Simple Summary
Over the past decade, cell-based immunotherapy has become a powerful strategy in solid cancer therapy. Tumor-infiltrating lymphocytes (TILs) are a group of intratumor lymphocytes. With the development of new technologies, the isolation and generation of TILs from tumor tissues have improved. Clinical use of TILs for solid tumor treatment showed good efficacy. In this review, we summarize the current strategies and challenges of TIL generation. We highlight the clinical trials where TIL therapy is used independently and in combination with other therapies for solid tumor treatment. Finally, the limitations, future potential, and directions of TIL therapy for solid tumor treatment are also discussed.
Abstract
Over the past decade, immunotherapy, especially cell-based immunotherapy, has provided new strategies for cancer therapy. Recent clinical studies demonstrated that adopting cell transfer of tumor-infiltrating lymphocytes (TILs) for advanced solid tumors showed good efficacy. TIL therapy is a type of cell-based immunotherapy using the patient’s own immune cells from the microenvironment of the solid tumor to kill tumor cells. In this review, we provide a comprehensive summary of the current strategies and challenges in TIL isolation and generation. Moreover, the current clinical experience of TIL therapy is summarized and discussed, with an emphasis on lymphodepletion regimen, the use of interleukin-2, and related toxicity. Furthermore, we highlight the clinical trials where TIL therapy is used independently and in combination with other types of therapy for solid cancers. Finally, the limitations, future potential, and directions of TIL therapy for solid tumor treatment are also discussed.
1. Introduction
In the past few decades, immunotherapy has been the focus of cancer research and various methods have emerged to treat cancers, such as immune checkpoint inhibitors (ICI) against the programmed cell death protein-1 (PD-1) or its ligand (PD-L1) and cytotoxic T lymphocyte associated antigen-4 (CTLA-4), vascular endothelial growth factor (VEGF) inhibitors, cytokines, vaccines, and adoptive cellular therapy (ACT). The primary form of ACT is chimeric antigen receptor (CAR) T-cell therapy. In this strategy, T cells are obtained from the patient’s own peripheral blood cells and are modified to express a receptor that binds to the tumor antigen. CAR-T cell therapy has been approved for the treatment of some patients with hematological malignancies, such as lymphoblastic leukemia and B cell lymphoma [1]. However, currently CAR-T cell therapy has not been proven to be effective in the treatment of solid tumors. Therefore, in order to benefit patients with various types of solid tumors, it is necessary to award further research to TIL therapy which precedes CAR-T cell therapy. In addition, TIL therapy is expected to solve the problem of drug resistance of solid tumors to PD-1 antibodies.
The TIL-ACT process begins by isolating the natural infiltrating lymphocytes from the tumor tissues, expanding them in vitro, and then infusing these cells with a high dose (HD) of IL-2 into patients to identify and kill tumor cells. Prior to infusion of the TIL products, patients receive a non-myeloablative (NMA) lymphodepletion regimen. Pioneering work in the TIL-ACT field has been undertaken by the research group of Dr. Steven Rosenberg. In 1986, Rosenberg et al. isolated TILs from murine tumors and amplified them alongside interleukin-2 (IL-2) in vitro. They then infused them back into the tumor-bearing hosts, which caused significant antitumor reactivity [2]. In addition, they found that the combination of IL-2 with the infusion of TILs could improve the therapeutic effect [3]. Rosenberg then applied TIL therapy to patients with melanoma and published the first report on the successful treatment of melanoma with TIL-ACT in 1988 [4]. At present, an increasing number of clinical centers worldwide have implemented TIL-ACT, and this therapy has shown impressive results in patients with metastatic melanoma [5,6]. Significantly, TIL therapy is also applicable to other solid tumors to a certain extent, and has achieved good responses [7,8,9,10,11,12]. In this review, we will provide an overview of the current status of TIL therapy in solid tumors, and focus on the preparation process of TIL, strategies of screening effective TILs, and treatment protocols, as well as the application of combined therapy with TILs.
2. Current Clinical State of TIL Therapy in Melanoma
Since Rosenberg and his colleagues successfully applied TIL therapy to patients with metastatic melanoma, a series of clinical trials have emerged. Dafni et al. reported that the objective response rate (ORR) of advanced cutaneous melanoma patients who received the treatment which combined TIL-ACT with IL-2 from 1988 to 2016 was 41%, and the overall complete response rate (CRR) was 12% [13]. In addition, the ORR and CRR of patients who received the regimen of high dose (HD) IL-2 was 43% and 14%, respectively, compared with 35% and 7% in the low dose (LD) IL-2 group [13]. This analysis ruled out patients with uveal melanoma. TIL therapy has been successful when applied to the rare and refractory uveal melanoma and was first reported in 2017 [14]. The complete response (CR) and partial response (PR) of patients who received TIL and HD IL-2 was 4.5% and 31.8%, respectively [14]. In 2021, Iovance successfully carried out phase II trials of TIL product Lifileucel (LN-144) for patients with advanced melanoma after progression on ICI [15]. Patients received a mean number of TILs of 2.73 × 1010, and the disease control rate (DCR) was 80%, the ORR was 36%, the CR was 3%, and the PR was 33%. Remarkably, these results indicate that TIL therapy is a potential new treatment for patients with advanced melanoma after progression on ICI. More recently, the observed objective clinical response rate in patients with advanced melanoma was 50%, including two complete response and three partial response patients [16]. At present, TIL therapy is being tested mainly as a second-line treatment, and melanoma is still the main type of tumor in most clinical trials (Table 1).

Table 1.
Clinical trials with TILs for melanoma from ClinicalTrials.gov between 2011 and 2021.
3. Successful Application of TIL Therapy in Other Solid Tumors
In addition to melanoma, TIL therapy has been proven to have impressive clinical benefits for patients with cervical cancer [8,17,18], and has also shown preliminary efficacy in colorectal cancer [10], cholangiocarcinoma [9], non-small cell lung cancer [11], and breast cancer [7] (Table 2).
Table 2.
Recent preliminary studies have shown that TIL therapy is effective in patients with other solid tumors.
Persistent infection with human papilloma viruses (HPVs) is essential in the pathogenesis of virtually all cervical cancers [19]. A clinical study was designed to determine whether the ACT with HPV-TILs can mediate the regression of metastatic cervical cancer, and nine eligible cervical patients were recruited [8]. The HPV-TILs were initiated and selected based on their reactivity against HPV E6 and E7. Patients received a single-dose infusion of HPV-TIL followed by a high dose of IL-2. Three patients experienced an objective tumor response, two patients had complete responses lasting over one year, and the rest had partial responses. Rosenberg et al. further screened tumor-specific antigens through a personalized immunogenomic approach including whole-exome sequencing [20]. In addition, they also conducted a phase II clinical trial to evaluate the efficacy of TIL therapy in the treatment of metastatic human HPV-associated cancers [17]. Although the overall response rate was modest, the results support the premise that TIL-ACT can mediate tumor regression, which provides a wealth of potential for the treatment of metastatic human HPV-associated cancers.
Remarkably, in a phase two clinical trial, the TIL product of LN-145 successfully treated patients with advanced cervical cancer. Patients received a mean number of TILs of 2.8 × 1010; the ORR was 44%, including one complete response, nine partial responses, and two unconfirmed partial responses [18]. To this end, the Food and Drug Administration (FDA) awarded the status of breakthrough treatment to LN-145. Ultimately, this pivotal study has demonstrated the feasibility and efficacy of this approach in patients with cervical cancer and will ensure further development of adoptive cell therapy in these kinds of malignancies.
In 2014, Tran et al. successfully amplified neoantigen-specific TILs from a patient with metastatic cholangiocarcinoma [9]. After the patient received an infusion of IL-2 and 42.4 billion TILs, which contained a large number of CD4+ neoantigen-specific T cells, the patient achieved dramatic regression of liver and lung metastases [9].
In a phase two clinical trial (NCT01174121), a 50-year old woman with metastatic colorectal cancer received a single infusion of 1.48 × 1011 TILs consisting of approximately 75% CD8+ T cells, which can specifically recognize the KRAS G12D mutant [10]. All metastatic lesions in this patient achieved regression and they achieved a nine-month partial response.
In 2017, Lee et al. successfully isolated and amplified TILs in vitro from patients with breast cancer which showed their reaction to autologous tumor cells [21]. Zacharakis et al. administrated TIL therapy to patients with hormone receptor-positive metastatic breast cancer in 2018 [7]. In short, the patient underwent complete and durable tumor regression after receiving a combination treatment of TIL therapy and anti-PD-1 monoclonal antibodies. It is worth noting that they screened out the TILs that were mainly CD4+ T cells (62.5%), which could recognize the mutant of four proteins (SLC3A2, KIAA0368, CADPS2, and CTSB) [7].
In 2020, it was reported that the combination of PD-1 inhibitor and TIL therapy showed preliminary efficacy in the treatment of NSCLC [11]. In this phase one trial (NCT03215810), patients with metastatic NSCLC who progressed after nivolumab treatment were treated with TILs. Patients received the infusion of TILs and IL-2, followed by nivolumab to augment the persistence of the TILs. Two of 13 evaluable patients achieved durable complete responses. The exciting results of this clinical trial offer hope for patients who are progressing after anti-PD-1 treatment, and indicate that TIL therapy combined with a PD-1 inhibitor may be a promising choice for patients with metastatic NSCLC. Currently, a clinical trial using ACT with TIL for the treatment of NSCLC is recruiting patients (NCT04614103), and several trials of TIL combined with PD-1 inhibitor for the treatment of NSCLC have been launched (NCT03903887, NCT03645928).
