

BRAF and MEK Targeted Therapies in Pediatric Central Nervous System Tumors
 , ,
, , 
 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Blood-Brain Barrier (BBB) and Brain Drug Delivery
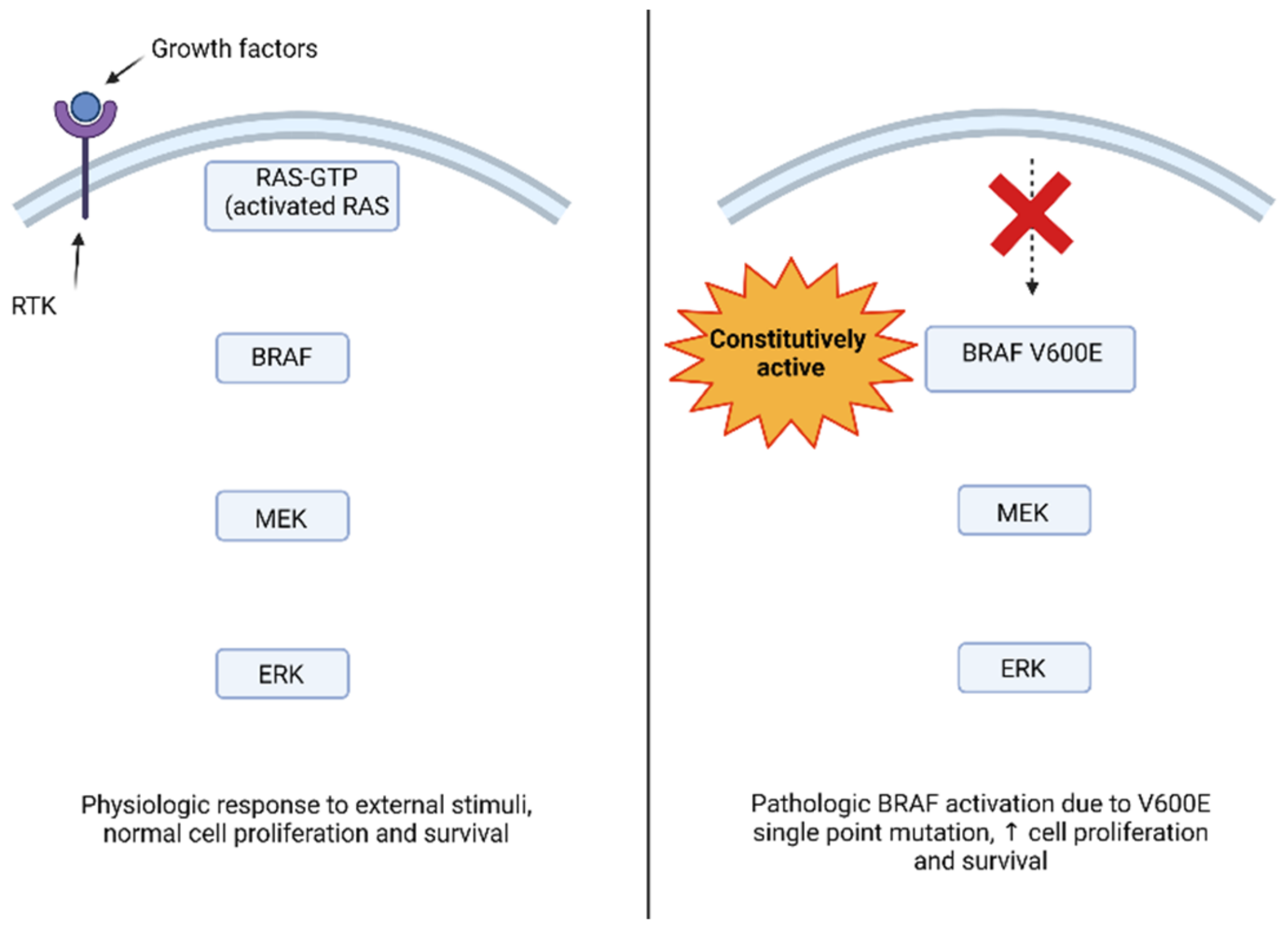
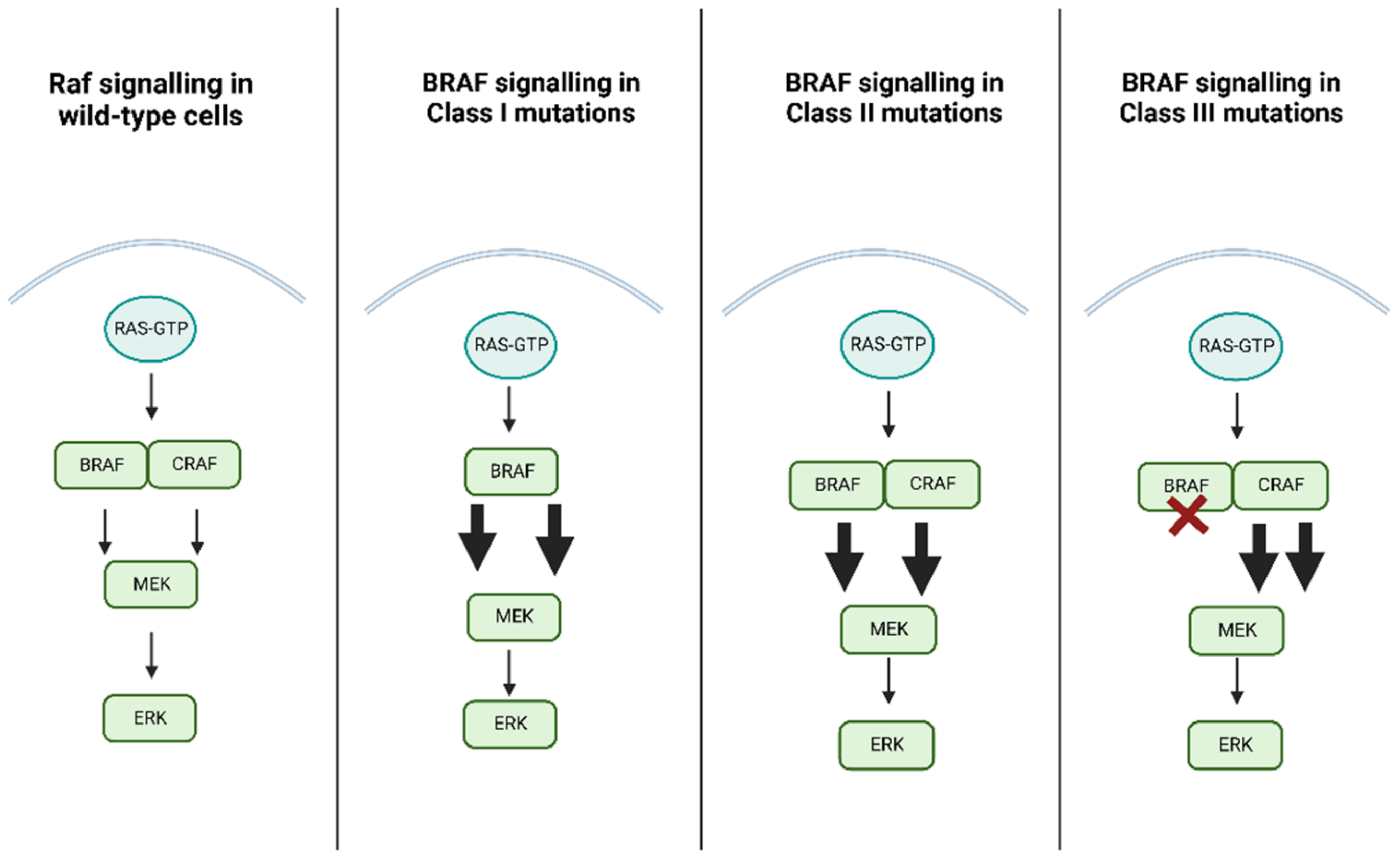
3. BRAF Function and Pathologic Activation
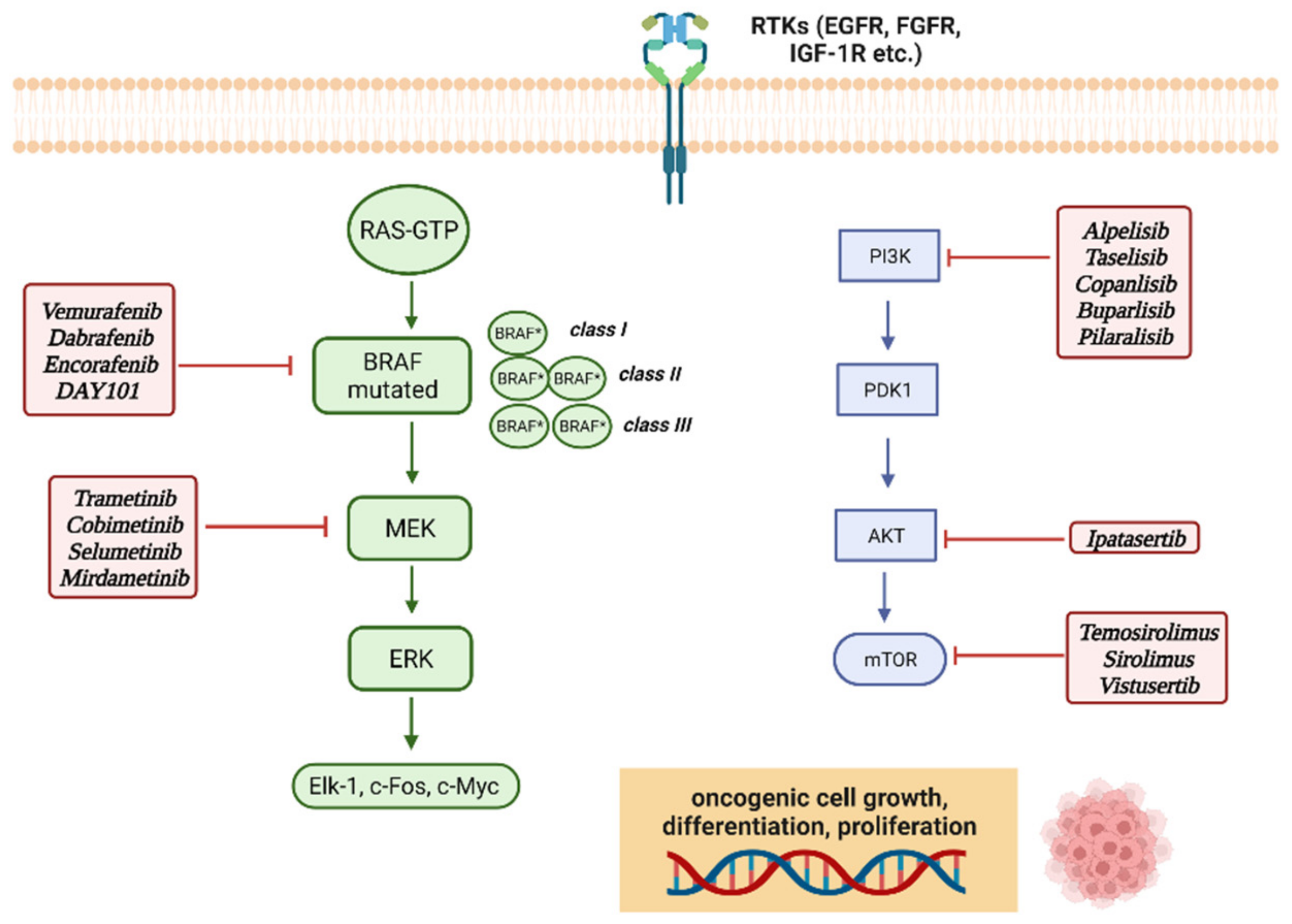
4. MAPK (Mitogen-Activated Protein Kinases) and mTOR Molecular Pathways
5. BRAF and MEK Inhibitors in Pediatric CNS Tumors
5.1. Pediatric Low-Grade Glioma (pLLG)
5.2. Pediatric High-Grade Glioma (pHGG)
5.3. Other Tumors
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Triarico, S.; Maurizi, P.; Mastrangelo, S.; Attinà, G.; Capozza, M.A.; Ruggiero, A. Improving the Brain Delivery of Chemotherapeutic Drugs in Childhood Brain Tumors. Cancers 2019, 11, 824. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child.-Educ. Pract. Ed. 2013, 98, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.stjude.org/research/translational-innovation/pediatric-cancer-genome-project.html (accessed on 6 June 2022).
