In PD-1+ human colon cancer cells NIVOLUMAB promotes survival and could protect tumor cells from conventional therapies. J. Immunother. Cancer 2022, 10, 4032. [Google Scholar] [CrossRef]
- Alankus, B.; Ecker, V.; Vahl, N.; Braun, M.; Weichert, W.; Macher-Göppinger, S.; Gehring, T.; Neumayer, T.; Zenz, T.; Buchner, M.; et al. Pathological RANK signaling in B cells drives autoimmunity and chronic lymphocytic leukemia. J. Exp. Med. 2021, 218, e20200517. [Google Scholar] [CrossRef]
- Li, C.; Ma, Y.; Fei, F.; Zheng, M.; Li, Z.; Zhao, Q.; Du, J.; Liu, K.; Lu, R.; Zhang, S. Critical role and its underlying molecular events of the plasminogen receptor, S100A10 in malignant tumor and non-tumor diseases. J. Cancer 2020, 11, 826. [Google Scholar] [CrossRef]
- Tantyo, N.A.; Karyadi, A.S.; Rasman, S.Z.; Salim, M.R.G.; Devina, A.; Sumarpo, A. The prognostic value of S100A10 expression in cancer. Oncol. Lett. 2019, 17, 1417–1424. [Google Scholar] [CrossRef]
- Huang, D.; Yang, Y.; Sun, J.; Dong, X.; Wang, J.; Liu, H.; Lu, C.; Hongchen, L.; Shao, J.; Yan, J. Annexin A2-S100A10 heterotetramer is upregulated by PML/RARα fusion protein and promotes plasminogen-dependent fibrinolysis and matrix invasion in acute promyelocytic leukemia. Front. Med. 2017, 11, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, X.Y.; Li, L.X.; Zhou, R.C.; Sikong, Y.; Gu, X.; Jin, B.Y.; Li, B.; Li, Y.Q.; Zuo, X.L. S100A10 Accelerates Aerobic Glycolysis and Malignant Growth by Activating mTOR-Signaling Pathway in Gastric Cancer. Front. Cell Dev. Biol. 2020, 8, 1430. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Iwasaki, T.; Sonoda, A.; Endo, A. Membranous overexpression of S100A10 is associated with a high-grade cellular status of breast carcinoma. Med. Mol. Morphol. 2020, 53, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Gonda, T.J. Role and potential for therapeutic targeting of MYB in leukemia. Leukemia 2013, 27, 269–277. [Google Scholar] [CrossRef]
- Ramsay, R.G.; Gonda, T.J. MYB function in normal and cancer cells. Nat. Rev. Cancer 2008, 8, 523–534. [Google Scholar] [CrossRef]
- Robetorye, R.S.; Bohling, S.D.; Morgan, J.W.; Fillmore, G.C.; Lim, M.S.; Elenitoba-Johnson, K.S.J. Microarray analysis of B-cell lymphoma cell lines with the t(14;18). J. Mol. Diagn. 2002, 4, 123–136. [Google Scholar] [CrossRef]
- Zhang, B.; Yuan, F.; Liu, J.; Li, Y.; Zhou, F.; Liu, X.; Hao, Z.; Li, Q.; Zheng, Y.; Wang, W. Hsa-miR-495 acts as a tumor suppressor gene in glioma via the negative regulation of MYB. Mol. Med. Rep. 2016, 14, 977–982. [Google Scholar] [CrossRef]
- Yu, L.; Ding, G.F.; He, C.; Sun, L.; Jiang, Y.F.; Zhu, L. MicroRNA-424 is down-regulated in hepatocellular carcinoma and suppresses cell migration and invasion through c-Myb. PLoS ONE 2014, 9, e91661. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Ferguson, K.M.; Abrams, C.S. Pleckstrin homology domains and the cytoskeleton. FEBS Lett. 2002, 513, 71–76. [Google Scholar] [CrossRef]
- Ingley, E.; Hemmings, B.A. Pleckstrin homology (PH) domains in signal transducton. J. Cell. Biochem. 1994, 56, 436–443. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Chen, K.-D.; Su, M.-C.; Chin, C.-H.; Chen, C.-J.; Liou, C.-W.; Chen, T.; Chang, Y.-C.; Huang, K.-T.; Wang, C.-C.; et al. Genome-wide gene expression array identifies novel genes related to disease severity and excessive daytime sleepiness in patients with obstructive sleep apnea. PLoS ONE 2017, 12, e176575. