
The Role of Programmed Necrosis in Colorectal Cancer
 , ,
, ,  and
and 
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
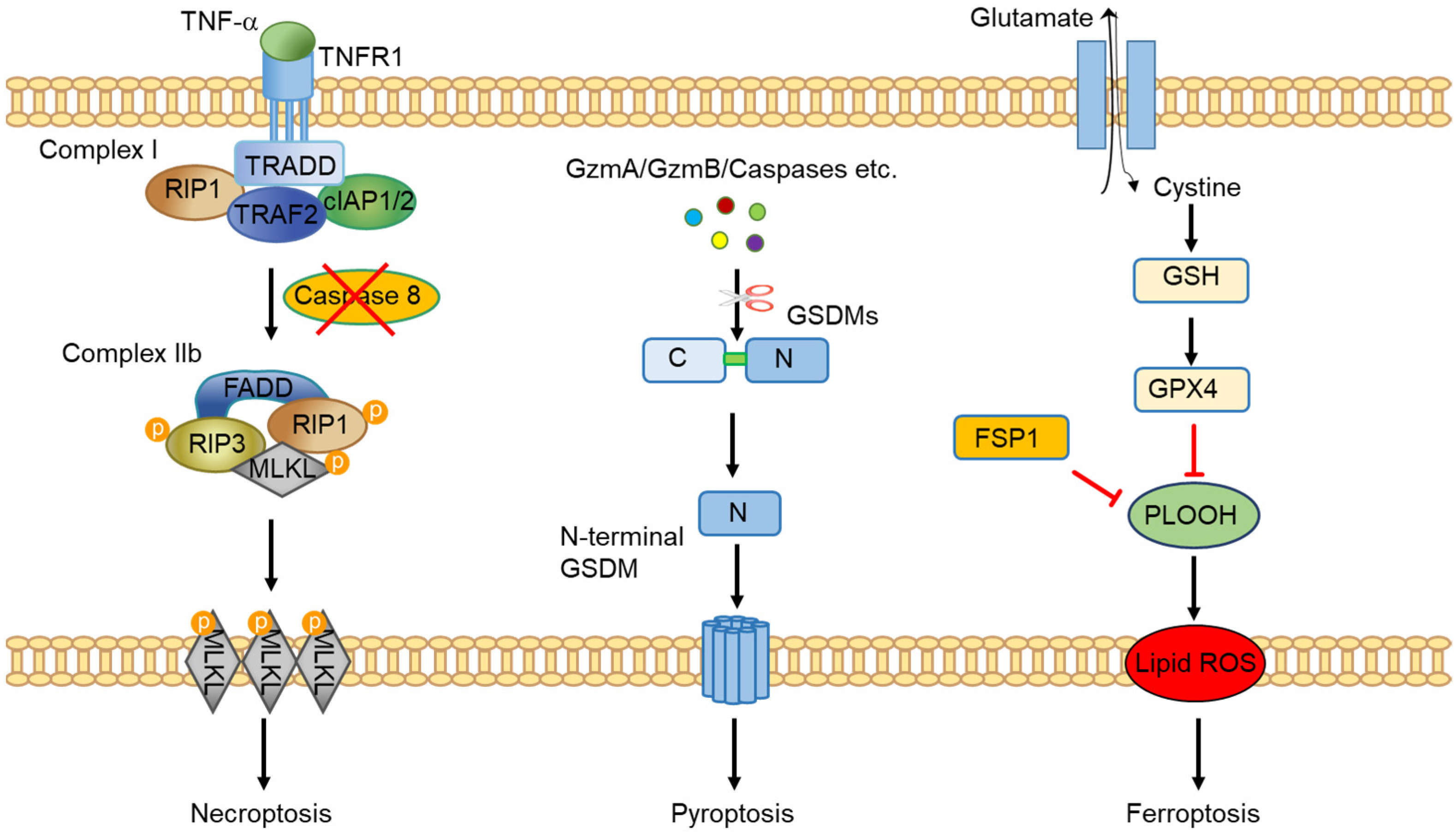
2. An Overview of Programmed Necrosis
3. Programmed Necrosis in Colorectal Tumor Cells
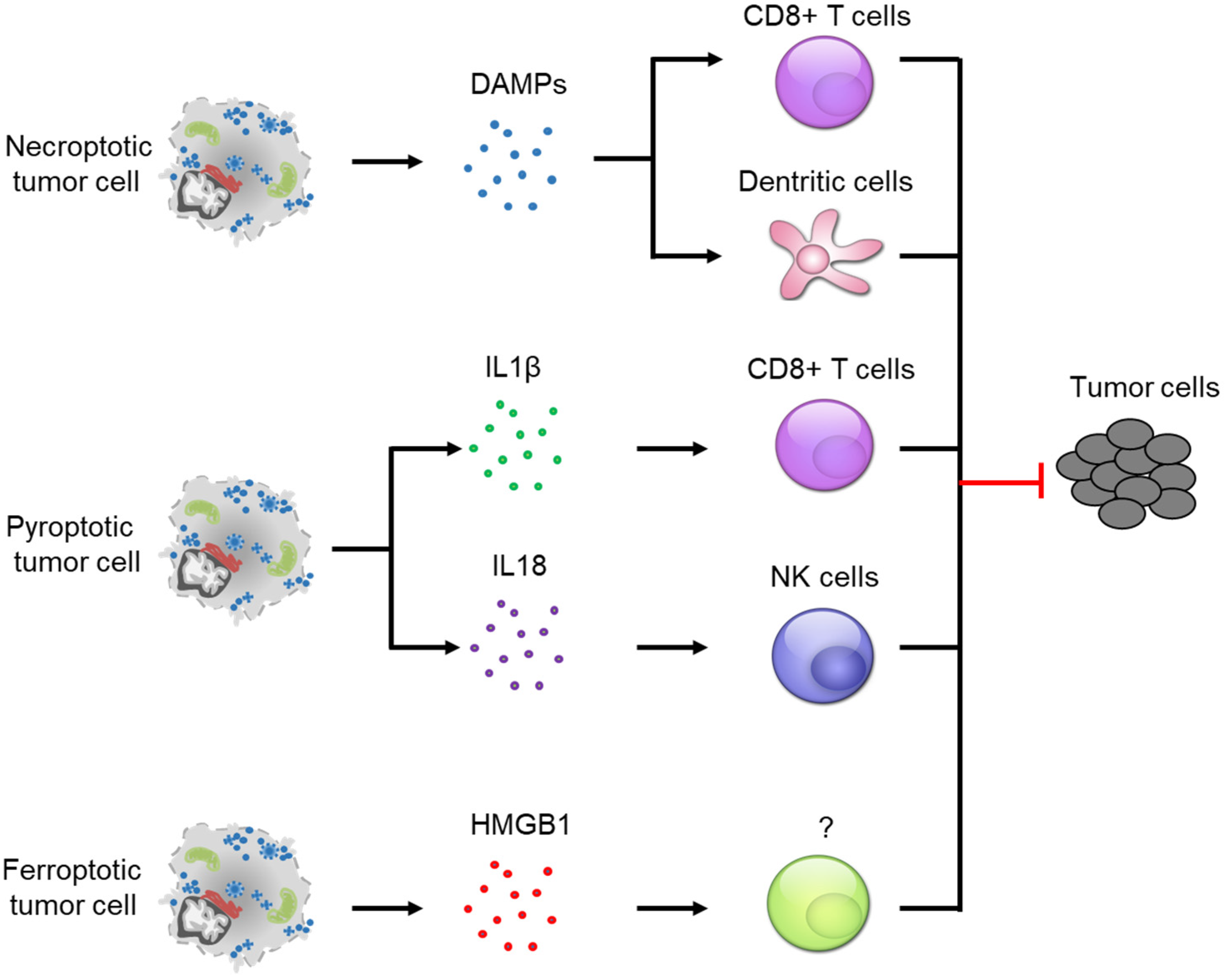
4. Crosstalk between Programmed Necrosis and Anti-Tumor Immunity
5. Targeting Programmed Necrosis in Colorectal Cancer
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability—A new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Patankar, J.V.; Becker, C. Cell death in the gut epithelium and implications for chronic inflammation. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 543–556. [Google Scholar] [CrossRef]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef]
- Koren, E.; Fuchs, Y. Modes of Regulated Cell Death in Cancer. Cancer Discov. 2021, 11, 245–265. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Caspase-8 in cancer biology and therapy. Cancer Lett. 2009, 281, 128–133. [Google Scholar] [CrossRef]
- Li, C.; Egloff, A.M.; Sen, M.; Grandis, J.R.; Johnson, D.E. Caspase-8 mutations in head and neck cancer confer resistance to death receptor-mediated apoptosis and enhance migration, invasion, and tumor growth. Mol. Oncol. 2014, 8, 1220–1230. [Google Scholar] [CrossRef]
- Fritsch, M.; Gunther, S.D.; Schwarzer, R.; Albert, M.C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef]
- Linkermann, A.; Brasen, J.H.; Darding, M.; Jin, M.K.; Sanz, A.B.; Heller, J.O.; De Zen, F.; Weinlich, R.; Ortiz, A.; Walczak, H.; et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2013, 110, 12024–12029. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Demon, D.; Bogaert, P.; Vandendriessche, B.; Goethals, A.; Depuydt, B.; Vuylsteke, M.; Roelandt, R.; Van Wonterghem, E.; Vandenbroecke, J.; et al. Simultaneous targeting of IL-1 and IL-18 is required for protection against inflammatory and septic shock. Am. J. Respir. Crit. Care Med. 2014, 189, 282–291. [Google Scholar] [CrossRef] [PubMed]
- He, G.W.; Gunther, C.; Thonn, V.; Yu, Y.Q.; Martini, E.; Buchen, B.; Neurath, M.F.; Sturzl, M.; Becker, C. Regression of apoptosis-resistant colorectal tumors by induction of necroptosis in mice. J. Exp. Med. 2017, 214, 1655–1662. [Google Scholar] [CrossRef]