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed]
- Belden, S.; Flaherty, K.T. MEK and RAF inhibitors for BRAF-mutated cancers. Expert Rev. Mol. Med. 2012, 14, e17. [Google Scholar] [CrossRef]
- Bouchè, V.; Aldegheri, G.; Donofrio, C.A.; Fioravanti, A.; Roberts-Thomson, S.; Fox, S.B.; Schettini, F.; Generali, D. BRAF Signaling Inhibition in Glioblastoma: Which Clinical Perspectives? Front. Oncol. 2021, 11, 772052. [Google Scholar] [CrossRef]
- Ryall, S.; Tabori, U.; Hawkins, C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol. Commun. 2020, 8, 30. [Google Scholar] [CrossRef]
- Pardridge, W.M. CSF, blood-brain barrier, and brain drug delivery. Expert Opin. Drug Deliv. 2016, 13, 963–975. [Google Scholar] [CrossRef]
- Gaillard, P.J.; Appeldoorn, C.C.M.; Dorland, R.; van Kregten, J.; Manca, F.; Vugts, D.J.; Windhorst, B.; van Dongen, G.A.M.S.; de Vries, H.E.; Maussang, D.; et al. Pharmacokinetics, brain delivery, and efficacy in brain tumor-bearing mice of glutathione pegylated liposomal doxorubicin (2B3-101). PLoS ONE 2014, 9, e82331. [Google Scholar]
- Lockman, P.R.; Mumper, R.J.; Khan, M.A.; Allen, D.D. Nanoparticle technology for drug delivery across the blood–brain barrier. Drug Dev. Ind. Pharm. 2002, 28, 1–13. [Google Scholar] [CrossRef]
- Pandit, R.; Chen, L.; Götz, J. The blood-brain barrier: Physiology and strategies for drug delivery. Adv. Drug Deliv. Rev. 2019, 165–166, 1–14. [Google Scholar] [CrossRef]
- Haumann, R.; Videira, J.C.; Kaspers, G.J.L.; van Vuurden, D.G.; Hulleman, E. Overview of Current Drug Delivery Methods Across the Blood–Brain Barrier for the Treatment of Primary Brain Tumors. CNS Drugs 2020, 34, 1121–1131. [Google Scholar] [CrossRef]
- De Gooijer, M.C.; Zhang, P.; Weijer, R.; Buil, L.C.M.; Beijnen, J.H.; van Tellingen, O. The impact of Pglycoprotein and breast cancer resistance protein on the brain pharmacokinetics and pharmacodynamics of a panel of MEK inhibitors. Int. J. Cancer 2018, 142, 381–391. [Google Scholar] [CrossRef]
- Meel, M.H.; De Gooijer, M.C.; Guillén Navarro, M.; Waranecki, P.; Breur, M.; Buil, L.; Wedekind, L.E.; Twisk, J.W.; Koster, J.; Hashizume, R.; et al. MELK Inhibition in Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2018, 24, 5645–5657. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef]
- Niault, T.S.; Baccarini, M. Targets of Raf in tumorigenesis. Carcinogenesis 2010, 31, 1165–1174. [Google Scholar] [CrossRef]
- Shaul, Y.D.; Gibor, G.; Plotnikov, A.; Seger, R. Specific phosphorylation and activation of ERK1c by MEK1b: A unique route in the ERK cascade. Genes Dev. 2009, 23, 1779–1790. [Google Scholar] [CrossRef][Green Version]
- Yao, Z.; Seger, R. The ERK signaling cascade—Views from different subcellular compartments. Biofactors 2009, 35, 407–416. [Google Scholar] [CrossRef]
- Maraka, S.; Janku, F. BRAF alterations in primary brain tumors. Discov. Med. 2018, 26, 51–60. [Google Scholar]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef]
- Flaherty, K.T.; McArthur, G. BRAF, a target in melanoma: Implications for solid tumor drug development. Cancer 2010, 116, 4902–4913. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Herter, S.; Tien, J.; Wong, L.; Berry, L.; Chan, J.; O’Brien, C.; Modrusan, Z.; Seshagiri, S.; Lackner, M.; et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAF V600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. 2009, 69, 3042–3051. [Google Scholar] [CrossRef] [PubMed]
- Schirripa, M.; Biason, P.; Lonardi, S.; Pella, N.; Pino, M.S.; Urbano, F.; Antoniotti, C.; Cremolini, C.; Corallo, S.; Pietrantonio, F.; et al. Class 1, 2, and 3 BRAF-Mutated Metastatic Colorectal Cancer: A Detailed Clinical, Pathologic, and Molecular Characterization. Clin. Cancer Res. 2019, 25, 3954–3961. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef]
