
Defining the Immune Checkpoint Landscape in Human Colorectal Cancer Highlights the Relevance of the TIGIT/CD155 Axis for Optimizing Immunotherapy
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
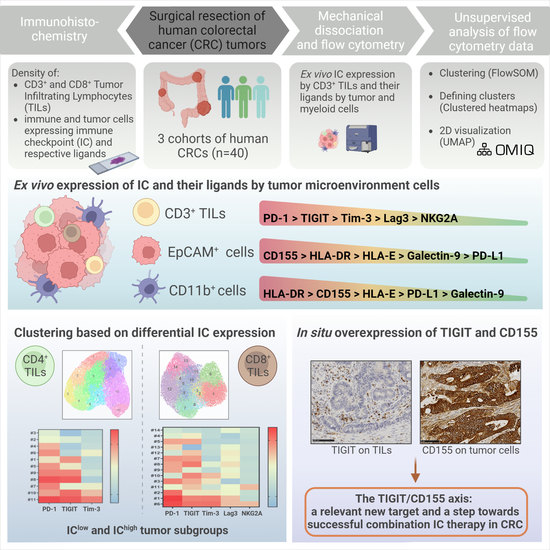
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. CMS Classification of CRC
2.3. Immunohistochemistry and Immunostaining Scores
2.4. Mechanical Dissociation of Colorectal Tissues
2.5. Ex vivo Flow Cytometry Staining
2.6. Supervised and Unsupervised Analyses of Flow Cytometry Data
2.7. Statistical Analyses
3. Results
3.1. IC Expression by T Cells Infiltrating CRC Tumors
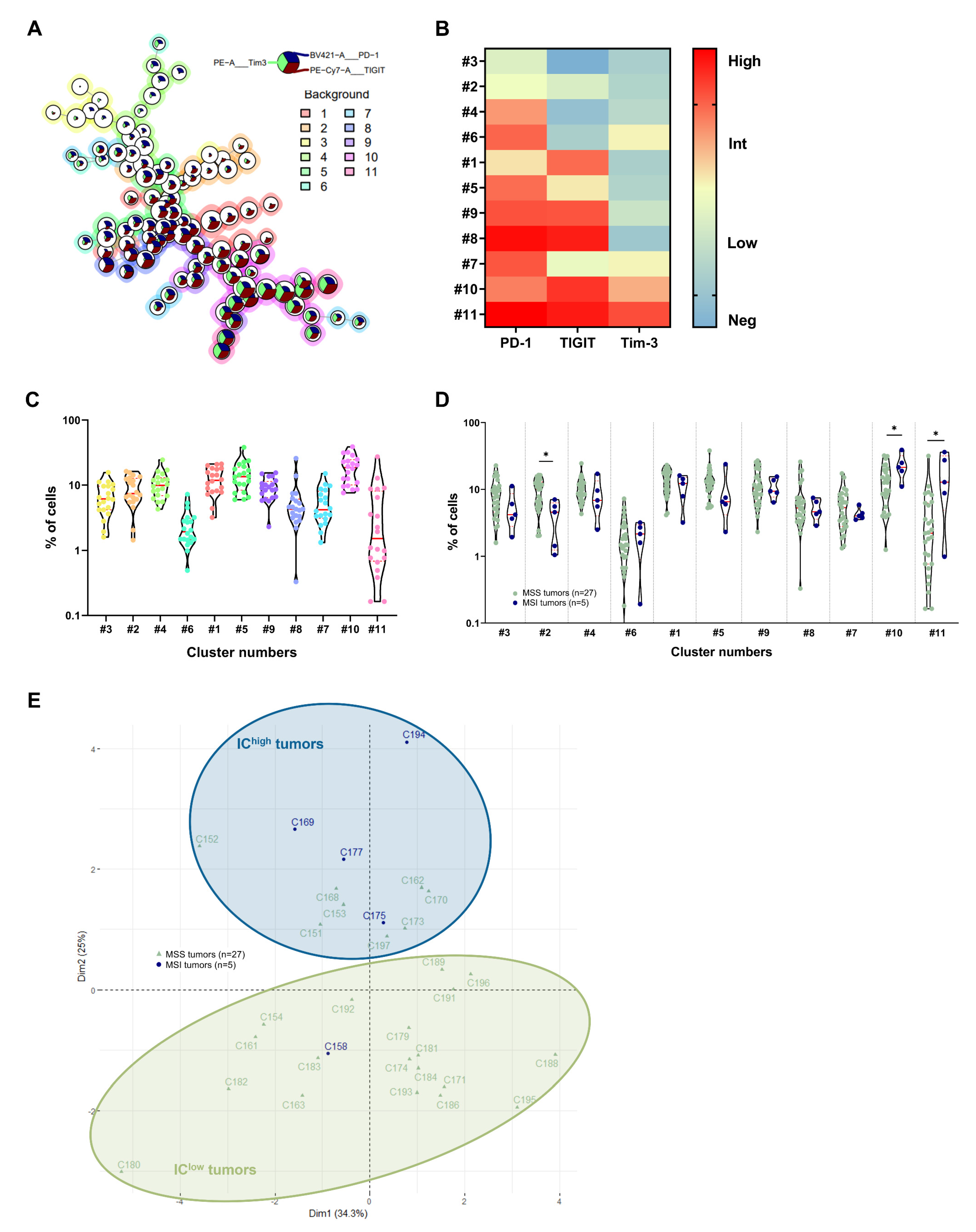
3.2. Identification of CRC Subgroups Based on IC Expression by CD4+ TILs
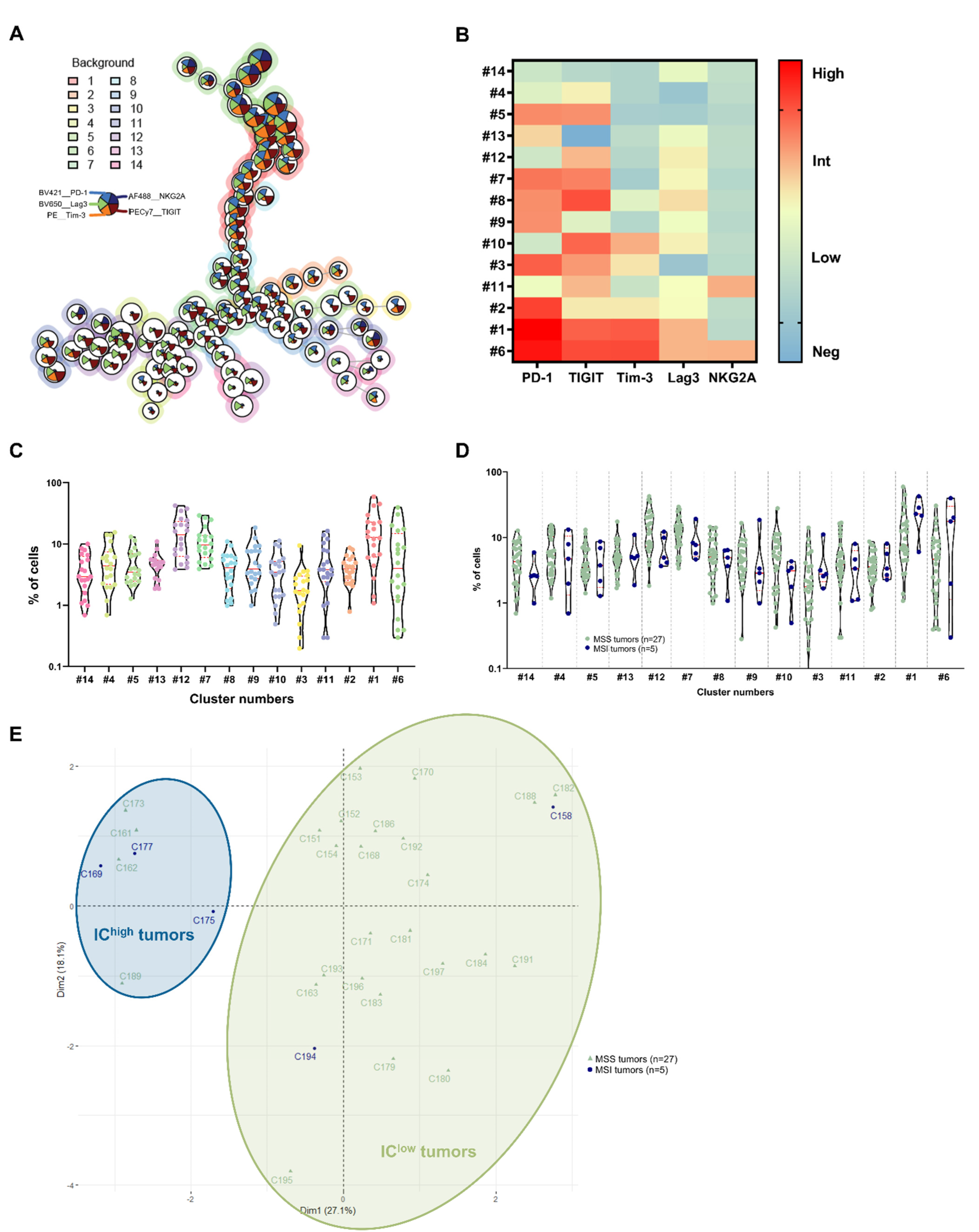
3.3. Identification of CRC Subgroups Based on IC Expression by CD8+ TILs
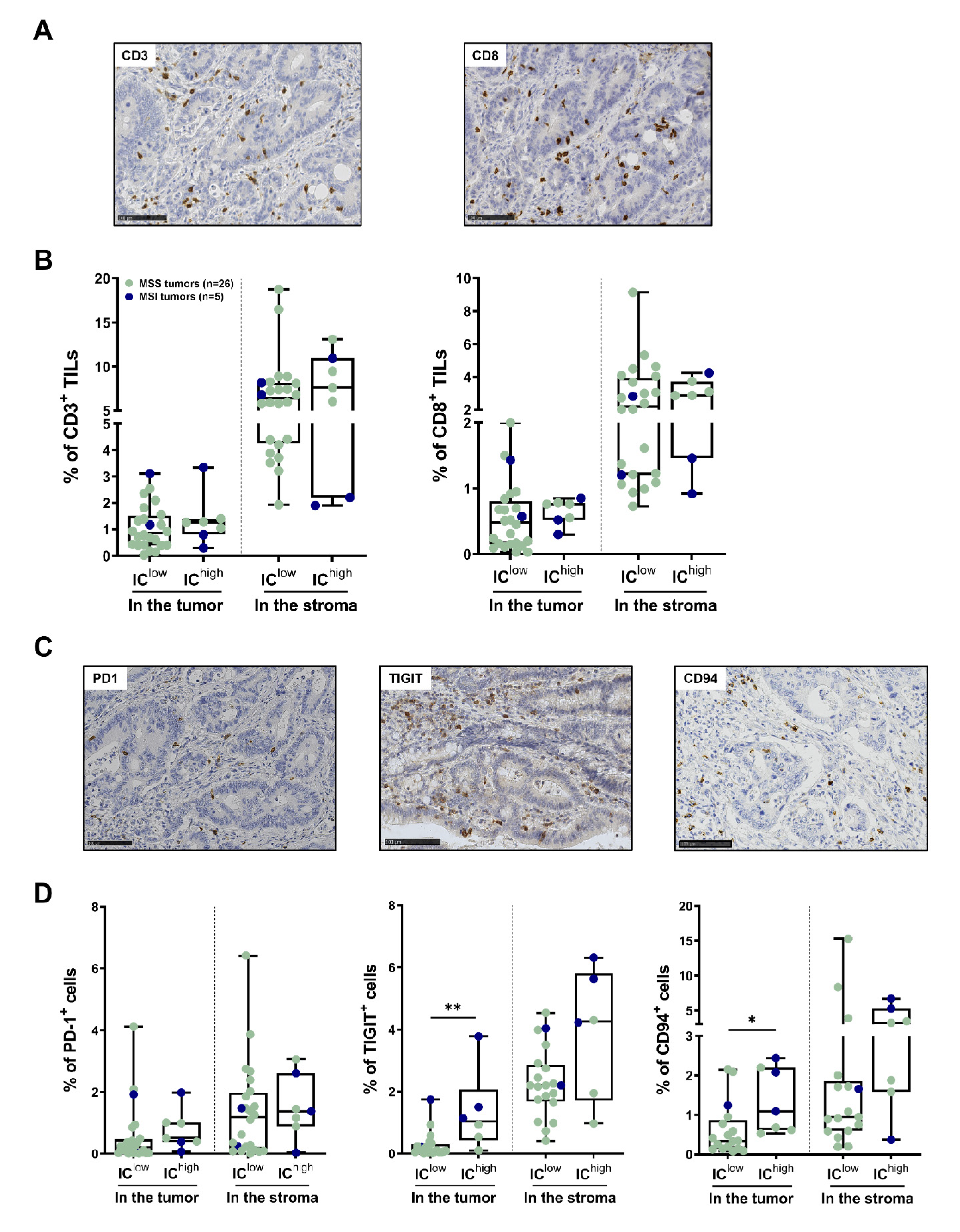
3.4. In Situ T-Cell Density and IC Expression by TILs in the Subgroups of CRC Identified Using Flow Cytometry
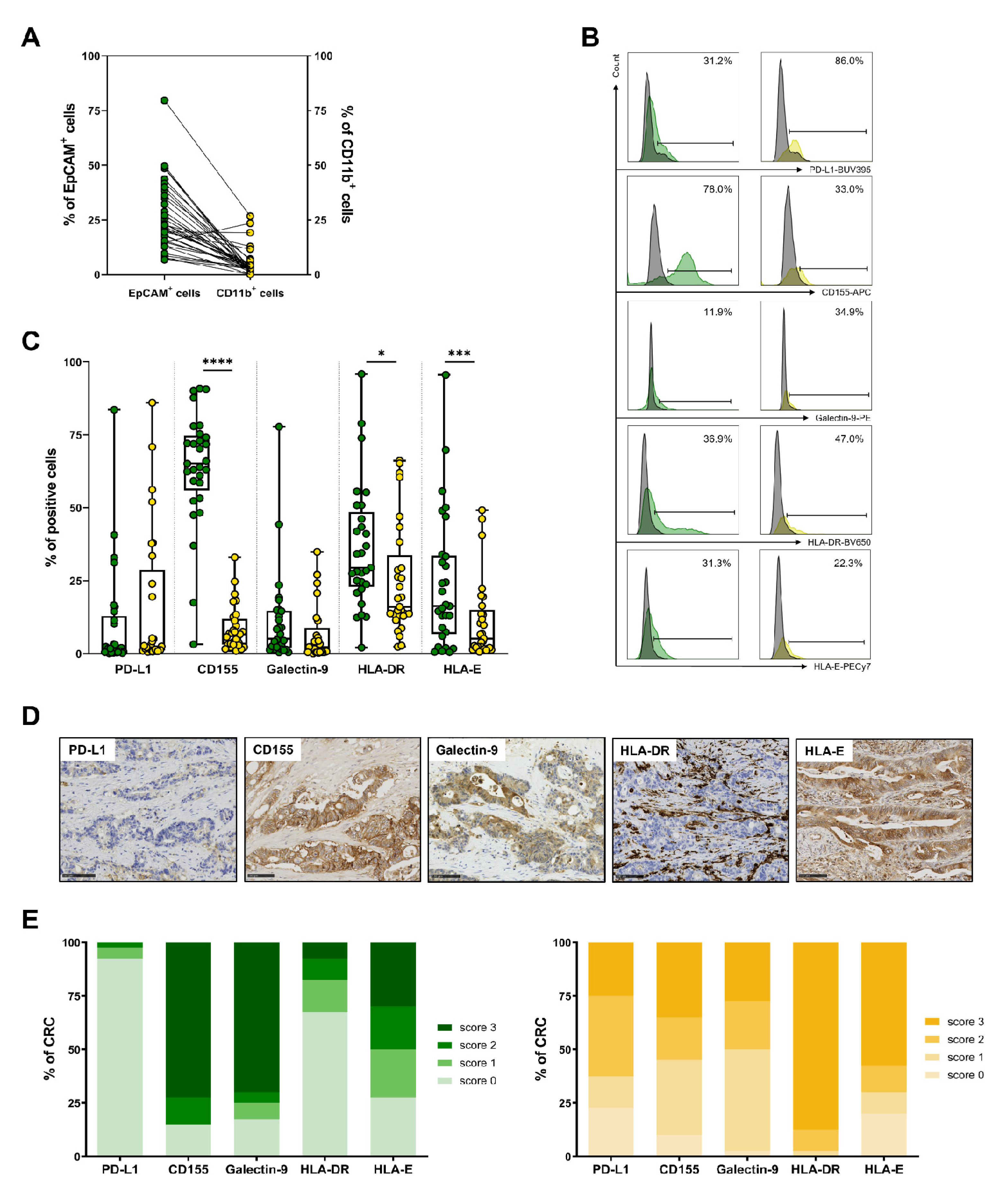
3.5. Expression of IC Ligands by Tumor Cells and Myeloid Cells in CRC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2020; Available online: https://gco.iarc.fr/today (accessed on 2 August 2021).
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Hoorn, S.T.; de Back, T.R.; Sommeijer, D.W.; Vermeulen, L. Clinical Value of Consensus Molecular Subtypes in Colorectal Cancer: A Systematic Review and Meta-Analysis. JNCI J. Natl. Cancer Inst. 2021, 114, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Galon, J.; Pagès, F.; Marincola, F.M.; Thurin, M.; Trinchieri, G.; Fox, A.B.; Gajewski, T.F.; Ascierto, A.P. The immune score as a new possible approach for the classification of cancer. J. Transl. Med. 2012, 10, 1–4. [Google Scholar] [CrossRef]
- Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Berger, A.; Bindea, G.; Meatchi, T.; Bruneval, P.; Trajanoski, Z.; Fridman, W.-H.; Pagès, F.; et al. Histopathologic-Based Prognostic Factors of Colorectal Cancers Are Associated with the State of the Local Immune Reaction. J. Clin. Oncol. 2011, 29, 610–618. [Google Scholar] [CrossRef]
- Williams, D.S.; Mouradov, D.; Jorissen, R.N.; Newman, M.R.; Amini, E.; Nickless, D.K.; Teague, J.A.; Fang, C.G.; Palmieri, M.J.; Parsons, M.; et al. Lymphocytic response to tumour and deficient DNA mismatch repair identify subtypes of stage II/III colorectal cancer associated with patient outcomes. Gut 2018, 68, 465–474. [Google Scholar] [CrossRef]
- Diaz, A.L.; Shiu, K.-K.; Kim, T.-W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): Final analysis of a randomised, open-label, phase 3 study. Lancet Oncol. 2022, 23, 659–670. [Google Scholar] [CrossRef]
- Lenz, H.-J.; Van Cutsem, E.; Limon, M.L.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; García-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.B.; et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J. Clin. Oncol. 2022, 40, 161–170. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The Vigorous Immune Microenvironment of Microsatellite Instable Colon Cancer Is Balanced by Multiple Counter-Inhibitory Checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Marisa, L.; Svrcek, M.; Collura, A.; Becht, E.; Cervera, P.; Wanherdrick, K.; Buhard, O.; Goloudina, A.; Jonchere, V.; Selves, J.; et