4. Preparation of TIL
Although TILs exist largely in the tumor microenvironment, this does not mean TILs can be successfully isolated and amplified from tumor tissues, nor does it mean tumor cells can be recognized and eliminated by isolated TILs. The ability of TILs to kill tumor cells will be inhibited by various factors in the tumor microenvironment, such as the existence of immunosuppressive subsets including regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages, as well as the increase in immunosuppressive molecules and metabolites [22,23]. Furthermore, the number of TILs in the tumor microenvironment is small and difficult to amplify. Therefore, it is essential to optimize the preparation process of TILs. In general, the generation of TILs can be divided into two steps: the pre-rapid expansion procedure (pre-REP), which is the initial growth stage of TILs from tumor tissue, and the large-scale expansion of the 14-day REP (Figure 1).
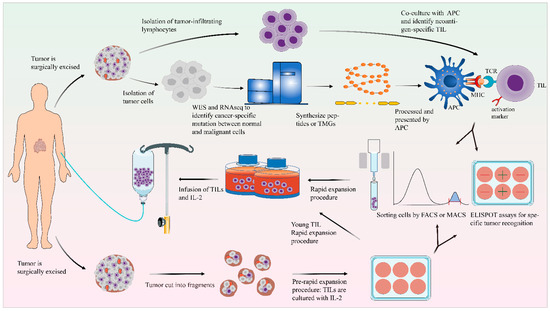
Figure 1.
General scheme of the preparation of TILs. After excision, the tumor tissue is fragmented or digested into a single-cell suspension and then cultured in cell culture plates with IL-2. Using the selected TIL method, the detection of tumor reactivity is based on the production of IFN-γ by co-culturing with autologous tumor cells. In the “young” TIL method, the assays for specific tumor recognition are omitted. TILs could be enriched by FACS or MACS. TIL cultures are then expanded to treatment levels by REP. These TILs are then infused back into the lymphodepleted patient. On the other side, the tumor cells and normal cells undergo WES and RNA-seq to identify mutations. Based on this information, TMG or peptides are synthesized. These TMG or peptides are then processed by autologous APC and presented to T cells which are co-cultured with APCs. The recognition of neoantigen-specific T cells depends on IFN-γ ELISPOT assays, and these identified T cells with activated surface markers (such as CD137) can be purified by FACS.
4.1. Pre-Rapid Expansion Procedure
Generally, fresh tumor tissue is obtained through surgery, and should be processed in a reasonable amount of time. For initial growth of TILs, tumor tissue can be cut into fragments or made into a single-cell suspension through enzyme digestion, physical disaggregation, or fine needle aspiration [24,25]. The tumor fragments or single-cell suspensions are then cultured in a complete medium with IL-2.
The tumor tissues for initial growth of TILs may be from breast [7], skin [6], metastatic lymph nodes [26], lung [11], colorectum [10], or other body parts. It is important to note that the tumor location may affect the production of TILs. TILs from the colorectal or skin regions may be contaminated with microbes, lymph nodes may contain a large number of ineffective T cells, and the fibroblasts may occupy the whole culture container [27].
4.2. Unselected/Young TILs
In most early studies, only the tumor-reactive cultures that detected interferon-gamma (IFN-γ) in co-culture with autologous tumor materials or tumor cells in vitro could be pre-selected for further outgrowth. However, this “selected” method largely limits the clinical application of TIL therapy, which adds to the time required for the TIL expansion, and the tumor cells or materials used for testing are difficult to obtain [28,29]. Remarkably, several studies showed that a shorter culture duration before beginning the REP is beneficial to clinical response [29,30,31].
In order to simplify the generation process of TILs and potentially improve the properties of TIL cultures, a modified TIL production protocol which uses “unselected” TILs to start the REP protocol was tested [32,33]. These unselected bulk TILs, called “young” TILs, have no pre-selection step for tumor reactivity based on IFN-γ (Figure 1). Crucially, the clinical response rate of the protocols which use “young” TILs is not low [34], which means the status of “young” TIL schemes are rapidly improving [35]. Studies demonstrated that “young” TILs have longer telomeres and higher levels of the costimulatory molecules CD27 and CD28 compared with the selected TILs, which means “young” TILs at a low level of differentiation are conducive to the proliferation, survival, and persistence of TILs in vivo [32,36].
4.3. TIL Selection Strategies
The TILs isolated from the complex tumor microenvironment are a heterogeneous cellular population. The effector T cells with anti-tumor activity are required, so it is necessary to screen the effector T cells. Isolation strategies have been proposed and developed for this program. Here, we attempt to summarize the known strategies for isolating tumor-reactive T cells.
4.3.1. Selection by Surface Markers
TILs could be enriched through selection markers (Figure 1). CD8+ TILs play a key role in immune response; they can specifically recognize and kill tumor cells [37], although CD4+ T cells also show the potential to kill tumors [9]. It is reported that the infusion of a large amount of CD8+ TILs is associated with an objective clinical response [34,38]. Therefore, the enrichment of CD8+ TILs is a feasible method to enhance tumor reactivity.
Studies have shown that CD103+ TILs indicate strong prognostic significance in ovarian and oral cancer [39,40]. In 2017, Ganesan et al. demonstrated that CD103 and CD8 are important phenotypes in the tissue-resident memory of T cells in lung cancers [41]. Furthermore, Simoni et al. reported that CD39 could be a marker for recognizing tumor-reactive CD8+ T cells [42]. In 2018, Doohen et al. proved that CD103+, CD39+, and CD8 TILs are unique tumor-reactive cells in the tumor microenvironment, which can be isolated from the tumor digests by immunomagnetic beads and fluorescence-activated cell sorting (FACS) [43]. The co-culture experiment of TILs and tumor cells showed that CD103+, CD39+, and CD8 TILs exhibited enhanced tumor reactivity [43]. In 2020, Kortekaas and his colleagues highlighted that CD39+ T cells have specific tumor reactivity, and have further proven the importance of CD39 in identifying and isolating tumor-reactive T cells [44]. However, CD39 was also highly expressed on exhausted CD8 TILs with low proliferation potential [45], and in order to improve the proliferation potential of tumor-specific CD8 TILs a new sorting strategy with negative CD39 was defined [45].
Although PD-1/CD279 has been reported as an immunosuppressive checkpoint, which is expressed on activated T cells and hijacked by tumors to escape immune surveillance and induce immune tolerance [46,47,48], it could be more accurately defined as a marker for tumor-reactive T cells to recognize tumor cells [49]. Inozume et al. pre-selected CD8+ PD-1+ T cells using the FACS method and immunomagnetic beads, and these cells showed greater tumor reactivity when compared to CD8+ PD-1− or non-selected TILs [50]. Fernandez-Poma et al. [51] found that the expression of PD-1 on CD8+ TILs accurately defined the tumor-reactive cells and evaluated the antitumor activity of CD8+ PD-1+ TILs in vivo, which provide direct support for this notion.
Current strategies also include selection by the co-stimulatory marker 4-1BB/CD137, which can be detected on all phenotypes of activated CD8+ T cells [52] and natural tumor-reactive TILs [53]. CD137, a costimulatory molecule, is induced by specific interactions between T cells and tumor cells [54,55]. The stimulation of tumor antigens can upregulate the expression of CD137 on T cells and mediates the activation and proliferation of T cells [53,56]. The screening scheme based on CD137 is widely advocated because it can recognize a wide range of antigens, including new antigens and shared tumor antigens [55]. Studies have shown that CD8+ TILs expressing 4-1BB seems to represent tumor-specific T cell subsets of melanoma [53,57]. Poch et al. [26] and Tavera et al. [58] proposed an amplification method of TILs co-stimulated by CD137 and IL-2, which rapidly increased the amplification rate of TILs in skin, uveal melanoma, and primary bladder cancer. However, as CD137 is a costimulatory molecule, long-term stimulation makes it over-differentiated, and its proliferative potential is affected. Parkhurst et al. proposed a strategy, which screen upregulates CD137 after antigen-specific stimulation to enrich tumor-reactive T cells and isolate TCRs, and then introduce it into less-differentiated PBLs for reinfusion therapy [59]. However, for most advanced metastatic patients, a lengthy preparation time is unreasonable. Seliktar-Ofir et al. proposed a simplified and efficient protocol. Briefly, the CD137 positive TIL was directly isolated from the co-culture system of TILs and tumor cells, and then the standard rapid amplification steps were carried out. TCR sequencing steps were omitted [55].
4.3.2. Selection by Neoantigens
TIL therapy depends largely on the recognition of specific tumor antigens, especially neoantigens. To this end, Rosenberg and his colleagues combined whole-exome sequencing (WES) technology and human leucocyte antigen (HLA) class I to identify and candidate the mutant proteins expressed in tumor cells that differ from healthy cells. These mutated epitopes are then synthesized and introduced to MHC matched antigen-presenting cells (APC). They are then co-cultured with the initial TIL, and the recognition ability of TILs was evaluated by performing ELISPOT assays of IFN-γ [60]. The identified neoantigen-specific CD8+ T cells can be further purified by FACS based on activated markers, such as CD137 [59], and then expanded in vitro. With the improvement of the tandem minigene (TMG) library, the ability to recognize and identify neoantigen-specific TILs has been further improved, because it omits the step of identifying and synthesizing a large number of peptides [20] (Figure 1). Using these techniques, neoantigen-specific effector TILs have been successfully performed in patients with melanoma, cholangiocarcinoma, colorectal cancer, breast cancer, and ovarian cancer [7,9,10,12,56]. However, this method also takes a lot of time to prepare the TIL for reinfusion, and requires complex equipment, making this unsuitable for most advanced patients. In addition, the proliferative potential of TILs will be also impaired by long-term antigen stimulation and amplification. Recently, Deniger and colleagues transferred the TP53 “hot spot” mutation-reactive T cell receptor to peripheral blood T cells, which may provide a potential solution [12].
4.4. Rapid Expansion Procedure
When an adequate quantity of the initial TIL has been obtained, it is then immediately cryopreserved or used directly for further REP, which usually takes approximately 14 days in the standard protocol. During the REP, TILs are stimulated and further expanded to treatment levels with IL-2, anti-CD3 antibody (which is added only at the start of REP), and irradiated feeder cells. The irradiated feeder cells originate from allogeneic or autologous peripheral blood mononuclear cells, which can activate and release growth factors to promote the expansion of TILs. During the REP, TILs are usually cultured in a culture flask and then transferred to gas-permeable bags to expand the cultures. In order to improve the growth rates of TILs, Jin et al. used gas-permeable flasks for the initial growth and REP of TILs [61].