- Freeman, A.K.; Ritt, D.A.; Morrison, D.K. The importance of Raf dimerization in cell signaling. Small GTPases 2013, 4, 180–185. [Google Scholar] [CrossRef]
- Gibney, G.T.; Messina, J.L.; Fedorenko, I.V.; Sondak, V.K.; Smalley, K.S.M. Paradoxical oncogenesis—The long-term effects of BRAF inhibition in melanoma. Nat. Rev. Clin. Oncol. 2013, 10, 390–399. [Google Scholar] [CrossRef]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Frisone, D.; Friedlaender, A.; Malapelle, U.; Banna, G.; Addeo, A. A BRAF new world. Crit. Rev. Oncol. Hematol. 2020, 152, 103008. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.; Puzanov, I.; Sosman, J.; Kim, K.; Ribas, A.; McArthur, G.; Lee, R.J.; Grippo, J.F.; Nolop, K.; Chapman, P. Phase I study of PLX4032: Proof of concept for V600E BRAF mutation as a therapeutic target in human cancer. J. Clin. Oncol. 2009, 27 (Suppl. S15), 9000. [Google Scholar] [CrossRef]
- Holderfield, M.; Nagel, T.E.; Stuart, D.D. Mechanism and consequences of RAF kinase activation by small-molecule inhibitors. Br. J. Cancer 2014, 111, 640–645. [Google Scholar] [CrossRef]
- Noeparast, A.; Giron, P.; De Brakeleer, S.; Eggermont, C.; De Ridder, U.; Teugels, E.; De Grève, J. Type II RAF inhibitor causes superior ERK pathway suppression compared to type I RAF inhibitor in cells expressing different BRAF mutant types recurrently found in lung cancer. Oncotarget 2018, 9, 16110–16123. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, K.; Zhu, X.; Lin, G.; Song, F.; Zhao, Y.; Piao, Y.; Liu, J.; Cheng, W.; Bi, X.; et al. Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAF V600E melanoma cells. Cancer Lett. 2016, 370, 332–344. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16 (Suppl. S2), S17–S27. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Das Thakur, M.; Stuart, D.D. Molecular pathways: Response and resistance to BRAF and MEK inhibitors in BRAF V600E tumors. Clin. Cancer Res. 2014, 20, 1074–1080. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, H. Current Development Status of MEK Inhibitors. Molecules 2017, 22, 1551. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kakefuda, R.; Tajima, N.; Sowa, Y.; Sakai, T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int. J. Oncol. 2011, 39, 23–31. [Google Scholar] [CrossRef]
- Yeh, T.C.; Marsh, V.; Bernat, B.A.; Ballard, J.; Colwell, H.; Evans, R.J.; Parry, J.; Smith, D.; Brandhuber, B.J.; Gross, S.; et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin. Cancer Res. 2007, 13, 1576–1583. [Google Scholar] [CrossRef]
- Fangusaro, J.R.; Onar-Thomas, A.; Young-Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.I.; Banerjee, A.; Packer, R.; Kilburn, L.B.; Pollack, I.; et al. A phase II prospective study of selumetinib in children with recurrent or refractory low-grade glioma (LGG): A Pediatric Brain Tumor Consortium (PBTC) study. J. Clin. Oncol. 2017, 19, iv34–iv35. [Google Scholar] [CrossRef]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 2003, 28, 573–576. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Memmott, R.M.; Dennis, P.A. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell. Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Polivka, J., Jr.; Janku, F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 2014, 142, 164–175. [Google Scholar] [CrossRef]
- Kieran, M.W.; Yao, X.; Macy, M.; Leary, S.; Cohen, K.; Macdonald, T.; Allen, J.; Boklan, J.; Smith, A.; Nazemi, K.; et al. Final results of a prospective multi-institutional phase II study of everolimus (RAD001), an mTOR inhibitor, in pediatric patients with recurrent or progressive low-grade glioma. A POETIC consortium trial. Neuro-Oncology 2014, 16, iii27. [Google Scholar] [CrossRef][Green Version]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncology 2020, 22 (Suppl. S1), iv1–iv96. [Google Scholar] [CrossRef]