- Tan, G.; Huang, C.; Chen, J.; Zhi, F. HMGB1 released from GSDME-mediated pyroptotic epithelial cells participates in the tumorigenesis of colitis-associated colorectal cancer through the ERK1/2 pathway. J. Hematol. Oncol. 2020, 13, 149. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, W.; Qi, Y.; Wang, S.; Li, L.; Li, W.; Xie, T.; Zhu, H.; Tang, Z.; Zhou, M. H2 S-Scavenged and Activated Iron Oxide-Hydroxide Nanospindles for MRI-Guided Photothermal Therapy and Ferroptosis in Colon Cancer. Small 2020, 16, e2001356. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Wong, S.W.; Martinez, J. Programmed Necrosis and Disease:We interrupt your regular programming to bring you necroinflammation. Cell Death Differ. 2019, 26, 25–40. [Google Scholar] [CrossRef]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef]
- Tonnus, W.; Belavgeni, A.; Beuschlein, F.; Eisenhofer, G.; Fassnacht, M.; Kroiss, M.; Krone, N.P.; Reincke, M.; Bornstein, S.R.; Linkermann, A. The role of regulated necrosis in endocrine diseases. Nat. Rev. Endocrinol. 2021, 17, 497–510. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [Green Version]
- Annibaldi, A.; Wicky John, S.; Vanden Berghe, T.; Swatek, K.N.; Ruan, J.; Liccardi, G.; Bianchi, K.; Elliott, P.R.; Choi, S.M.; Van Coillie, S.; et al. Ubiquitin-Mediated Regulation of RIPK1 Kinase Activity Independent of IKK and MK2. Mol. Cell 2018, 69, 566–580.e565. [Google Scholar] [CrossRef] [PubMed]
- Gunther, C.; He, G.W.; Kremer, A.E.; Murphy, J.M.; Petrie, E.J.; Amann, K.; Vandenabeele, P.; Linkermann, A.; Poremba, C.; Schleicher, U.; et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J. Clin. Investig. 2016, 126, 4346–4360. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Tanaka, S.; Fujii, T.; Aoki, A.; Komiyama, H.; Ezawa, K.; Sumiyama, K.; Sagai, T.; Shiroishi, T. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics 2007, 89, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Liu, X.; Xia, S.; Zhang, Z.; Wu, H.; Lieberman, J. Channelling inflammation: Gasdermins in physiology and disease. Nat. Rev. Drug. Discov. 2021, 20, 384–405. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef]
- Burgener, S.S.; Leborgne, N.G.F.; Snipas, S.J.; Salvesen, G.S.; Bird, P.I.; Benarafa, C. Cathepsin G Inhibition by Serpinb1 and Serpinb6 Prevents Programmed Necrosis in Neutrophils and Monocytes and Reduces GSDMD-Driven Inflammation. Cell Rep. 2019, 27, 3646–3656.e3645. [Google Scholar] [CrossRef]
- Karmakar, M.; Minns, M.; Greenberg, E.N.; Diaz-Aponte, J.; Pestonjamasp, K.; Johnson, J.L.; Rathkey, J.K.; Abbott, D.W.; Wang, K.; Shao, F.; et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1beta release independently of plasma membrane pores and pyroptosis. Nat. Commun. 2020, 11, 2212. [Google Scholar] [CrossRef]