5. Role of Lymphodepletion
A non-myeloablative (NMA) lymphodepletion regimen with chemotherapy or total body irradiation (TBI) before TIL infusion is an important milestone in TIL therapy. The application of the lymphodepletion regimen comes from a murine model in which the efficiency of TILs is improved after lymphodepletion [3]. It is reported that the lymphodepletion regimen can enhance the effect of TILs through several potential mechanisms, such as the elimination of Tregs, by increasing host homeostatic cytokines (including IL-7 and IL-15), and by decreasing endogenous lymphocytes, which compete for these trophic cytokines [62,63]. Moreover, lymphodepleting can enhance the activation of antigen presenting cells (APC) which play an important role in regulating adoptively transferred T cells [64].
In 1994, Rosenberg et al. showed that the ORR in patients with metastatic melanoma treated with and without lymphodepletion were 35% and 31%, respectively [65]. Similarly, in 2002, Dudley and his colleagues demonstrated that the adoptive transfer of TILs could achieve a better ORR after lymphodepletion of cyclophosphamide (Cy) 60 mg/kg/day for two days and fludarabine (Flu) 25 mg/m2/day for five days in patients with metastatic melanoma [66]. Some clinical studies suggested that adding TBI to the NMA chemotherapy regimen could improve the response rate [6,67]. However, there was no obvious effect in a randomized trial [68]. Recently, a study summarized different NMA regimens, and suggested that the regimen of Cy (30 mg/kg for two days) and Flu (25 mg/m2 for five days) is a better choice [69], which has maximum effect on lymphodepletion with minimum toxicity compared to other doses and regimens. Notably, most patients have experienced adverse effects caused by the NMA lymphodepletion regimen, such as neutropenia, lymphopenia, and coagulopathy, which are hematological side effects [5,70]. Non-hematological events include nausea, headaches, loss of appetite, neutropenic fever, diarrhea, and hyperbilirubinemia [5,69], which are mostly manageable by standard supportive treatment. Interestingly, in order to avoid the severe toxicity of the NMA lymphodepletion regimen, Santos et al. designed a regimen which used oncolytic adenoviruses to replace NMA lymphodepleting which resulted in great efficacy [71].
6. Role of Interleukin-2
IL-2 is an important cytokine; it stimulates effector T cell growth and survival [72]. Rosenberg and his colleagues found that the addition of IL-2 to cultured lymphocytes promoted lymphocytes to lyse autologous tumor cells [73,74]. In addition, they found that the combination of IL-2 with an infusion of TILs could improve their therapeutic effect [3]. Rosenberg then applied TIL therapy combined with IL-2 to patients with melanoma and published the first report on the successful treatment of melanoma with TIL-ACT in 1988 [4].
However, it is reported that IL-2 is an essential cytokine for the development and function of Tregs [75,76], which would compete with CD8+ T cells for IL-2 and suppress the response of the CD8+ T cells [76]. Liu et al. have proved that IL-2 could regulate tumor-reactive CD8+ T cell exhaustion [77]. Briefly, high levels of IL-2 upregulate the inhibitory receptors of CD8+ T cells and reduce the production of cytokines and effector molecules, resulting in the dysfunction of CD8+ T cells [77]. IL-2 could lead to terminal differentiation and T cell exhaustion, while IL-7, IL-15, and IL-21 can produce poorly differentiated T cells and enhanced proliferation [78]. Compared with the use of IL-2 alone, the addition of cytokines IL-15 and IL-21 to the culture medium could increase the number of TILs cultured from lung and colorectal tumors [79,80,81]. However, the current clinical trials mainly use IL-2 to expand and maintain TILs, and further clinical trials are needed to determine whether these cytokines can replace IL-2.
The optimal dose of IL-2 infusion is yet to be determined. Since Rosenberg and his colleagues published their research, the regimen of TILs combined with IL-2 has been used in the treatment of patients with metastatic melanoma for decades [6]. It has been reported that the HD IL-2 (≥720,000 IU/kg) regimen, which when administrated after TIL infusion, can continuously support the growth and activity of infused TILs [68]. However, HD IL-2 is associated with severe adverse events, include capillary leakage syndrome, which is characterized by hypotension, oliguria, edema, and even a hypovolemic shock [82,83]. Therefore, LD IL-2 (<720,000 IU/kg) combined with a TIL infusion is worthy of investigation. A study from Copenhagen demonstrated that LD IL-2 combined with a TIL infusion resulted in two out of six patients with metastatic melanoma having durable objective responses [84]. Furthermore, Ullenhag et al. showed that five out of 24 patients had objective clinical responses after LD IL-2 treatment [85]. In another clinical trial, the ORR of the patients who received attenuated doses of IL2 was 42% [86]. Recently, in a phase two clinical trial, 12 patients treated with TILs followed by a LD IL-2 regimen (125,000 IU/kg/day over 12 days) experienced no grade five IL-2-related adverse events, but did show the expected blood toxicities caused by lymphodepletion. Only three patients had a partial clinical response, and no patients achieved a complete response. Nguyen et al. believes that this unsatisfactory result may be caused by the LD IL-2 regimen [87].
There is not yet sufficient evidence to suggest that LD IL-2 can achieve a sustained response comparable to that of HD IL-2 regimens. However, these results are promising; they must be urgently addressed and further studied in clinical trials (Table 1). Recently, Hsu et al. have engineered an IL-2 prodrug to solve the problem of high toxicity in vivo and short half-life [88].
7. Combination Therapy with TILs
7.1. Immune Checkpoint Inhibitors
The combination of TIL therapy and anti-PD-1/PD-L1 antibody therapy has shown preliminary favorable results in some recent trials [11,89]. The immune checkpoint receptors (e.g., CTLA-4 and PD-1/PD-L1) are expressed on the surface of T cells, which is the self-protective mechanism of the immune system. In cancer patients, the CTLA-4 and PD-1 molecules on effector T cells are upregulated and, respectively, bind to B7-1/B7-2 and PD-L1 of antigen-presenting cells or tumor cells. This results in inhibited T cell function, which can be blocked by anti-CTLA-4 and anti-PD-1 antibodies [90]. In addition, studies have demonstrated that after long-term exposure to tumor antigens, CD8+ T cells will show apoptosis, or enter a state of abnormal differentiation with high express inhibitory receptors and almost no response to specific tumor antigens, which could be remedied with a checkpoint inhibitor [91,92,93]. In addition, the in vivo treatment models also proved these results [94,95]. These mechanisms provide a theoretical basis for TILs combined with ICIs. Therefore, to obtain increased tumor-reactive TILs, the ICIs should be administrated before the resection of tumor tissues and during the initial growth of TILs [96,97,98,99]. In order to make infused TILs more tumoricidal, ICIs were also administered after TIL infusion [5,7,11,97]. The effect of TILs combined with anti-PD-1 therapy as a first-line treatment is still in the clinical trial stage, and the additive effect of TILs and anti-PD-1 cannot be evaluated until the preliminary results are released. Recent studies have found that in addition to tumor cells, dendritic cells (DC) also express high levels of PD-L1, which can weaken the activation of T cells and inhibit anti-tumor activity [100,101]. This will provide a theoretical basis for TILs combined with PD-L1 inhibitors in the treatment of cancer patients with high PD-L1 expression.
7.2. BRAF Inhibitor
The BRAF gene plays an important role in cell growth and differentiation. In some cancers, BRAF mutations change the cascade of ERK/MAPK signals, resulting in increased cell proliferation [102]. BRAF mutation is the most common mutation that causes overactivation of the MAPK pathway and has been found to carry it in approximately half of cutaneous melanomas [103]. The activating BRAF mutation (Mainly V600E) can induce immune-escape mechanisms, which make them “dull” in immunity and gives them the ability to evade T-cell immune responses [104]. Studies have shown that the BRAF inhibitor vemurafenib can reduce related immunosuppressive signals, facilitate infiltration of lymphocytes, and reduce immunosuppressive cells, as well as enhance the presentation of melanoma antigens [105,106] (Figure 2). The ORR of BRAF inhibitor Vemurafenib in the treatment of BRAFV600E mutant melanoma is up to 50%, which improves the progression-free and overall survival rate [107]. However, the clinical response of BRAF inhibitors is usually short-lived [108], and as the disease progresses, alternative treatments available to patients is limited. Peiffer and colleagues indicated that BRAF/MEK inhibitors can promote the proliferation of melanoma-specific T cells in patients with melanoma [109]. Recently, in a clinical trial, seven of 11 patients with metastatic melanoma who received a combination treatment of TILs, HD IL-2, and vemurafenib experienced an objective clinical response, two of which achieved a complete response [110].
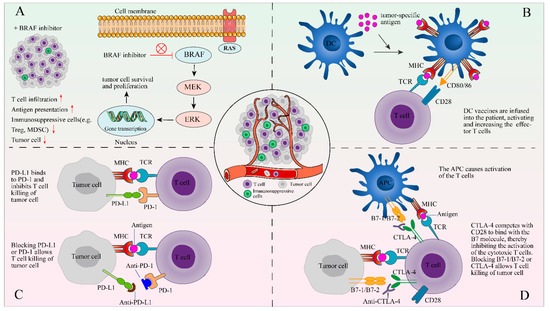
Figure 2.
The combination of TIL therapy and other therapies. The following four methods of combined therapy are described in the form of figures: (A) a BRAF inhibitor can reduce immunosuppressive signals and facilitate tumor infiltration of lymphocytes; (B) DCs are stimulated by signals and loaded with tumor-specific antigens on MHC to activate the antigen-specific T-cells; (C) PD-L1 or PD-1 blockers may prevent the interaction of PD-1 and PDL-1 between TILs and tumor cells; and (D) blocking CTLA-4, which competes with CD28 to bind the B7 molecule, allows T cell killing of tumor cell.