- Krishnatry, R.; Zhukova, N.; Guerreiro Stucklin, A.S.; Pole, J.D.; Mistry, M.; Fried, I.; Ramaswamy, V.; Bartels, U.; Huang, A.; Laperriere, N.; et al. Clinical and treatment factors determining long-term outcomes for adult survivors of childhood low-grade glioma: A population-based study. Cancer 2016, 122, 1261–1269. [Google Scholar] [CrossRef]
- De Blank, P.; Bandopadhayay, P.; Haas-Kogan, D.; Fouladi, M.; Fangusaro, J. Management of pediatric low-grade glioma. Curr. Opin. Pediatr. 2019, 31, 21–27. [Google Scholar] [CrossRef]
- The St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar] [CrossRef]
- Fangusaro, J.; Onar-Thomas, A.; Poussaint, T.Y.; Wu, S.; Ligon, A.H.; Lindeman, N.I.; Banerjee, A.; Packer, R.; Kilburn, L.B.; Pollack, I.F.; et al. LTBK-01. Updates on the phase II and re-treatment study of AZD6244 (Selumetinib) for children with recurrent or refractory pediatric low-grade glioma: A pediatric brain tumor consortium (PBTC) study. Neuro-Oncology 2018, 20 (Suppl. S2), i214. [Google Scholar] [CrossRef]
- Schreck, K.C.; Grossman, S.A.; Pratilas, C.A. BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers 2019, 11, 1262. [Google Scholar] [CrossRef]
- Geoerger, B.; Moertel, C.L.; Whitlock, J.; McCowage, G.B.; Kieran, M.W.; Broniscer, A.; Hargrave, D.R.; Hingorani, P.; Kilburn, L.B.; Mueller, S.; et al. Phase 1 trial of trametinib alone and in combination with dabrafenib in children and adolescents with relapsed solid tumors or neurofibromatosis type 1 (NF1) progressive plexiform neurofibromas (PN). J. Clin. Oncol. 2018, 36, 10537. [Google Scholar] [CrossRef]
- Rankin, A.; Johnson, A.; Roos, A.; Kannan, G.; Knipstein, J.; Britt, N.; Rosenzweig, M.; Haberberger, J.; Pavlick, D.; Severson, E.; et al. Targetable BRAF and RAF1 Alterations in Advanced Pediatric Cancers. Oncologist 2021, 26, e153–e163. [Google Scholar] [CrossRef]
- Karajannis, M.A.; Legault, G.; Fisher, M.J.; Milla, S.S.; Cohen, K.J.; Wisoff, J.H.; Harter, D.H.; Goldberg, J.D.; Hochman, T.; Merkelson, A.; et al. Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro-Oncology 2014, 16, 1408–1416. [Google Scholar] [CrossRef] [PubMed]
- Sievert, A.J.; Lang, S.S.; Boucher, K.L.; Madsen, P.J.; Slaunwhite, E.; Choudhari, N.; Kellet, M.; Storm, P.B.; Resnick, A.C. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc. Natl. Acad. Sci. USA 2013, 110, 5957–5962. [Google Scholar] [CrossRef]
- Davies, K.G.; Maxwell, R.E.; Seljeskog, E.; Sung, J.H. Pleomorphic xanthoastrocytoma—Report of four cases, with MRI scan appearances and literature review. Br. J. Neurosurg. 1994, 8, 681–689. [Google Scholar] [CrossRef]
- Srinivasa, K.; Cross, K.A.; Dahiya, S. BRAF Alteration in Central and Peripheral Nervous System Tumors. Front. Oncol. 2020, 10, 574974. [Google Scholar] [CrossRef] [PubMed]
- Schindler, G.; Capper, D.; Meyer, J.; Janzarik, W.; Omran, H.; Herold-Mende, C.; Schmieder, K.; Wesseling, P.; Mawrin, C.; Hasselblatt, M.; et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011, 121, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Ida, C.M.; Rodriguez, F.J.; Burger, P.C.; Caron, A.A.; Jenkins, S.M.; Spears, G.M.; Aranguren, D.L.; Lachance, D.H.; Giannini, C. Pleomorphic Xanthoastrocytoma: Natural History and Long-Term Follow-Up. Brain Pathol. 2015, 25, 575–586. [Google Scholar] [CrossRef]
- Pediatric Low-Grade Glioma—MEKinhibitor TRIal Vs. Chemotherapy (PLGG—MEKTRIC). Available online: https://clinicaltrials.gov/ct2/show/NCT05180825 (accessed on 6 June 2022).