al. The Balance Between Cytotoxic T-cell Lymphocytes and Immune Checkpoint Expression in the Prognosis of Colon Tumors. JNCI J. Natl. Cancer Inst. 2017, 110, 68–77. [Google Scholar] [CrossRef]
- Zhou, E.; Huang, Q.; Wang, J.; Fang, C.; Yang, L.; Zhu, M.; Chen, J.; Chen, L.; Dong, M. Up-regulation of Tim-3 is associated with poor prognosis of patients with colon cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 8018–8027. [Google Scholar] [PubMed]
- Kuai, W.; Xu, X.; Yan, J.; Zhao, W.; Li, Y.; Wang, B.; Yuan, N.; Li, Z.; Jia, Y. Prognostic Impact of PD-1 and Tim-3 Expression in Tumor Tissue in Stage I-III Colorectal Cancer. BioMed Res. Int. 2020, 2020, 5294043. [Google Scholar] [CrossRef]
- Zhou, X.; Ding, X.; Li, H.; Yang, C.; Ma, Z.; Xu, G.; Yang, S.; Zhang, D.; Xie, X.; Xin, L.; et al. Upregulation of TIGIT and PD-1 in Colorectal Cancer with Mismatch-repair Deficiency. Immunol. Investig. 2020, 50, 338–355. [Google Scholar] [CrossRef]
- Liang, R.; Zhu, X.; Lan, T.; Ding, D.; Zheng, Z.; Chen, T.; Huang, Y.; Liu, J.; Yang, X.; Shao, J.; et al. TIGIT promotes CD8+T cells exhaustion and predicts poor prognosis of colorectal cancer. Cancer Immunol. Immunother. 2021, 70, 2781–2793. [Google Scholar] [CrossRef]
- Xu, J.; Shen, D.; Zhang, T.; Wang, J.; De, W.; Zhang, J. Lymphocyte-activated gene-3 (LAG3) protein expressed in tumor-infiltrating lymphocytes of colorectal cancer. Pol. J. Pathol. 2021, 72, 324–330. [Google Scholar] [CrossRef]