7.3. Other Combination Therapy with TILs
Notably, the DC vaccine can induce an immune response and can activate and increase the number of TILs [111,112] (Figure 2), and its combination with TIL therapy is being evaluated in clinical trials (NCT01946373, Table 1). The combination of TIL therapy and oncolytic virus is also being explored [113,114]. The virus can combat tumor immunosuppression by producing cytokines that promote the anti-tumor effect of TILs [115]. In a clinical trial of TIL therapy combined with adenovirus in the treatment of metastatic melanoma, five of 13 patients achieved objective responses, of which three achieved complete responses [115].
8. Limitations of TIL Therapy and Future Perspectives
TIL therapy has been successfully applied to patients with metastatic melanoma and other solid tumors. Compared with other ACTs (such as CAR-T and TCR-T), TIL therapy has its unique advantages. TILs are composed of T cells with multiple TCR clones, which can not only directly act against shared self-antigens [116], but can also act against tumor-specific neoantigens [117], making it more effective in response to tumor heterogeneity. TILs usually contain a large number of effector memory T cells and express chemokine receptors after being stimulated by the tumor antigen in vivo, which makes it easier to locate in the tumor tissue after transfusion. In addition, TILs come from patients themselves without gene modification, meaning the method has low toxicity.
However, TIL therapy obviously also has its limitations. Firstly, in order to achieve durable anti-tumor responses, effector T cells with anti-tumor activity must be present in the tumor, which is not the case for many solid tumors. In contrast, the NK cells have higher feasibility for manufacturing. However, NK cells are generally inhibited in cancer patients and are easily affected by a series of inhibitory factors in the cancer microenvironment [118,119]. Another potential translational anti-tumor cell population are tumor infiltrating γδT cells, which usually perform their anti-tumor activity by secreting interferon (IFN)γ and tumor necrotic factor (TNF), but they are not dependent on tumor-associated antigens [120]. To overcome the homing challenges of γδT cells, many strategies, including bispecific molecules [121], topical injection [122], and combination use with checkpoint inhibitors [123], have been tried for solid tumor treatment.
Secondly, although the selection strategies of TILs have made great progress, due to the inability to efficiently identify and isolate neoantigen-specific lymphocytes, as well as the obstacle of the immunosuppressive tumor microenvironment, the wide application of TIL therapy in a variety of cancers is still a challenge. Unlike TIL therapy, T cell receptor-engineered T cell (TCR-T) therapy modifies the TCRs on the surface of endogenous T cells to recognize tumor-specific neoantigens [124]. Therefore, TCR-T therapy is free from the limit of surface antigen expression of the target cells, which is a potential cellular immunotherapy for the treatment of various cancers. Significantly, the TCRs that respond to TP53 mutations identified by Deniger et al. may provide a new treatment for patients who carry these mutations and express the corresponding HLA types [12]. Moreover, as CAR-T cells are not required in APC presentation to act against antigen-positive cancer cells [125], CAR technology is a potential strategy for modifying TILs to recognize tumor associated antigens or neoantigens and enhance the therapy effect of TILs. A recent study by Mills et.al. designed dual-specific TILs by transducing a CAR to act against the common tumor antigen Her2 (anti-Her2 CAR-TILs); further studies showed that anti-Her2 CAR-TILs could act against Her-2 positive tumors in vitro and in vivo [126]. However, as tumor cells usually show different types of gene mutations which will generate many kinds of neoantigens, so it is difficult to design universal CARs-TILs to eliminate cancer cells.
Meanwhile, the immunosuppressive tumor microenvironment may induce the exhaustion of infiltrating cytotoxic T cells, which in turn results in reducing the elimination ability of cancer cells. Thus, new specific exhaustion markers must be explored. The development of new technologies, such as single-cell level analytic technologies, could help to unravel the characteristics of heterogeneous TILs; new TIL subsets could be identified with therapeutic potentials or new T cell markers as potential targets to act against solid cancers [127]. Using single-cell RNA sequencing, Singer et.al. separated the activation and dysfunction gene modules in dysfunctional CD8+ TILs, further investigation showed that the zinc-finger transcription factor Gata-3 is a regulator of CD8+ TILs dysfunction [128]. Using single-cell mass cytometry, Wagner et al. found high-grade ER+ breast tumors infiltrated with high levels of PD-L1+ exhausted T cells [129].
In addition, the infused TIL has a short survival time in vivo. In order to improve the survival and tumor-homing ability of TILs after reinfusion into patients, the modification technology of TILs has been explored [130,131]. Remarkably, researchers from the Jean-Philippe Girard research group of the University of Toulouse in France have found that the tumor-associated high endothelial venules (TA-HEVs) are the main sites that mediate the entry of lymphocytes into tumors. Increasing the density and maturity of TA-HEV endothelial cells (TA-HECs) can promote tumor-specific CD8+T cell infiltration and improve the efficacy of ICI. Moreover, TA-HEVs are closely related to the efficacy of ICI in patients with melanoma, and can be used as a powerful predictor of clinical treatment [132] and a potential target of combined TIL therapy.
9. Conclusions
In conclusion, TIL therapy has some unique advantages in the treatment of solid tumors, but it still faces a series of challenges. The tumor immunosuppressive microenvironment is still the main obstacle of TIL therapy. Furthermore, there is still considerable room for improvement in the isolation and expansion of effective tumor-reactive T cells, and alternative combined therapies still need to be explored.
Author Contributions
Conceptualization, Y.Z., Z.X. and J.L.; literature investigation, Y.Z., J.D. and S.R.; figures, Y.Z., J.D., S.G. and J.L.; writing—original draft preparation, Y.Z., J.D. and S.R.; writing—review and editing, S.G., J.S., F.D., X.W., Y.C., M.L., M.C., X.L., W.L., L.G., Y.S., Z.Z., Q.W., Z.X. and J.L.; supervision, Y.Z., Z.X. and J.L.; funding acquisition, Y.Z. and Z.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant number 81972643, and by the Science and Technology Strategic Cooperation Programs of Luzhou Municipal People’s Government and Southwest Medical University, grant number 2019LZXNYDJ45.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
ACT: adoptive cell therapy; APC: antigen presenting cell; BRAF: v-raf murine sarcoma viral oncogene homolog B1; b.i.d.: bis in die; CD: cluster of differentiation; Cy: cyclophosphamide; d: day; CXCR: c-x-c chemokine receptor; CRR: complete response rate; CTLA-4: cytotoxic T lymphocyte associated antigen-4; DC: dendritic cells; Durva: durvalumab; FACS: fluorescence-activated cell sorting; flu: fludarabine; FDA: food and drug administration; gy: gray; HD: high dose; HPVs: human papilloma viruses; hr: hour; i.d.: intradermal; i.v.: intravenous; IU: international unit; IL-2: interleukin-2; IFN-γ: interferon-gamma; ICI: immune checkpoint inhibitors; kg: kilogram; LD: low dose; Ipili: ipilimumab; max: maximum; m: milligram; MHC: histocompatibility complex; NGFR: nerve growth factor receptor; nivo: nivolumab; NMA: non-myeloablative; NK: natural killer; NSCLC: non-small cell lung cancer; ORR: objective response rate; pembro: pembrolizumab; PD-1: programmed cell death protein-1; PD-L1: programmed cell death protein ligand-1; qd.: quaque die; REP: rapid expansion procedure; RNA: ribonucleic acid; RCC: renal cell carcinoma; s.c.: subcutaneous; TMG: tandem minigene; t.i.d.: ter in die; TBI: total body irradiation; tremeli: tremelimumab; TILs: tumor-infiltrating lymphocytes; tregs: regulatory T cells; tisleli: tislelizumab; vem: vemurafenib; VEGF: vascular endothelial growth factor; WES: whole-exome sequencing; w: week; x: times.