- Evavold, C.L.; Hafner-Bratkovic, I.; Devant, P.; D′Andrea, J.M.; Ngwa, E.M.; Borsic, E.; Doench, J.G.; LaFleur, M.W.; Sharpe, A.H.; Thiagarajah, J.R.; et al. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 2021, 184, 4495–4511.e4419. [Google Scholar] [CrossRef]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O′Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, 7548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Zhou, B.; Sun, R.Y.; Ai, Y.L.; Cheng, K.; Li, F.N.; Wang, B.R.; Liu, F.J.; Jiang, Z.H.; Wang, W.J.; et al. The metabolite alpha-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell. Res. 2021, 31, 980–997. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.W.; You, Y.; Hsu, J.M.; Nie, L.; Chen, Y.; Wang, Y.C.; Liu, C.; et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell. Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef] [PubMed]
- LaRock, D.L.; Johnson, A.F.; Wilde, S.; Sands, J.S.; Monteiro, M.P.; LaRock, C.N. Group A Streptococcus induces GSDMA-dependent pyroptosis in keratinocytes. Nature 2022, 605, 527–531. [Google Scholar] [CrossRef]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Muller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kossl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell. 2019, 51, 575–586.e574. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef]
- Liu, Z.G.; Jiao, D. Necroptosis, tumor necrosis and tumorigenesis. Cell. Stress 2019, 4, 1–8. [Google Scholar] [CrossRef]
- Feng, X.; Song, Q.; Yu, A.; Tang, H.; Peng, Z.; Wang, X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma 2015, 62, 592–601. [Google Scholar] [CrossRef]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Orlowski, G.M.; Chan, F.K. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015, 6, e1636. [Google Scholar] [CrossRef]
- Li, X.; Guo, J.; Ding, A.P.; Qi, W.W.; Zhang, P.H.; Lv, J.; Qiu, W.S.; Sun, Z.Q. Association of Mixed Lineage Kinase Domain-Like Protein Expression with Prognosis in Patients with Colon Cancer. Technol. Cancer Res. Treat. 2017, 16, 428–434. [Google Scholar] [CrossRef] [Green Version]