- Neupane, P.; Mimura, K.; Nakajima, S.; Okayama, H.; Ito, M.; Min, A.K.T.; Saito, K.; Onozawa, H.; Fujita, S.; Sakamoto, W.; et al. The Expression of Immune Checkpoint Receptors and Ligands in the Colorectal Cancer Tumor Microenvironment. Anticancer. Res. 2021, 41, 4895–4905. [Google Scholar] [CrossRef]
- Borst, L.; van der Burg, S.H.; van Hall, T. The NKG2A–HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 5549–5556. [Google Scholar] [CrossRef]
- Ducoin, K.; Oger, R.; Mutala, L.B.; Deleine, C.; Jouand, N.; Desfrançois, J.; Podevin, J.; Duchalais, E.; Cruard, J.; Benlalam, H.; et al. Targeting NKG2A to boost anti-tumor CD8 T-cell responses in human colorectal cancer. OncoImmunology 2022, 11, 2046931. [Google Scholar] [CrossRef]
- Ott, E.; Bilonda, L.; Dansette, D.; Deleine, C.; Duchalais, E.; Podevin, J.; Volteau, C.; Bennouna, J.; Touchefeu, Y.; Fourquier, P.; et al. The density of Tbet+ tumor-infiltrating T lymphocytes reflects an effective and druggable preexisting adaptive antitumor immune response in colorectal cancer, irrespective of the microsatellite status. OncoImmunology 2019, 8, e1562834. [Google Scholar] [CrossRef] [PubMed]
- Eugène, J.; Jouand, N.; Ducoin, K.; Dansette, D.; Oger, R.; Deleine, C.; Leveque, E.; Meurette, G.; Podevin, J.; Matysiak, T.; et al. The inhibitory receptor CD94/NKG2A on CD8+ tumor-infiltrating lymphocytes in colorectal cancer: A promising new druggable immune checkpoint in the context of HLAE/β2m overexpression. Mod. Pathol. 