References
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Spiess, P.J.; Yang, J.C.; Rosenberg, S.A. In vivo antitumor activity of tumor-infiltrating lymphocytes expanded in recombinant interleukin-2. J. Natl. Cancer Inst. 1987, 79, 1067–1075. [Google Scholar] [PubMed]
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Besser, M.J.; Shapira-Frommer, R.; Itzhaki, O.; Treves, A.J.; Zippel, D.B.; Levy, D.; Kubi, A.; Shoshani, N.; Zikich, D.; Ohayon, Y.; et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: Intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin. Cancer Res. 2013, 19, 4792–4800. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.-C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef]
- Stevanović, S.; Draper, L.M.; Langhan, M.M.; Campbell, T.E.; Kwong, M.L.; Wunderlich, J.R.; Dudley, M.E.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1543–1550. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Creelan, B.C.; Wang, C.; Teer, J.K.; Toloza, E.M.; Yao, J.; Kim, S.; Landin, A.M.; Mullinax, J.E.; Saller, J.J.; Saltos, A.N.; et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: A phase 1 trial. Nat. Med. 2021, 27, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-cell Responses to TP53 "Hotspot" Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin. Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef] [PubMed]
- Dafni, U.; Michielin, O.; Lluesma, S.M.; Tsourti, Z.; Polydoropoulou, V.; Karlis, D.; Besser, M.J.; Haanen, J.; Svane, I.M.; Ohashi, P.S.; et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: A systematic review and meta-analysis. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1902–1913. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.S.; Somerville, R.P.T.; Yang, J.C.; Sherry, R.M.; Klebanoff, C.A.; Goff, S.L.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.; Paria, B.C.; et al. Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: A single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 792–802. [Google Scholar] [CrossRef]
- Sarnaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Thomas, S.S.; Domingo-Musibay, E.; Pavlick, A.C.; Whitman, E.D.; et al. Lifileucel, a Tumor-Infiltrating Lymphocyte Therapy, in Metastatic Melanoma. J. Clin. Oncol. 2021, 39, 2656–2666. [Google Scholar] [CrossRef]
- Van den Berg, J.H.; Heemskerk, B.; van Rooij, N.; Gomez-Eerland, R.; Michels, S.; van Zon, M.; de Boer, R.; Bakker, N.A.M.; Jorritsma-Smit, A.; van Buuren, M.M.; et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: Boosting of neoantigen-specific T cell reactivity and long-term follow-up. J. Immunother. Cancer 2020, 8, e000848. [Google Scholar] [CrossRef]
- Stevanović, S.; Helman, S.R.; Wunderlich, J.R.; Langhan, M.M.; Doran, S.L.; Kwong, M.L.M.; Somerville, R.P.T.; Klebanoff, C.A.; Kammula, U.S.; Sherry, R.M.; et al. A Phase II Study of Tumor-infiltrating Lymphocyte Therapy for Human Papillomavirus-associated Epithelial Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 1486–1493. [Google Scholar] [CrossRef]
- Jazaeri, A.A.; Zsiros, E.; Amaria, R.N.; Artz, A.S.; Edwards, R.P.; Wenham, R.M.; Slomovitz, B.M.; Walther, A.; Thomas, S.S.; Chesney, J.A.; et al. Safety and efficacy of adoptive cell transfer using autologous tumor infiltrating lymphocytes (LN-145) for treatment of recurrent, metastatic, or persistent cervical carcinoma. J. Clin. Oncol. 2019, 37 (Suppl. 15), 2538. [Google Scholar] [CrossRef]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Stevanović, S.; Pasetto, A.; Helman, S.R.; Gartner, J.J.; Prickett, T.D.; Howie, B.; Robins, H.S.; Robbins, P.F.; Klebanoff, C.A.; Rosenberg, S.A.; et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 2017, 356, 200–205. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, Y.-A.; Sim, C.K.; Heo, S.-H.; Song, I.H.; Park, H.S.; Park, S.Y.; Bang, W.S.; Park, I.A.; Lee, M.; et al. Expansion of tumor-infiltrating lymphocytes and their potential for application as adoptive cell transfer therapy in human breast cancer. Oncotarget 2017, 8, 113345–113359. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Pearce, E.L. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 2016, 17, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Yen, P.H.; Nie, J.; Liadis, N.; Ghazarian, D.; Al-Habeeb, A.; Easson, A.; Leong, W.; Lipa, J.; McCready, D.; et al. Expansion and characterization of human melanoma tumor-infiltrating lymphocytes (TILs). PLoS ONE 2010, 5, e13940. [Google Scholar] [CrossRef]
- Poch, M.; Hall, M.; Joerger, A.; Kodumudi, K.; Beatty, M.; Innamarato, P.P.; Bunch, B.L.; Fishman, M.N.; Zhang, J.; Sexton, W.J.; et al. Expansion of tumor infiltrating lymphocytes (TIL) from bladder cancer. Oncoimmunology 2018, 7, e1476816. [Google Scholar] [CrossRef]
- Wickström, S.; Lövgren, T. Expansion of Tumor-Infiltrating Lymphocytes from Melanoma Tumors. Methods Mol. Biol. 2019, 1913, 105–118. [Google Scholar]
- Wu, R.; Forget, M.-A.; Chacon, J.; Bernatchez, C.; Haymaker, C.; Chen, J.Q.; Hwu, P.; Radvanyi, L.G. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: Current status and future outlook. Cancer J. 2012, 18, 160–175. [Google Scholar] [CrossRef]
- Besser, M.J.; Shapira-Frommer, R.; Treves, A.J.; Zippel, D.; Itzhaki, O.; Hershkovitz, L.; Levy, D.; Kubi, A.; Hovav, E.; Chermoshniuk, N.; et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2010, 16, 2646–2655. [Google Scholar] [CrossRef]
- Aebersold, P.; Hyatt, C.; Johnson, S.; Hines, K.; Korcak, L.; Sanders, M.; Lotze, M.; Topalian, S.; Yang, J.; Rosenberg, S.A. Lysis of autologous melanoma cells by tumor-infiltrating lymphocytes: Association with clinical response. J. Natl. Cancer Inst. 1991, 83, 932–937. [Google Scholar] [CrossRef]
- Schwartzentruber, D.J.; Hom, S.S.; Dadmarz, R.; White, D.E.; Yannelli, J.R.; Steinberg, S.M.; Rosenberg, S.A.; Topalian, S.L. In vitro predictors of therapeutic response in melanoma patients receiving tumor-infiltrating lymphocytes and interleukin-2. J. Clin. Oncol. 1994, 12, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.Q.; Zhou, J.; Durflinger, K.H.; Langhan, M.M.; Shelton, T.E.; Wunderlich, J.R.; Robbins, P.F.; Rosenberg, S.A.; Dudley, M.E. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J. Immunother. 2008, 31, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; Junker, N.; Ellebaek, E.; Andersen, M.H.; Straten, P.T.; Svane, I.M. Characterization and comparison of ‘standard’ and ‘young’ tumour-infiltrating lymphocytes for adoptive cell therapy at a Danish translational research institution. Scand. J. Immunol. 2012, 75, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Gross, C.A.; Langhan, M.M.; Garcia, M.R.; Sherry, R.M.; Yang, J.C.; Phan, G.Q.; Kammula, U.S.; Hughes, M.S.; Citrin, D.E.; et al. CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin. Cancer Res. 2010, 16, 6122–6131. [Google Scholar] [CrossRef]
- Itzhaki, O.; Hovav, E.; Ziporen, Y.; Levy, D.; Kubi, A.; Zikich, D.; Hershkovitz, L.; Treves, A.J.; Shalmon, B.; Zippel, D.; et al. Establishment and large-scale expansion of minimally cultured “young” tumor infiltrating lymphocytes for adoptive transfer therapy. J. Immunother. 2011, 34, 212–220. [Google Scholar] [CrossRef]