- Bozec, D.; Iuga, A.C.; Roda, G.; Dahan, S.; Yeretssian, G. Critical function of the necroptosis adaptor RIPK3 in protecting from intestinal tumorigenesis. Oncotarget 2016, 7, 46384–46400. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Guo, J.; Cheng, X.; Liao, Y.; Bi, Y.; Gong, Y.; Zhang, X.; Guo, Y.; Wang, X.; Yu, W.; et al. RIPK3 Suppresses the Progression of Spontaneous Intestinal Tumorigenesis. Front. Oncol. 2021, 11, 664927. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Diaz, S.; Preaudet, A.; Samson, A.L.; Nguyen, P.M.; Fung, K.Y.; Garnham, A.L.; Alexander, W.S.; Strasser, A.; Ernst, M.; Putoczki, T.L.; et al. Necroptosis is dispensable for the development of inflammation-associated or sporadic colon cancer in mice. Cell Death Differ. 2021, 28, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Martens, S.; Bridelance, J.; Roelandt, R.; Vandenabeele, P.; Takahashi, N. MLKL in cancer: More than a necroptosis regulator. Cell Death Differ. 2021, 28, 1757–1772. [Google Scholar] [CrossRef]
- He, S.; Wang, X. RIP kinases as modulators of inflammation and immunity. Nat. Immunol. 2018, 19, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Thi Nguyen, D.; Hattori, T.; Manh Le, T.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 2091. [Google Scholar] [CrossRef]
- Miguchi, M.; Hinoi, T.; Shimomura, M.; Adachi, T.; Saito, Y.; Niitsu, H.; Kochi, M.; Sada, H.; Sotomaru, Y.; Ikenoue, T.; et al. Gasdermin C Is Upregulated by Inactivation of Transforming Growth Factor beta Receptor Type II in the Presence of Mutated Apc, Promoting Colorectal Cancer Proliferation. PLoS ONE 2016, 11, e0166422. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; He, Y.; Lin, L.; Chen, P.; Chen, M.; Zhang, S. The emerging role of ferroptosis in intestinal disease. Cell Death Dis. 2021, 12, 289. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mo, J.; Dai, J.; Ye, C.; Cen, W.; Zheng, X.; Jiang, L.; Ye, L. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 2021, 12, 1079. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yu, C.; Luo, M.; Cen, C.; Qiu, J.; Zhang, S.; Hu, K. Ferroptosis in Cancer Treatment: Another Way to Rome. Front. Oncol. 2020, 10, 571127. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef]
- Zhang, X.; Sui, S.; Wang, L.; Li, H.; Zhang, L.; Xu, S.; Zheng, X. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J. Cell. Physiol. 2020, 235, 3425–3437. [Google Scholar] [CrossRef]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin′s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef]
- Serebriiskii, I.G.; Connelly, C.; Frampton, G.; Newberg, J.; Cooke, M.; Miller, V.; Ali, S.; Ross, J.S.; Handorf, E.; Arora, S.; et al. Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat. Commun. 2019, 10, 3722. [Google Scholar] [CrossRef]