2019, 33, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; Chen, H.; Poidinger, M.; Chen, J.; de Magalhães, J.P.; Larbi, A. flowAI: Automatic and interactive anomaly discerning tools for flow cytometry data. Bioinformatics 2016, 32, 2473–2480. [Google Scholar] [CrossRef] [PubMed]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T.; Saeys, Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part A 2015, 87, 636–645. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, P.; Li, L.; Shi, L.; Chang, P.; Liang, T.; Yang, Q.; Liu, Y.; Wang, L.; Hu, L. Co-expression of TIM-3 and CEACAM1 promotes T cell exhaustion in colorectal cancer patients. Int. Immunopharmacol. 2017, 43, 210–218. [Google Scholar] [CrossRef]
- Shao, Q.; Wang, L.; Yuan, M.; Jin, X.; Chen, Z.; Wu, C. TIGIT Induces (CD3+) T Cell Dysfunction in Colorectal Cancer by Inhibiting Glucose Metabolism. Front. Immunol. 2021, 12, 688961. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, X.; Zheng, L.; Zhang, Y.; Li, Y.; Fang, Q.; Gao, R.; Kang, B.; Zhang, Q.; Huang, J.Y.; et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018, 564, 268–272. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 Impair Tumor Antigen–Specific CD8+ T Cells in Melanoma Patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Van Montfoort, N.; Borst, L.; Korrer, M.J.; Sluijter, M.; Marijt, K.A.; Santegoets, S.J.; van Ham, V.J.; Ehsan, I.; Charoentong, P.; André, P.; et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell 2018, 175, 1744–1755. [Google Scholar] [CrossRef]
- Yu, M.; Lu, B.; Liu, Y.; Me, Y.; Wang, L.; Zhang, P. Tim-3 is upregulated in human colorectal carcinoma and associated with tumor progression. Mol. Med. Rep. 2016, 15, 689–695. [Google Scholar] [CrossRef] [Green Version]