- Geukes Foppen, M.H.; Donia, M.; Svane, I.M.; Haanen, J.B.A.G. Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol. Oncol. 2015, 9, 1918–1935. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol. Rev. 2006, 211, 214–224. [Google Scholar] [CrossRef]
- Radvanyi, L.G.; Bernatchez, C.; Zhang, M.; Fox, P.S.; Miller, P.; Chacon, J.; Wu, R.; Lizee, G.; Mahoney, S.; Alvarado, G.; et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2012, 18, 6758–6770. [Google Scholar] [CrossRef]
- Webb, J.R.; Milne, K.; Nelson, B.H. PD-1 and CD103 Are Widely Coexpressed on Prognostically Favorable Intraepithelial CD8 T Cells in Human Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 926–935. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, H.; Mao, L.; Yang, Q.C.; Fu, L.Q.; Wu, C.C.; Liu, B.; Sun, Z.J. CD103+ T and Dendritic Cells Indicate a Favorable Prognosis in Oral Cancer. J. Dent. Res. 2019, 98, 1480–1487. [Google Scholar] [CrossRef]
- Ganesan, A.P.; Clarke, J.; Wood, O.; Garrido-Martin, E.M.; Chee, S.J.; Mellows, T.; Samaniego-Castruita, D.; Singh, D.; Seumois, G.; Alzetani, A.; et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017, 18, 940–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.-L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8 T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; de Miranda, N.F.; Goodall, C.P.; Blair, T.C.; Fox, B.A.; McDermott, J.E.; et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018, 9, 2724. [Google Scholar] [CrossRef]
- Kortekaas, K.E.; Santegoets, S.J.; Sturm, G.; Ehsan, I.; van Egmond, S.L.; Finotello, F.; Trajanoski, Z.; Welters, M.J.P.; van Poelgeest, M.I.E.; van der Burg, S.H. CD39 Identifies the CD4 Tumor-Specific T-cell Population in Human Cancer. Cancer Immunol. Res. 2020, 8, 1311–1321. [Google Scholar] [CrossRef]
- Martinez-Usatorre, A.; Carmona, S.J.; Godfroid, C.; Yacoub Maroun, C.; Labiano, S.; Romero, P. Enhanced Phenotype Definition for Precision Isolation of Precursor Exhausted Tumor-Infiltrating CD8 T Cells. Front. Immunol. 2020, 11, 340. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef]
- Inozume, T.; Hanada, K.-I.; Wang, Q.J.; Ahmadzadeh, M.; Wunderlich, J.R.; Rosenberg, S.A.; Yang, J.C. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J. Immunother. 2010, 33, 956–964. [Google Scholar] [CrossRef]
- Fernandez-Poma, S.M.; Salas-Benito, D.; Lozano, T.; Casares, N.; Riezu-Boj, J.I.; Mancheño, U.; Elizalde, E.; Alignani, D.; Zubeldia, N.; Otano, I.; et al. Expansion of Tumor-Infiltrating CD8(+) T cells Expressing PD-1 Improves the Efficacy of Adoptive T-cell Therapy. Cancer Res. 2017, 77, 3672–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfl, M.; Kuball, J.; Ho, W.Y.; Nguyen, H.; Manley, T.J.; Bleakley, M.; Greenberg, P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood 2007, 110, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Song, D.-G.; Poussin, M.; Yamamoto, T.; Best, A.; Li, C.; Coukos, G.; Powell, D.J. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin. Cancer Res. 2014, 20, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou-Thompson, D.; Forget, M.-A.; Creasy, C.; Bernard, V.; Zhao, L.; Kim, Y.U.; Hurd, M.W.; Uraoka, N.; Parra, E.R.; Kang, Y.; et al. 4-1BB Agonist Focuses CD8 Tumor-Infiltrating T-Cell Growth into a Distinct Repertoire Capable of Tumor Recognition in Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 7263–7275. [Google Scholar] [CrossRef]
- Seliktar-Ofir, S.; Merhavi-Shoham, E.; Itzhaki, O.; Yunger, S.; Markel, G.; Schachter, J.; Besser, M.J. Selection of Shared and Neoantigen-Reactive T Cells for Adoptive Cell Therapy Based on CD137 Separation. Front. Immunol. 2017, 8, 1211. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Crystal, J.S.; Cohen, C.J.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Yao, X.; Wang, R.; Gros, A.; Li, Y.F.; et al. Durable Complete Response from Metastatic Melanoma after Transfer of Autologous T Cells Recognizing 10 Mutated Tumor Antigens. Cancer Immunol. Res. 2016, 4, 669–678. [Google Scholar] [CrossRef]
- Chacon, J.A.; Sarnaik, A.A.; Chen, J.Q.; Creasy, C.; Kale, C.; Robinson, J.; Weber, J.; Hwu, P.; Pilon-Thomas, S.; Radvanyi, L. Manipulating the tumor microenvironment ex vivo for enhanced expansion of tumor-infiltrating lymphocytes for adoptive cell therapy. Clin. Cancer Res. 2015, 21, 611–621. [Google Scholar] [CrossRef]
- Tavera, R.J.; Forget, M.A.; Kim, Y.U.; Sakellariou-Thompson, D.; Creasy, C.A.; Bhatta, A.; Fulbright, O.J.; Ramachandran, R.; Thorsen, S.T.; Flores, E.; et al. Utilizing T-cell Activation Signals 1, 2, and 3 for Tumor-infiltrating Lymphocytes (TIL) Expansion: The Advantage Over the Sole Use of Interleukin-2 in Cutaneous and Uveal Melanoma. J. Immunother. 2018, 41, 399–405. [Google Scholar] [CrossRef]
- Parkhurst, M.; Gros, A.; Pasetto, A.; Prickett, T.; Crystal, J.S.; Robbins, P.; Rosenberg, S.A. Isolation of T-Cell Receptors Specifically Reactive with Mutated Tumor-Associated Antigens from Tumor-Infiltrating Lymphocytes Based on CD137 Expression. Clin. Cancer Res. 2017, 23, 2491–2505. [Google Scholar] [CrossRef]
- Robbins, P.F.; Lu, Y.-C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Jin, J.; Sabatino, M.; Somerville, R.; Wilson, J.R.; Dudley, M.E.; Stroncek, D.F.; Rosenberg, S.A. Simplified method of the growth of human tumor infiltrating lymphocytes in gas-permeable flasks to numbers needed for patient treatment. J. Immunother. 2012, 35, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.C.; Chan, C.-C.; Klebanoff, C.A.; Overwijk, W.W.; et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Finkelstein, S.E.; Klebanoff, C.A.; Antony, P.A.; Palmer, D.C.; Spiess, P.J.; Hwang, L.N.; Yu, Z.; Wrzesinski, C.; Heimann, D.M.; et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005, 202, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.L.; Cole, D.J. Dendritic cell recovery post-lymphodepletion: A potential mechanism for anti-cancer adoptive T cell therapy and vaccination. Cancer Immunol. Immunother. 2010, 59, 341–353. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef]
- Wrzesinski, C.; Paulos, C.M.; Kaiser, A.; Muranski, P.; Palmer, D.C.; Gattinoni, L.; Yu, Z.; Rosenberg, S.A.; Restifo, N.P. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J. Immunother. 2010, 33, 1–7. [Google Scholar] [CrossRef]
- Goff, S.L.; Dudley, M.E.; Citrin, D.E.; Somerville, R.P.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J. Clin. Oncol. 2016, 34, 2389–2397. [Google Scholar] [CrossRef]
- Nissani, A.; Lev-Ari, S.; Meirson, T.; Jacoby, E.; Asher, N.; Ben-Betzalel, G.; Itzhaki, O.; Shapira-Frommer, R.; Schachter, J.; Markel, G.; et al. Comparison of non-myeloablative lymphodepleting preconditioning regimens in patients undergoing adoptive T cell therapy. J. Immunother. Cancer 2021, 9, e001743. [Google Scholar] [CrossRef]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive cell therapy for patients with metastatic melanoma: Evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef]