- Chen, P.; Li, X.; Zhang, R.; Liu, S.; Xiang, Y.; Zhang, M.; Chen, X.; Pan, T.; Yan, L.; Feng, J.; et al. Combinative treatment of beta-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics 2020, 10, 5107–5119. [Google Scholar] [CrossRef]
- Yang, W.H.; Ding, C.C.; Sun, T.; Rupprecht, G.; Lin, C.C.; Hsu, D.; Chi, J.T. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell. Rep. 2019, 28, 2501–2508 e2504. [Google Scholar] [CrossRef]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell. 2010, 19, 491–505. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Chakraborty, B.; Safi, R.; Kazmin, D.; Chang, C.Y.; McDonnell, D.P. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat. Commun. 2021, 12, 5103. [Google Scholar] [CrossRef] [PubMed]
- Ferroptosis Is Inhibited in Lymph, Promoting Metastasis of Melanoma. Cancer Discov. 2020, 10, 1621. [CrossRef]
- Tasdogan, A.; Ubellacker, J.M.; Morrison, S.J. Redox Regulation in Cancer Cells during Metastasis. Cancer Discov. 2021, 11, 2682–2692. [Google Scholar] [CrossRef] [PubMed]
- Riera Romo, M. Cell death as part of innate immunity: Cause or consequence? Immunology 2021, 163, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell. Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef]
- Snyder, A.G.; Hubbard, N.W.; Messmer, M.N.; Kofman, S.B.; Hagan, C.E.; Orozco, S.L.; Chiang, K.; Daniels, B.P.; Baker, D.; Oberst, A. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci. Immunol. 2019, 4, 2004. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Zheng, M.; Li, Y.M.; Fan, X.Y.; Wang, J.C.; Li, Z.C.; Yang, H.J.; Yu, J.M.; Cui, J.; Jiang, J.L.; et al. RIP3 promotes colitis-associated colorectal cancer by controlling tumor cell proliferation and CXCL1-induced immune suppression. Theranostics 2019, 9, 3659–3673. [Google Scholar] [CrossRef]
- Moriwaki, K.; Balaji, S.; McQuade, T.; Malhotra, N.; Kang, J.; Chan, F.K. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity 2014, 41, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Orning, P.; Lien, E.; Fitzgerald, K.A. Gasdermins and their role in immunity and inflammation. J. Exp. Med. 2019, 216, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Malik, A.; Guy, C.S.; Karki, R.; Vogel, P.; Kanneganti, T.D. Pyrin Inflammasome Regulates Tight Junction Integrity to Restrict Colitis and Tumorigenesis. Gastroenterology 2018, 154, 948–964.e948. [Google Scholar] [CrossRef]