- Toor, S.M.; Murshed, K.; Al-Dhaheri, M.; Khawar, M.; Abu Nada, M.; Elkord, E. Immune Checkpoints in Circulating and Tumor-Infiltrating CD4+ T Cell Subsets in Colorectal Cancer Patients. Front. Immunol. 2019, 10, 2936. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef]
- Picard, E.; Verschoor, C.P.; Ma, G.W.; Pawelec, G. Relationships Between Immune Landscapes, Genetic Subtypes and Responses to Immunotherapy in Colorectal Cancer. Front. Immunol. 2020, 11, 369. [Google Scholar] [CrossRef]
- Keshinro, A.; Vanderbilt, C.; Kim, J.K.; Firat, C.; Chen, C.-T.; Yaeger, R.; Ganesh, K.; Segal, N.H.; Gonen, M.; Shia, J.; et al. Tumor-Infiltrating Lymphocytes, Tumor Mutational Burden, and Genetic Alterations in Microsatellite Unstable, Microsatellite Stable, or Mutant POLE/POLD1 Colon Cancer. JCO Precis. Oncol. 2021, 5, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan Nair, V.; Toor, S.M.; Taha, R.Z.; Shaath, H.; Elkord, E. DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clin. Epigenetics 2018, 10, 104. [Google Scholar] [CrossRef]
- Valentini, A.M.; Di Pinto, F.; Cariola, F.; Guerra, V.; Giannelli, G.; Caruso, M.L.; Pirrelli, M. PD-L1 expression in colorectal cancer defines three subsets of tumor immune microenvironments. Oncotarget 2018, 9, 8584–8596. [Google Scholar] [CrossRef]
- Bossard, C.; Bézieau, S.; Matysiak-Budnik, T.; Volteau, C.; Laboisse, C.L.; Jotereau, F.; Mosnier, J.-F. HLA-E/β2 microglobulin overexpression in colorectal cancer is associated with recruitment of inhibitory immune cells and tumor progression. Int. J. Cancer 2011, 131, 855–863. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair–Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef]