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xue, M.; Cao, D.; Qin, L.; Wang, Y.; Miao, Z.; Wang, P.; Hu, X.; Shen, J.; Xiong, B. Multi-omics characterization of WNT pathway reactivation to ameliorate BET inhibitor resistance in liver cancer cells. Genomics 2021, 113, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Ekmekci, S.S.; Emrence, Z.; Abacı, N.; Sarıman, M.; Salman, B.; Ekmekci, C.G.; Güleç, Ç. LEF1 Induces DHRS2 Gene Expression in Human Acute Leukemia Jurkat T-Cells. Turk. J. Hematol. 2020, 37, 226. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, L.; Ban, X.; Zeng, T.; Zhu, Y.; Li, M.; Guan, X.-Y.; Li, Y. DHRS2 inhibits cell growth and motility in esophageal squamous cell carcinoma. Oncogene 2018, 37, 1086. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, Z.; Sun, S.; Zhang, Z.; Liu, J.; Jin, X.; Wu, P.; Ji, T.; Ding, W.; Wang, B.; et al. Decreased DHRS2 expression is associated with HDACi resistance and poor prognosis in ovarian cancer. Epigenetics 2020, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Peltekova, V.D.; Lemire, M.; Qazi, A.; Zaidi, S.H.E.; Trinh, Q.M.; Bielecki, R.; Rogers, M.; Hodgson, L.; Wang, M.; D’Souza, D.J.A.; et al. Identification of genes expressed by immune cells of the colon that are regulated by colorectal cancer-associated variants. Int. J. Cancer 2014, 134, 2330–2341. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Song, B.; Wang, J.; Shao, C.; Xu, Y.; Jiang, H. Genome-Wide Association and Transcriptome-Wide Association Studies Identify Novel Susceptibility Genes Contributing to Colorectal Cancer. J. Immunol. Res. 2022, 2022, 5794055. [Google Scholar] [CrossRef]
- Taher, L.; Beck, J.; Liu, W.; Roolf, C.; Soller, J.T.; Rütgen, B.C.; Hammer, S.E.; Chodisetti, M.; Sender, S.; Sterenczak, K.A.; et al. Comparative High-Resolution Transcriptome Sequencing of Lymphoma Cell Lines and de novo Lymphomas Reveals Cell-Line-Specific Pathway Dysregulation. Sci. Rep. 2018, 8, 6279. [Google Scholar] [CrossRef]
- Mudaliar, M.A.V.; Haggart, R.D.; Miele, G.; Sellar, G.; Tan, K.A.L.; Goodlad, J.R.; Milne, E.; Vail, D.M.; Kurzman, I.; Crowther, D.; et al. Comparative Gene Expression Profiling Identifies Common Molecular Signatures of NF-κB Activation in Canine and Human Diffuse Large B Cell Lymphoma (DLBCL). PLoS ONE 2013, 8, e72591. [Google Scholar] [CrossRef]
- Teater, M.; Dominguez, P.M.; Redmond, D.; Chen, Z.; Ennishi, D.; Scott, D.W.; Cimmino, L.; Ghione, P.; Chaudhuri, J.; Gascoyne, R.D.; et al. AICDA drives epigenetic heterogeneity and accelerates germinal center-derived lymphomagenesis. Nat. Commun. 2018, 9, 222. [Google Scholar] [CrossRef]
- Bochman, M.L.; Schwacha, A. The Mcm Complex: Unwinding the Mechanism of a Replicative Helicase. Microbiol. Mol. Biol. Rev. 2009, 73, 652. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.-A.; Shin, S.M.; Namkoong, H.; Lee, H.; Cho, G.W.; Hur, S.Y.; Kim, T.E.; Kim, J.W. Cancer-Associated Expression of Minichromosome Maintenance 3 Gene in Several Human Cancers and Its Involvement in Tumorigenesis. Clin. Cancer Res. 2004, 10, 8386–8395. [Google Scholar] [CrossRef] [PubMed]
- Obermann, E.C.; Went, P.; Zimpfer, A.; Tzankov, A.; Wild, P.J.; Stoehr, R.; Pileri, S.A.; Dirnhofer, S. Expression of minichromosome maintenance protein 2 as a marker for proliferation and prognosis in diffuse large B-cell lymphoma: A tissue microarray and clinico-pathological analysis. BMC Cancer 2005, 5, 162. [Google Scholar] [CrossRef]