- Dupaul-Chicoine, J.; Arabzadeh, A.; Dagenais, M.; Douglas, T.; Champagne, C.; Morizot, A.; Rodrigue-Gervais, I.G.; Breton, V.; Colpitts, S.L.; Beauchemin, N.; et al. The Nlrp3 Inflammasome Suppresses Colorectal Cancer Metastatic Growth in the Liver by Promoting Natural Killer Cell Tumoricidal Activity. Immunity 2015, 43, 751–763. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Lin, K.H.; Chang, L.S.; Tian, C.Y.; Yeh, Y.C.; Chen, Y.J.; Chuang, T.H.; Liu, S.J.; Leng, C.H. Carboxyl-terminal fusion of E7 into Flagellin shifts TLR5 activation to NLRC4/NAIP5 activation and induces TLR5-independent anti-tumor immunity. Sci. Rep. 2016, 6, 24199. [Google Scholar] [CrossRef]
- Garaude, J.; Kent, A.; van Rooijen, N.; Blander, J.M. Simultaneous targeting of toll- and nod-like receptors induces effective tumor-specific immune responses. Sci. Transl. Med. 2012, 4, 120ra116. [Google Scholar] [CrossRef]
- Lee, P.H.; Yamamoto, T.N.; Gurusamy, D.; Sukumar, M.; Yu, Z.; Hu-Li, J.; Kawabe, T.; Gangaplara, A.; Kishton, R.J.; Henning, A.N.; et al. Host conditioning with IL-1beta improves the antitumor function of adoptively transferred T cells. J. Exp. Med. 2019, 216, 2619–2634. [Google Scholar] [CrossRef]
- Derangere, V.; Chevriaux, A.; Courtaut, F.; Bruchard, M.; Berger, H.; Chalmin, F.; Causse, S.Z.; Limagne, E.; Vegran, F.; Ladoire, S.; et al. Liver X receptor beta activation induces pyroptosis of human and murine colon cancer cells. Cell Death Differ. 2014, 21, 1914–1924. [Google Scholar] [CrossRef]
- Yamazaki, T.; Hannani, D.; Poirier-Colame, V.; Ladoire, S.; Locher, C.; Sistigu, A.; Prada, N.; Adjemian, S.; Catani, J.P.; Freudenberg, M.; et al. Defective immunogenic cell death of HMGB1-deficient tumors: Compensatory therapy with TLR4 agonists. Cell Death Differ. 2014, 21, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, K.; Borchert, A.; Kuhn, H. Inverse regulation of lipid-peroxidizing and hydroperoxyl lipid-reducing enzymes by interleukins 4 and 13. FASEB J. 1999, 13, 143–154. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell. Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, A.; Tanaka, S.; Cicka, M.; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Investig. 2019, 129, 2293–2304. [Google Scholar] [CrossRef]
- Allam, R.; Kumar, S.V.; Darisipudi, M.N.; Anders, H.J. Extracellular histones in tissue injury and inflammation. J. Mol. Med. 2014, 92, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Cederquist, L.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Engstrom, P.F.; et al. NCCN Guidelines Insights: Colon Cancer, Version 2.2018. J. Natl. Compr. Canc. Netw. 2018, 16, 359–369. [Google Scholar] [CrossRef]