- Niu, J.; Maurice-Dror, C.; Lee, D.; Kim, D.-W.; Nagrial, A.; Voskoboynik, M.; Chung, H.; Mileham, K.; Vaishampayan, U.; Rasco, D.; et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer. Ann. Oncol. 2021, 33, 169–180. [Google Scholar] [CrossRef]
- Cho, B.C.; Abreu, D.R.; Hussein, M.; Cobo, M.; Patel, A.J.; Secen, N.; Lee, K.H.; Massuti, B.; Hiret, S.; Yang, J.C.H.; et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): Primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 2022, 23, 781–792. [Google Scholar] [CrossRef]
- Hermel, D.J.; Sigal, D. The Emerging Role of Checkpoint Inhibition in Microsatellite Stable Colorectal Cancer. J. Pers. Med. 2019, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort 1 (n = 8) | Cohort 2 (n = 20) | Cohort 3 (n = 12) | |
|---|---|---|---|
| 7 MSS, 1 MSI | 16 MSS, 4 MSI | 11 MSS, 1 MSI | |
| Age: mean (range) | 61.1 (43–75) | 69.8 (40–88) | 66.3 (36–85) |
| Gender | |||
| Men | 4 | 10 | 9 |
| Women | 4 | 10 | 3 |
| Tumor location | |||
| Right colon | 1 | 9 | 3 |
| Left colon | 6 | 9 | 8 |
| Rectum | 1 | 2 | 1 |
| pTNM stage (IUCC) | |||
| I | 1 | 3 | 2 |
| II | 4 | 11 | 5 |
| III | 3 | 6 | 5 |
| IV | 0 | 0 | 0 |
| Histological subtypes | |||
| Adenocarcinoma NOS | 6 | 18 | 8 |
| Mucinous | 2 | 2 | 4 |
| CMS classification | |||
| CMS1 | 3 | 4 | 0 |
| CMS2 | 1 | 8 | 3 |
| CMS3 | 1 | 5 | 0 |
| CMS4 | 1 | 1 | 1 |
| Unclassified | 0 | 1 | 2 |
| ND | 2 | 1 | 6 |
| Score | Positive Tumor Cells | Positive Immune Cells of the Stroma |
|---|---|---|
| 0 | <5% | Isolated cells or <4 clusters of stained cells/cm2 |