- Phase II Pediatric Study with Dabrafenib in Combination with Trametinib in Patients with HGG and LGG. Available online: https://clinicaltrials.gov/ct2/show/NCT02684058 (accessed on 6 June 2022).
- Kaley, T.; Touat, M.; Subbiah, V.; Hollebecque, A.; Rodon, J.; Lockhart, A.C.; Keedy, V.; Bielle, F.; Hofheinz, R.D.; Joly, F.; et al. BRAF Inhibition in BRAFV600-Mutant Gliomas: Results From the VE-BASKET Study. J. Clin. Oncol. 2018, 36, 3477–3484. [Google Scholar] [CrossRef]
- Ellison, D.W.; Hawkins, C.; Jones, D.T.W.; Onar-Thomas, A.; Pfister, S.M.; Reifenberger, G.; Louis, D.N. cIMPACT-NOW update 4: Diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF. Acta Neuropathol. 2019, 137, 683–687. [Google Scholar] [CrossRef]
- Suri, V.; Jha, P.; Agarwal, S.; Pathak, P.; Sharma, M.C.; Sharma, V.; Shukla, S.; Somasundaram, K.; Mahapatra, A.K.; Kale, S.S.; et al. Molecular profile of oligodendrogliomas in young patients. Neuro-Oncology 2011, 13, 1099–1106. [Google Scholar] [CrossRef]
- Evaluation of Hippocampal-Avoidance Using Proton Therapy in Low-Grade Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT04065776 (accessed on 6 June 2022).
- SJ901: Evaluation of Mirdametinib in Children, Adolescents, and Young Adults with Low-Grade Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT04923126 (accessed on 6 June 2022).
- Jones, C.; Perryman, L.; Hargrave, D. Paediatric and adult malignant glioma: Close relatives or distant cousins? Nat. Rev. Clin. Oncol. 2012, 9, 400–413. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Molinari, V.; Jones, D.T.W.; Izquierdo, E.; Brouwer-Visser, J.; Giangaspero, F.; Haberler, C.; Pietsch, T.; Jacques, T.S.; et al. Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 2018, 33, 829–842.e5. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. The 2021 WHO classification of tumors, 5th edition, central nervous system tumors: The 10 basic principles. Brain Tumor Pathol. 2022, 39, 47–50. [Google Scholar] [CrossRef]
- George, E.; Settler, A.; Connors, S.; Greenfield, J.P. Pediatric Gliomatosis Cerebri: A Review of 15 Years. J. Child. Neurol. 2016, 31, 378–387. [Google Scholar] [CrossRef]
- Herrlinger, U.; Jones, D.T.W.; Glas, M.; Hattingen, E.; Gramatzki, D.; Stuplich, M.; Felsberg, J.; Bähr, O.; Gielen, G.H.; Simon, M.; et al. Gliomatosis cerebri: No evidence for a separate brain tumor entity. Acta Neuropathol. 2016, 131, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Kline, C.; Felton, E.; Allen, I.E.; Tahir, P.; Mueller, S. Survival outcomes in pediatric recurrent high-grade glioma: Results of a 20-year systematic review and meta-analysis. J. Neurooncol. 2018, 137, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Behling, F.; Barrantes-Freer, A.; Skardelly, M.; Nieser, M.; Christians, A.; Stockhammer, F.; Rohde, V.; Tatagiba, M.; Hartmann, C.; Stadelmann, C.; et al. Frequency of BRAF V600E mutations in 969 central nervous system neoplasms. Diagn. Pathol. 2016, 11, 55. [Google Scholar] [CrossRef]
- Sturm, D.; Pfister, S.M.; Jones, D.T.W. Pediatric Gliomas: Current Concepts on Diagnosis, Biology, and Clinical Management. J. Clin. Oncol. 2017, 35, 2370–2377. [Google Scholar] [CrossRef]
- Khalid, S.I.; Kelly, R.; Adogwa, O.; Carlton, A.; Tam, E.; Naqvi, S.; Kushkuley, J.; Ahmad, S.; Woodward, J.; Khanna, R.; et al. Pediatric Brainstem Gliomas: A Retrospective Study of 180 Patients from the SEER Database. Pediatr. Neurosurg. 2019, 54, 151–164. [Google Scholar] [CrossRef]
- Dabrafenib Combined with Trametinib after Radiation Therapy in Treating Patients with Newly-Diagnosed High-Grade Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT03919071 (accessed on 6 June 2022).