- Santos, J.M.; Cervera-Carrascon, V.; Havunen, R.; Zafar, S.; Siurala, M.; Sorsa, S.; Anttila, M.; Kanerva, A.; Hemminki, A. Adenovirus Coding for Interleukin-2 and Tumor Necrosis Factor Alpha Replaces Lymphodepleting Chemotherapy in Adoptive T Cell Therapy. Mol. Ther. 2018, 26, 2243–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.E.; Ohlén, C.; Nelson, B.H.; Greenberg, P.D. Enhanced signaling through the IL-2 receptor in CD8+ T cells regulated by antigen recognition results in preferential proliferation and expansion of responding CD8+ T cells rather than promotion of cell death. Proc. Natl. Acad. Sci. USA 2002, 99, 3001–3006. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Grimm, E.A.; Mazumder, A.; Strausser, J.L.; Rosenberg, S.A. Lysis of fresh and cultured autologous tumor by human lymphocytes cultured in T-cell growth factor. Cancer Res. 1981, 41 Pt 1, 4420–4425. [Google Scholar] [PubMed]
- Grimm, E.A.; Mazumder, A.; Zhang, H.Z.; Rosenberg, S.A. Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J. Exp. Med. 1982, 155, 1823–1841. [Google Scholar] [CrossRef] [PubMed]
- Malek, T.R. The main function of IL-2 is to promote the development of T regulatory cells. J. Leukoc. Biol. 2003, 74, 961–965. [Google Scholar] [CrossRef]
- Kalia, V.; Sarkar, S. Regulation of Effector and Memory CD8 T Cell Differentiation by IL-2-A Balancing Act. Front. Immunol. 2018, 9, 2987. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, N.; Zhou, L.; Wang, J.; Zhou, Y.; Zhang, T.; Fang, Y.; Deng, J.; Gao, Y.; Liang, X.; et al. IL-2 regulates tumor-reactive CD8 T cell exhaustion by activating the aryl hydrocarbon receptor. Nat. Immunol. 2021, 22, 358–369. [Google Scholar] [CrossRef]
- Hinrichs, C.S.; Spolski, R.; Paulos, C.M.; Gattinoni, L.; Kerstann, K.W.; Palmer, D.C.; Klebanoff, C.A.; Rosenberg, S.A.; Leonard, W.J.; Restifo, N.P. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 2008, 111, 5326–5333. [Google Scholar] [CrossRef]
- Meng, Q.; Liu, Z.; Rangelova, E.; Poiret, T.; Ambati, A.; Rane, L.; Xie, S.; Verbeke, C.; Dodoo, E.; Del Chiaro, M.; et al. Expansion of Tumor-reactive T Cells From Patients With Pancreatic Cancer. J. Immunother. 2016, 39, 81–89. [Google Scholar] [CrossRef]
- Liu, Z.; Meng, Q.; Bartek, J.; Poiret, T.; Persson, O.; Rane, L.; Rangelova, E.; Illies, C.; Peredo, I.H.; Luo, X.; et al. Tumor-infiltrating lymphocytes (TILs) from patients with glioma. Oncoimmunology 2017, 6, e1252894. [Google Scholar] [CrossRef]
- Frank, L.S.A.; Lotze, M.; Ritthipichai, K.; Mosychuk, C. The T-cell Growth Factor Cocktail IL-2/IL-15/IL-21 Enhances Expansion and Effector Function of Tumor-Infiltrating T cells in a Novel Process Developed by Iovance. In Proceedings of the SITC Annual Meeting, National Harbor, MD, USA, 8 November 2017. [Google Scholar]
- Marabondo, S.; Kaufman, H.L. High-dose interleukin-2 (IL-2) for the treatment of melanoma: Safety considerations and future directions. Expert Opin. Drug. Saf. 2017, 16, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Acquavella, N.; Kluger, H.; Rhee, J.; Farber, L.; Tara, H.; Ariyan, S.; Narayan, D.; Kelly, W.; Sznol, M. Toxicity and activity of a twice daily high-dose bolus interleukin 2 regimen in patients with metastatic melanoma and metastatic renal cell cancer. J. Immunother. 2008, 31, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Ellebaek, E.; Iversen, T.Z.; Junker, N.; Donia, M.; Engell-Noerregaard, L.; Met, Ö.; Hölmich, L.R.; Andersen, R.S.; Hadrup, S.R.; Andersen, M.H.; et al. Adoptive cell therapy with autologous tumor infiltrating lymphocytes and low-dose Interleukin-2 in metastatic melanoma patients. J. Transl. Med. 2012, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Ullenhag, G.J.; Sadeghi, A.M.; Carlsson, B.; Ahlström, H.; Mosavi, F.; Wagenius, G.; Tötterman, T.H. Adoptive T-cell therapy for malignant melanoma patients with TILs obtained by ultrasound-guided needle biopsy. Cancer Immunol. Immunother. 2012, 61, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.; Donia, M.; Ellebaek, E.; Borch, T.H.; Kongsted, P.; Iversen, T.Z.; Hölmich, L.R.; Hendel, H.W.; Met, Ö.; Andersen, M.H.; et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin. Cancer Res. 2016, 22, 3734–3745. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Saibil, S.D.; Sotov, V.; Le, M.X.; Khoja, L.; Ghazarian, D.; Bonilla, L.; Majeed, H.; Hogg, D.; Joshua, A.M.; et al. Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol. Immunother. 2019, 68, 773–785. [Google Scholar] [CrossRef]
- Hsu, E.J.; Cao, X.; Moon, B.; Bae, J.; Sun, Z.; Liu, Z.; Fu, Y.-X. A cytokine receptor-masked IL2 prodrug selectively activates tumor-infiltrating lymphocytes for potent antitumor therapy. Nat. Commun. 2021, 12, 2768. [Google Scholar] [CrossRef]
- Wang, C.; Li, M.; Wei, R.; Wu, J. Adoptive transfer of TILs plus anti-PD1 therapy: An alternative combination therapy for treating metastatic osteosarcoma. J. Bone Oncol. 2020, 25, 100332. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef]
- Kumar, A.; Chamoto, K. Immune metabolism in PD-1 blockade-based cancer immunotherapy. Int. Immunol. 2021, 33, 17–26. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Chamoto, K.; Kumar, A.; Honjo, T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8+ T Cells and Facilitates Anti–PD-1 Therapy. Cancer Immunol. Res. 2018, 6, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [PubMed]
- Kodumudi, K.N.; Siegel, J.; Weber, A.M.; Scott, E.; Sarnaik, A.A.; Pilon-Thomas, S. Immune Checkpoint Blockade to Improve Tumor Infiltrating Lymphocytes for Adoptive Cell Therapy. PLoS ONE 2016, 11, e0153053. [Google Scholar] [CrossRef] [PubMed]
- Pilon-Thomas, S.; Mackay, A.; Vohra, N.; Mulé, J.J. Blockade of programmed death ligand 1 enhances the therapeutic efficacy of combination immunotherapy against melanoma. J. Immunol. 2010, 184, 3442–3449. [Google Scholar] [CrossRef]
- Friese, C.; Harbst, K.; Borch, T.H.; Westergaard, M.C.W.; Pedersen, M.; Kverneland, A.; Jönsson, G.; Donia, M.; Svane, I.M.; Met, Ö. CTLA-4 blockade boosts the expansion of tumor-reactive CD8+ tumor-infiltrating lymphocytes in ovarian cancer. Sci. Rep. 2020, 10, 3914. [Google Scholar] [CrossRef]
- Kverneland, A.H.; Pedersen, M.; Westergaard, M.C.W.; Nielsen, M.; Borch, T.H.; Olsen, L.R.; Aasbjerg, G.; Santegoets, S.J.; van der Burg, S.H.; Milne, K.; et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget 2020, 11, 2092–2105. [Google Scholar] [CrossRef]
- Hall, M.; Liu, H.; Malafa, M.; Centeno, B.; Hodul, P.J.; Pimiento, J.; Pilon-Thomas, S.; Sarnaik, A.A. Expansion of tumor-infiltrating lymphocytes (TIL) from human pancreatic tumors. J. Immunother. Cancer 2016, 4, 61. [Google Scholar] [CrossRef]
- Mullinax, J.E.; Hall, M.; Prabhakaran, S.; Weber, J.; Khushalani, N.; Eroglu, Z.; Brohl, A.S.; Markowitz, J.; Royster, E.; Richards, A.; et al. Combination of Ipilimumab and Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes for Patients with Metastatic Melanoma. Front. Oncol. 2018, 8, 44. [Google Scholar] [CrossRef]
- Peng, Q.; Qiu, X.; Zhang, Z.; Zhang, S.; Zhang, Y.; Liang, Y.; Guo, J.; Peng, H.; Chen, M.; Fu, Y.-X.; et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 2020, 11, 4835. [Google Scholar] [CrossRef]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaav7431. [Google Scholar] [CrossRef]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef] [PubMed]