| 1 | 5–25% | 4–5 clusters of stained cells/cm2 |
| 2 | 26–50% | >5 clusters of stained cells/cm2 |
| 3 | >51% | Diffuse staining of numerous positive cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ducoin, K.; Bilonda-Mutala, L.; Deleine, C.; Oger, R.; Duchalais, E.; Jouand, N.; Bossard, C.; Jarry, A.; Gervois-Segain, N. Defining the Immune Checkpoint Landscape in Human Colorectal Cancer Highlights the Relevance of the TIGIT/CD155 Axis for Optimizing Immunotherapy. Cancers 2022, 14, 4261. https://doi.org/10.3390/cancers14174261
Ducoin K, Bilonda-Mutala L, Deleine C, Oger R, Duchalais E, Jouand N, Bossard C, Jarry A, Gervois-Segain N. Defining the Immune Checkpoint Landscape in Human Colorectal Cancer Highlights the Relevance of the TIGIT/CD155 Axis for Optimizing Immunotherapy. Cancers. 2022; 14(17):4261. https://doi.org/10.3390/cancers14174261
Chicago/Turabian StyleDucoin, Kathleen, Linda Bilonda-Mutala, Cécile Deleine, Romain Oger, Emilie Duchalais, Nicolas Jouand, Céline Bossard, Anne Jarry, and Nadine Gervois-Segain. 2022. "Defining the Immune Checkpoint Landscape in Human Colorectal Cancer Highlights the Relevance of the TIGIT/CD155 Axis for Optimizing Immunotherapy" Cancers 14, no. 17: 4261. https://doi.org/10.3390/cancers14174261
APA StyleDucoin, K., Bilonda-Mutala, L., Deleine, C., Oger, R., Duchalais, E., Jouand, N., Bossard, C., Jarry, A., & Gervois-Segain, N. (2022). Defining the Immune Checkpoint Landscape in Human Colorectal Cancer Highlights the Relevance of the TIGIT/CD155 Axis for Optimizing Immunotherapy. Cancers, 14(17), 4261. https://doi.org/10.3390/cancers14174261