- Brown, J.M.; Attardi, L.D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar] [CrossRef]
- Jebelli, A.; Baradaran, B.; Mosafer, J.; Baghbanzadeh, A.; Mokhtarzadeh, A.; Tayebi, L. Recent developments in targeting genes and pathways by RNAi-based approaches in colorectal cancer. Med. Res. Rev. 2021, 41, 395–434. [Google Scholar] [CrossRef]
- Nehs, M.A.; Lin, C.I.; Kozono, D.E.; Whang, E.E.; Cho, N.L.; Zhu, K.; Moalem, J.; Moore, F.D., Jr.; Ruan, D.T. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery 2011, 150, 1032–1039. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Zhu, Y.; Li, N.; Li, W.; Shang, C.; Song, G.; Li, S.; Cong, J.; Li, T.; et al. Apoptin induces pyroptosis of colorectal cancer cells via the GSDME-dependent pathway. Int. J. Biol. Sci. 2022, 18, 717–730. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Chen, C.; Du, W.; Jing, W.; Sun, P.; Shi, C.; Zhang, S.; Liu, Y.; Cui, P.; Li, A.; Zhang, R.; et al. Leveraging tumor cell ferroptosis for colorectal cancer treatment via nanoelicitor-activated tumoricidal immunity. Chem. Eng. J. 2022, 430, 132983. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion during Chronic Viral Infection and Cancer. Ann. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef]
- Yu, J.; Zhong, B.; Xiao, Q.; Du, L.; Hou, Y.; Sun, H.S.; Lu, J.J.; Chen, X. Induction of programmed necrosis: A novel anti-cancer strategy for natural compounds. Pharmacol. Ther. 2020, 214, 107593. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.-Q.; Gamez-Belmonte, R.; Patankar, J.V.; Liebing, E.; Becker, C. The Role of Programmed Necrosis in Colorectal Cancer. Cancers 2022, 14, 4295. https://doi.org/10.3390/cancers14174295
Yu Y-Q, Gamez-Belmonte R, Patankar JV, Liebing E, Becker C. The Role of Programmed Necrosis in Colorectal Cancer. Cancers. 2022; 14(17):4295. https://doi.org/10.3390/cancers14174295
Chicago/Turabian StyleYu, Yu-Qiang, Reyes Gamez-Belmonte, Jay V. Patankar, Eva Liebing, and Christoph Becker. 2022. "The Role of Programmed Necrosis in Colorectal Cancer" Cancers 14, no. 17: 4295. https://doi.org/10.3390/cancers14174295
APA StyleYu, Y.-Q., Gamez-Belmonte, R., Patankar, J. V., Liebing, E., & Becker, C. (2022). The Role of Programmed Necrosis in Colorectal Cancer. Cancers, 14(17), 4295. https://doi.org/10.3390/cancers14174295