- Kuske, M.; Westphal, D.; Wehner, R.; Schmitz, M.; Beissert, S.; Praetorius, C.; Meier, F. Immunomodulatory effects of BRAF and MEK inhibitors: Implications for Melanoma therapy. Pharmacol. Res. 2018, 136, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Cooper, Z.A.; Frederick, D.T.; Juneja, V.R.; Sullivan, R.J.; Lawrence, D.P.; Piris, A.; Sharpe, A.H.; Fisher, D.E.; Flaherty, K.T.; Wargo, J.A. BRAF inhibition is associated with increased clonality in tumor-infiltrating lymphocytes. Oncoimmunology 2013, 2, e26615. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, K.M.; Correa, I.; Josephs, D.H.; Karagiannis, P.; Egbuniwe, I.U.; Cafferkey, M.J.; Spicer, J.F.; Harries, M.; Nestle, F.O.; Lacy, K.E.; et al. Effects of BRAF mutations and BRAF inhibition on immune responses to melanoma. Mol. Cancer Ther. 2014, 13, 2769–2783. [Google Scholar] [CrossRef]
- Sloot, S.; Zager, J.S.; Kudchadkar, R.R.; Messina, J.L.; Benedict, J.J.; Gonzalez, R.J.; DeConti, R.; Turner, L.M.; McCardle, T.; Smalley, K.S.; et al. BRAF inhibition for advanced locoregional BRAF V600E mutant melanoma: A potential neoadjuvant strategy. Melanoma Res. 2016, 26, 83–87. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef]
- Peiffer, L.; Farahpour, F.; Sriram, A.; Spassova, I.; Hoffmann, D.; Kubat, L.; Stoitzner, P.; Gambichler, T.; Sucker, A.; Ugurel, S.; et al. BRAF and MEK inhibition in melanoma patients enables reprogramming of tumor infiltrating lymphocytes. Cancer Immunol. Immunother. CII 2021, 70, 1635–1647. [Google Scholar] [CrossRef]
- Deniger, D.C.; Kwong, M.L.M.; Pasetto, A.; Dudley, M.E.; Wunderlich, J.R.; Langhan, M.M.; Lee, C.-C.R.; Rosenberg, S.A. A Pilot Trial of the Combination of Vemurafenib with Adoptive Cell Therapy in Patients with Metastatic Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 351–362. [Google Scholar] [CrossRef]
- van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; de Vries, I.J.M.; Bol, K.F. Dendritic Cell Cancer Therapy: Vaccinating the Right Patient at the Right Time. Front. Immunol. 2018, 9, 2265. [Google Scholar] [CrossRef]
- Zhang, R.; Yuan, F.; Shu, Y.; Tian, Y.; Zhou, B.; Yi, L.; Zhang, X.; Ding, Z.; Xu, H.; Yang, L. Personalized neoantigen-pulsed dendritic cell vaccines show superior immunogenicity to neoantigen-adjuvant vaccines in mouse tumor models. Cancer Immunol. Immunother. CII 2020, 69, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.M.; Heiniö, C.; Cervera-Carrascon, V.; Quixabeira, D.C.A.; Siurala, M.; Havunen, R.; Butzow, R.; Zafar, S.; de Gruijl, T.; Lassus, H.; et al. Oncolytic adenovirus shapes the ovarian tumor microenvironment for potent tumor-infiltrating lymphocyte tumor reactivity. J. Immunother. Cancer 2020, 8, e000188. [Google Scholar] [CrossRef] [PubMed]
- Feist, M.; Zhu, Z.; Dai, E.; Ma, C.; Liu, Z.; Giehl, E.; Ravindranathan, R.; Kowalsky, S.J.; Obermajer, N.; Kammula, U.S.; et al. Oncolytic virus promotes tumor-reactive infiltrating lymphocytes for adoptive cell therapy. Cancer Gene Ther. 2021, 28, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Khammari, A.; Nguyen, J.M.; Saint-Jean, M.; Knol, A.C.; Pandolfino, M.C.; Quereux, G.; Brocard, A.; Peuvrel, L.; Saiagh, S.; Bataille, V.; et al. Adoptive T cell therapy combined with intralesional administrations of TG1042 (adenovirus expressing interferon-γ) in metastatic melanoma patients. Cancer Immunol. Immunother. CII 2015, 64, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Kvistborg, P.; Shu, C.J.; Heemskerk, B.; Fankhauser, M.; Thrue, C.A.; Toebes, M.; van Rooij, N.; Linnemann, C.; van Buuren, M.M.; Urbanus, J.H.; et al. TIL therapy broadens the tumor-reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunology 2012, 1, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Vitale, M.; Cantoni, C.; Della Chiesa, M.; Ferlazzo, G.; Carlomagno, S.; Pende, D.; Falco, M.; Pessino, A.; Muccio, L.; De Maria, A.; et al. An Historical Overview: The Discovery of How NK Cells Can Kill Enemies, Recruit Defense Troops, and More. Front. Immunol. 2019, 10, 1415. [Google Scholar] [CrossRef]
- Du, N.; Guo, F.; Wang, Y.; Cui, J. NK Cell Therapy: A Rising Star in Cancer Treatment. Cancers 2021, 13, 4129. [Google Scholar] [CrossRef]
- Sebestyen, Z.; Prinz, I.; Déchanet-Merville, J.; Silva-Santos, B.; Kuball, J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat. Rev. Drug Discov. 2020, 19, 169–184. [Google Scholar] [CrossRef]
- de Bruin, R.C.G.; Veluchamy, J.P.; Lougheed, S.M.; Schneiders, F.L.; Lopez-Lastra, S.; Lameris, R.; Stam, A.G.; Sebestyen, Z.; Kuball, J.; Molthoff, C.F.M.; et al. A bispecific nanobody approach to leverage the potent and widely applicable tumor cytolytic capacity of Vγ9Vδ2-T cells. Oncoimmunology 2017, 7, e1375641. [Google Scholar] [CrossRef]
- Lamb, L.S., Jr.; Bowersock, J.; Dasgupta, A.; Gillespie, G.Y.; Su, Y.; Johnson, A.; Spencer, H.T. Engineered drug resistant γδ T cells kill glioblastoma cell lines during a chemotherapy challenge: A strategy for combining chemo- and immunotherapy. PLoS ONE 2013, 8, e51805. [Google Scholar] [CrossRef] [PubMed]
- de Weerdt, I.; Hofland, T.; Lameris, R.; Endstra, S.; Jongejan, A.; Moerland, P.D.; de Bruin, R.C.G.; Remmerswaal, E.B.M.; Ten Berge, I.J.M.; Liu, N.; et al. Improving CLL Vγ9Vδ2-T-cell fitness for cellular therapy by ex vivo activation and ibrutinib. Blood 2018, 132, 2260–2272. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, X.; Zhang, F.; Zhang, X.; Tang, F.; Han, Z.; Li, Y. TCR-T Immunotherapy: The Challenges and Solutions. Front. Oncol. 2022, 11, 794183. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.K.; Henderson, M.A.; Giuffrida, L.; Petrone, P.; Westwood, J.A.; Darcy, P.K.; Neeson, P.J.; Kershaw, M.H.; Gyorki, D.E. Generating CAR T cells from tumor-infiltrating lymphocytes. Ther. Adv. Vaccines Immunother. 2021, 9, 25151355211017119. [Google Scholar] [CrossRef]
- Hossain, M.A.; Liu, G.; Dai, B.; Si, Y.; Yang, Q.; Wazir, J.; Birnbaumer, L.; Yang, Y. Reinvigorating exhausted CD8(+) cytotoxic T lymphocytes in the tumor microenvironment and current strategies in cancer immunotherapy. Med. Res. Rev. 2021, 41, 156–201. [Google Scholar] [CrossRef]
- Singer, M.; Wang, C.; Cong, L.; Marjanovic, N.D.; Kowalczyk, M.S.; Zhang, H.; Nyman, J.; Sakuishi, K.; Kurtulus, S.; Gennert, D.; et al. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 2016, 166, 1500–1511.e09. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef]
- Sapoznik, S.; Ortenberg, R.; Galore-Haskel, G.; Kozlovski, S.; Levy, D.; Avivi, C.; Barshack, I.; Cohen, C.J.; Besser, M.J.; Schachter, J.; et al. CXCR1 as a novel target for directing reactive T cells toward melanoma: Implications for adoptive cell transfer immunotherapy. Cancer Immunol. Immunother. CII 2012, 61, 1833–1847. [Google Scholar] [CrossRef]
- Heemskerk, B.; Liu, K.; Dudley, M.E.; Johnson, L.A.; Kaiser, A.; Downey, S.; Zheng, Z.; Shelton, T.E.; Matsuda, K.; Robbins, P.F.; et al. Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2. Hum. Gene Ther. 2008, 19, 496–510. [Google Scholar] [CrossRef]
- Asrir, A.; Tardiveau, C.; Coudert, J.; Laffont, R.; Blanchard, L.; Bellard, E.; Veerman, K.; Bettini, S.; Lafouresse, F.; Vina, E.; et al. Tumor-associated high endothelial venules mediate lymphocyte entry into tumors and predict response to PD-1 plus CTLA-4 combination immunotherapy. Cancer Cell 2022, 40, 318–334.e9. [Google Scholar] [CrossRef] [PubMed]
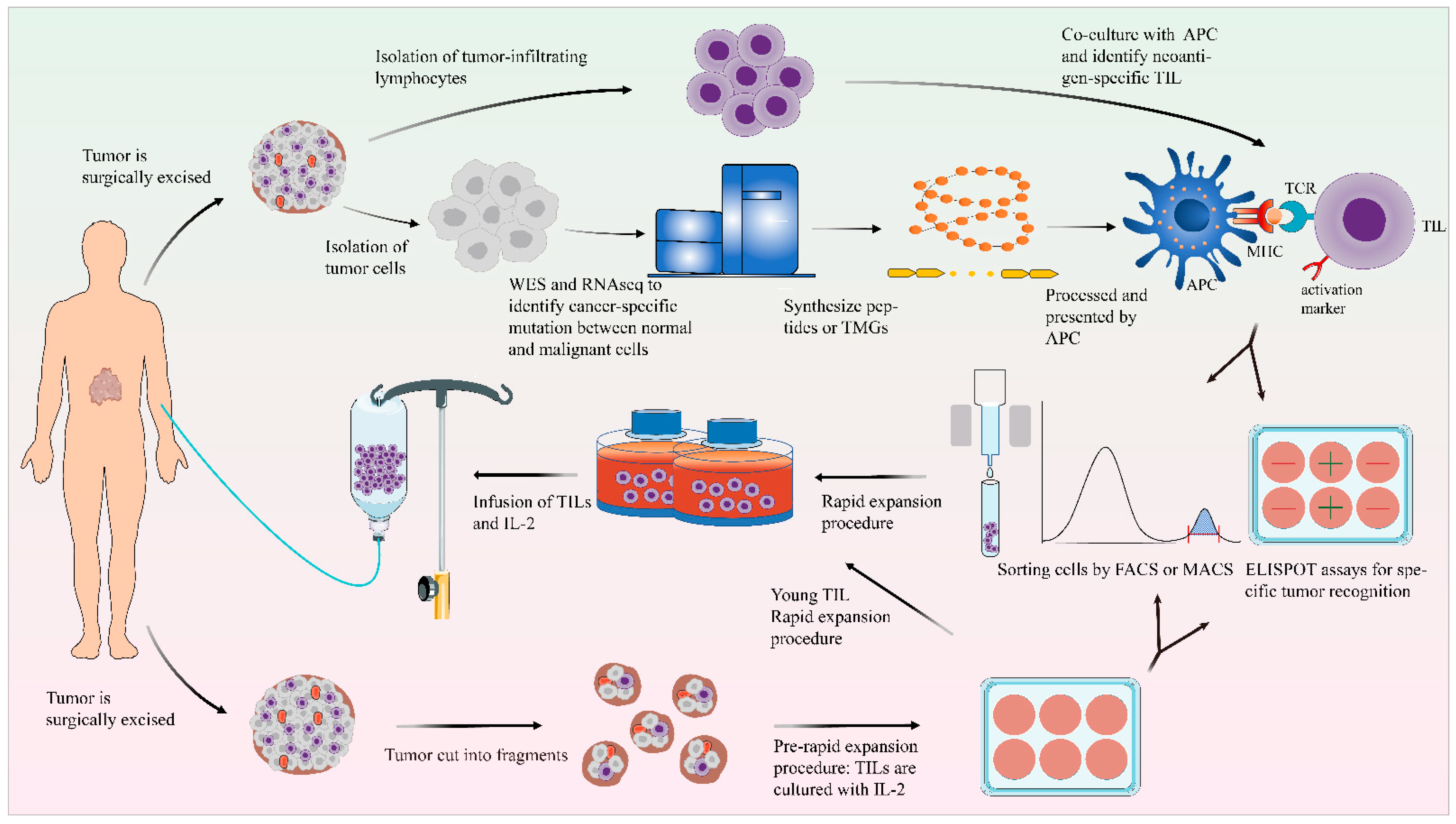
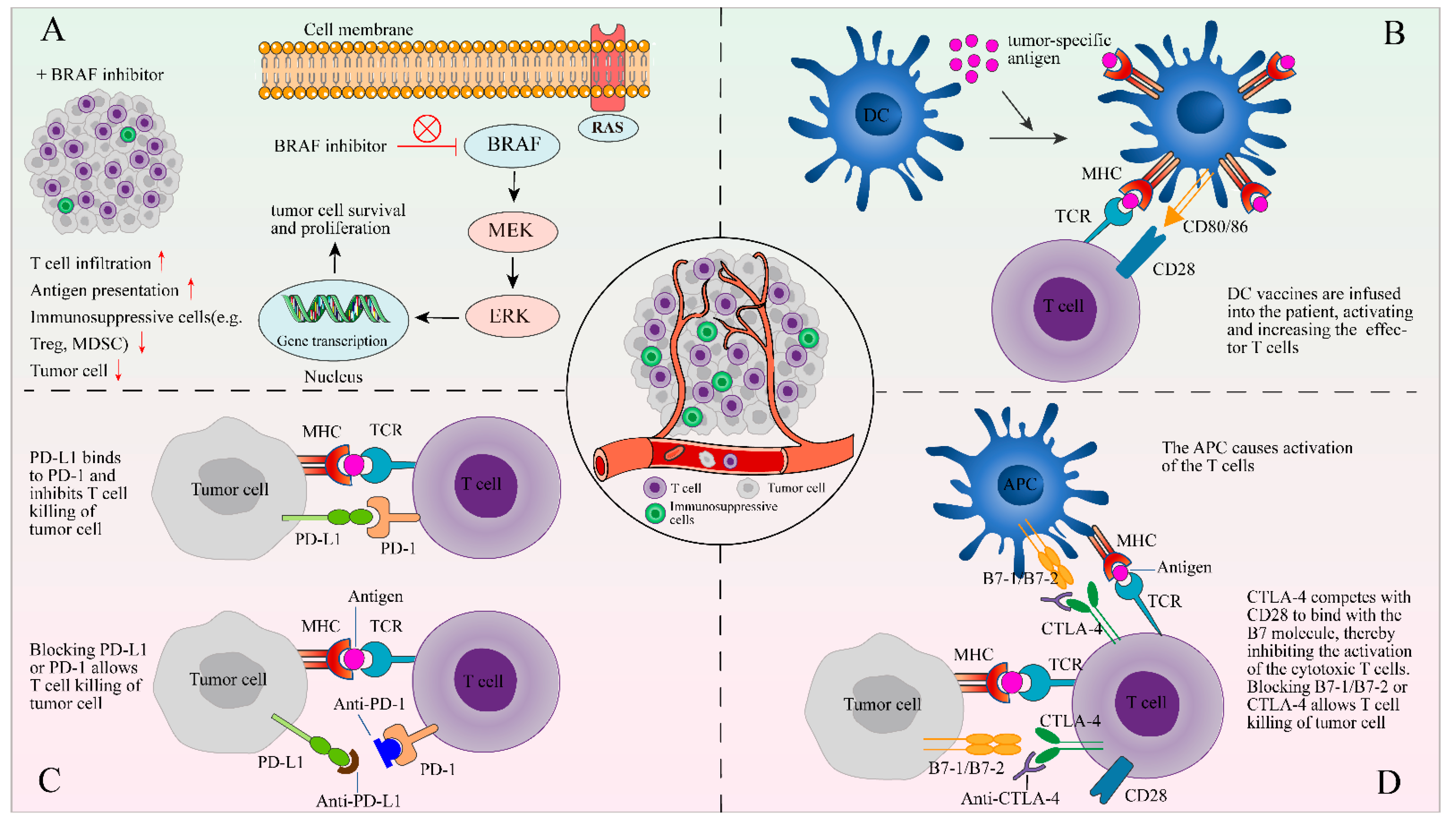
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).