- Mio, C.; Lavarone, E.; Conzatti, K.; Baldan, F.; Toffoletto, B.; Puppin, C.; Filetti, S.; Durante, C.; Russo, D.; Orlacchio, A.; et al. MCM5 as a target of BET inhibitors in thyroid cancer cells. Endocr. Relat. Cancer 2016, 23, 335. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zheng, W. Upregulation of CDC7 Associated with Cervical Cancer Incidence and Development. BioMed Res. Int. 2021, 2021, 6663367. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.N.; Jiang, S.S.; Fan, C.-C.; Lo, Y.-K.; Kuo, C.-Y.; Chen, C.-H.; Liu, Y.-L.; Lee, C.-C.; Chen, W.-S.; Huang, T.-S.; et al. Increased Cdc7 expression is a marker of oral squamous cell carcinoma and overexpression of Cdc7 contributes to the resistance to DNA-damaging agents. Cancer Lett. 2013, 337, 218–225. [Google Scholar] [CrossRef]
- Liu, R.; Huang, Y. CDC7 as a novel biomarker and druggable target in cancer. Clin. Transl. Oncol. 2022, 24, 1856–1864. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, H.-Q.; Fu, K.; Zhang, H.-L.; Qian, Z.-Z.; Qiu, L.-H.; Li, W.; Zhou, S.-Y.; Li, L.-F.; Hao, X.-S. Expression of Cdc7 and mcm2 as a marker for proliferation and prognosis in diffuse large B cell lymphoma. Chin. J. Oncol. 2011, 33, 911–915. [Google Scholar] [CrossRef]
- Ouyang, F.; Liu, J.; Xia, M.; Lin, C.; Wu, X.; Ye, L.; Song, L.; Li, J.; Wang, J.; Guo, P.; et al. GINS2 is a novel prognostic biomarker and promotes tumor progression in early-stage cervical cancer. Oncol. Rep. 2017, 37, 2652–2662. [Google Scholar] [CrossRef]
- Ahmad, M.; Hameed, Y.; Khan, M.; Usman, M.; Rehman, A.; Abid, U.; Asif, R.; Ahmed, H.; Hussain, M.S.; Rehman, J.U.; et al. Up-regulation of GINS1 highlighted a good diagnostic and prognostic potential of survival in three different subtypes of human cancer. Braz. J. Biol. 2021, 84. [Google Scholar] [CrossRef]
- Ilves, I.; Petojevic, T.; Pesavento, J.J.; Botchan, M.R. Activation of the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol. Cell 2010, 37, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Kusunoki, S.; Ishimi, Y. Protein interaction and cellular localization of human CDC45. J. Biochem. 2013, 153, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.S.; Kang, Y.H. The Human Replicative Helicase, the CMG Complex, as a Target for Anti-cancer Therapy. Front. Mol. Biosci. 2018, 5, 26. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sender, S.; Sultan, A.W.; Palmer, D.; Koczan, D.; Sekora, A.; Beck, J.; Schuetz, E.; Taher, L.; Brenig, B.; Fuellen, G.; et al. Evaluation of the Synergistic Potential of Simultaneous Pan- or Isoform-Specific BET and SYK Inhibition in B-Cell Lymphoma: An In Vitro Approach. Cancers 2022, 14, 4691. https://doi.org/10.3390/cancers14194691
Sender S, Sultan AW, Palmer D, Koczan D, Sekora A, Beck J, Schuetz E, Taher L, Brenig B, Fuellen G, et al. Evaluation of the Synergistic Potential of Simultaneous Pan- or Isoform-Specific BET and SYK Inhibition in B-Cell Lymphoma: An In Vitro Approach. Cancers. 2022; 14(19):4691. https://doi.org/10.3390/cancers14194691
Chicago/Turabian StyleSender, Sina, Ahmad Wael Sultan, Daniel Palmer, Dirk Koczan, Anett Sekora, Julia Beck, Ekkehard Schuetz, Leila Taher, Bertram Brenig, Georg Fuellen, and et al. 2022. "Evaluation of the Synergistic Potential of Simultaneous Pan- or Isoform-Specific BET and SYK Inhibition in B-Cell Lymphoma: An In Vitro Approach" Cancers 14, no. 19: 4691. https://doi.org/10.3390/cancers14194691
APA StyleSender, S., Sultan, A. W., Palmer, D., Koczan, D., Sekora, A., Beck, J., Schuetz, E., Taher, L., Brenig, B., Fuellen, G., Junghanss, C., & Murua Escobar, H. (2022). Evaluation of the Synergistic Potential of Simultaneous Pan- or Isoform-Specific BET and SYK Inhibition in B-Cell Lymphoma: An In Vitro Approach. Cancers, 14(19), 4691. https://doi.org/10.3390/cancers14194691