- Vemurafenib in Treating Patients With Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphoma, or Histiocytic Disorders With BRAF V600 Mutations (A Pediatric MATCH Treatment Trial). Available online: https://clinicaltrials.gov/ct2/show/NCT03220035 (accessed on 6 June 2022).
- Safety and Pharmacokinetics of Cobimetinib in Pediatric and Young Adult Participants with Previously Treated Solid Tumors (iMATRIXcobi). Available online: https://clinicaltrials.gov/ct2/show/NCT02639546 (accessed on 6 June 2022).
- Dahiya, S.; Haydon, D.H.; Alvarado, D.; Gurnett, C.A.; Gutmann, D.H.; Leonard, J.R. BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol. 2013, 125, 901–910. [Google Scholar] [CrossRef]
- Tiwari, S.; Yadav, T.; Pamnani, J.; Mathew, J.M.; Elhence, P.; Praneeth, K.; Vedant, D.; Khera, P.S.; Garg, P.; Vyas, V. Diffuse Leptomeningeal Glioneuronal Tumor: A Unique Leptomeningeal Tumor Entity. World Neurosurg. 2020, 135, 297–300. [Google Scholar] [CrossRef]
- Huse, J.T.; Snuderl, M.; Jones, D.T.; Brathwaite, C.D.; Altman, N.; Lavi, E.; Saffery, R.; Sexton-Oates, A.; Blumcke, I.; Capper, D.; et al. Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): An epileptogenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic alterations involving the MAP kinase pathway. Acta Neuropathol. 2017, 133, 417–429. [Google Scholar] [CrossRef]
- Deng, M.Y.; Sill, M.; Chiang, J.; Schittenhelm, J.; Ebinger, M.; Schuhmann, M.U.; Monoranu, C.M.; Milde, T.; Wittmann, A.; Hartmann, C.; et al. Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol. 2018, 136, 239–253. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Class I | Class II | Class III |
|---|---|---|
| p.V600D/E/K/M/R | p.G464E/V; p. G469A/R/V; p. L597Q/V; p.K601E/N/T; gene fusion | p.D287H; p.V459L; p.G466A/E/V; p.S467L; p.G469E; p.N581I/S; p.D594A/G/H/N; p.F595L; p.G596D/R |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talloa, D.; Triarico, S.; Agresti, P.; Mastrangelo, S.; Attinà, G.; Romano, A.; Maurizi, P.; Ruggiero, A. BRAF and MEK Targeted Therapies in Pediatric Central Nervous System Tumors. Cancers 2022, 14, 4264. https://doi.org/10.3390/cancers14174264
Talloa D, Triarico S, Agresti P, Mastrangelo S, Attinà G, Romano A, Maurizi P, Ruggiero A. BRAF and MEK Targeted Therapies in Pediatric Central Nervous System Tumors. Cancers. 2022; 14(17):4264. https://doi.org/10.3390/cancers14174264
Chicago/Turabian StyleTalloa, Dario, Silvia Triarico, Pierpaolo Agresti, Stefano Mastrangelo, Giorgio Attinà, Alberto Romano, Palma Maurizi, and Antonio Ruggiero. 2022. "BRAF and MEK Targeted Therapies in Pediatric Central Nervous System Tumors" Cancers 14, no. 17: 4264. https://doi.org/10.3390/cancers14174264
APA StyleTalloa, D., Triarico, S., Agresti, P., Mastrangelo, S., Attinà, G., Romano, A., Maurizi, P., & Ruggiero, A. (2022). BRAF and MEK Targeted Therapies in Pediatric Central Nervous System Tumors. Cancers, 14(17), 4264. https://doi.org/10.3390/cancers14174264