
DNMT3A/miR-129-2-5p/Rac1 Is an Effector Pathway for SNHG1 to Drive Stem-Cell-like and Invasive Behaviors of Advanced Bladder Cancer Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents, Antibodies, and Plasmids
2.2. Cell Lines, Culture, and Transfection
2.3. Sphere Formation Assay
2.4. Cell Invasion Assay
2.5. Luciferase Reporter Assay
2.6. Immunoblotting Analysis
2.7. Chromatin Immunoprecipitation (ChIP) Assay
2.8. RNA Immunoprecipitation (RIP)
2.9. Quantitative Real-Time PCR
2.10. DNA Extraction, Bisulfite DNA Modification, and Methylation-Specific PCR
2.11. RNA Pull-Down
2.12. Statistical Analysis
3. Results
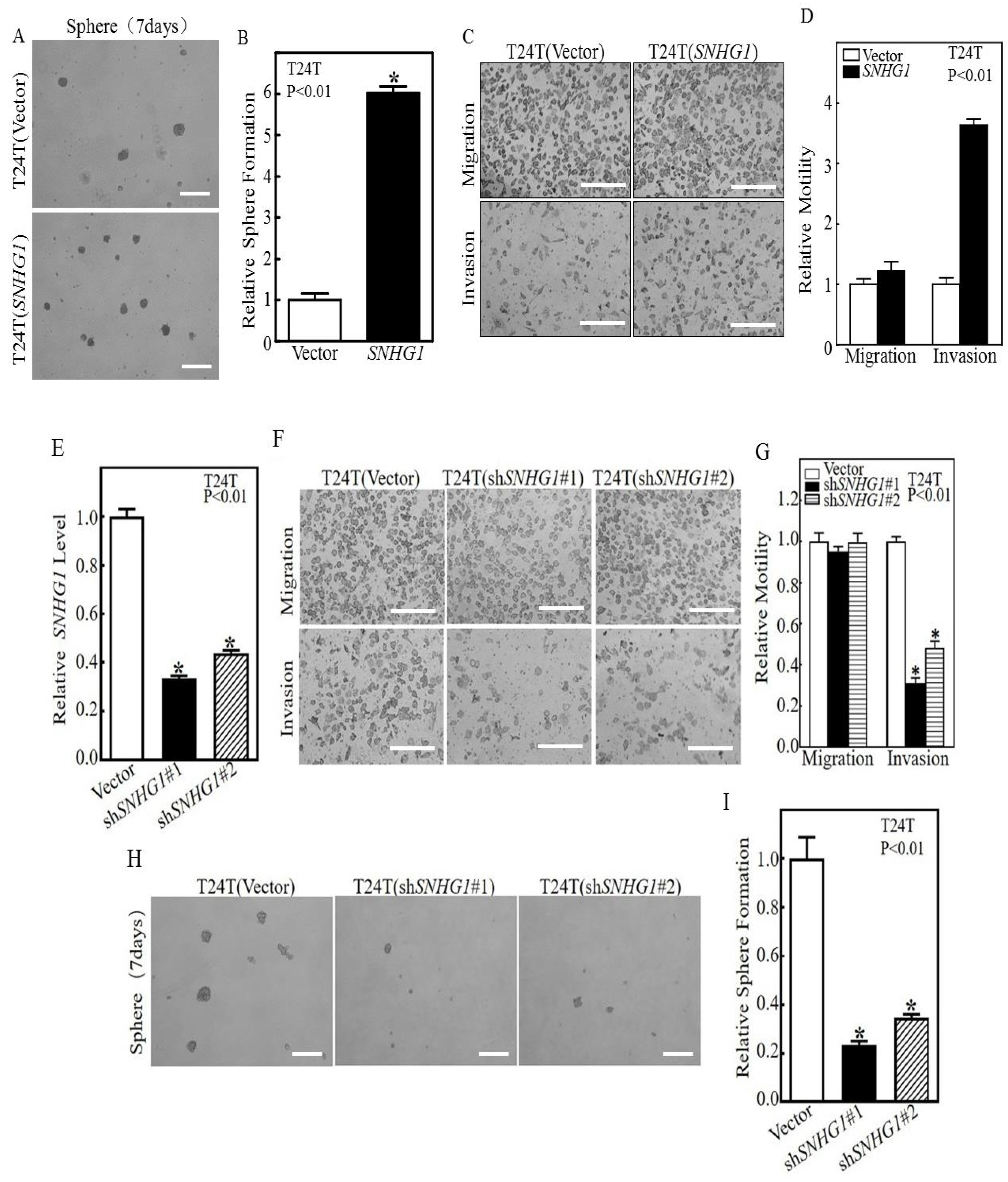
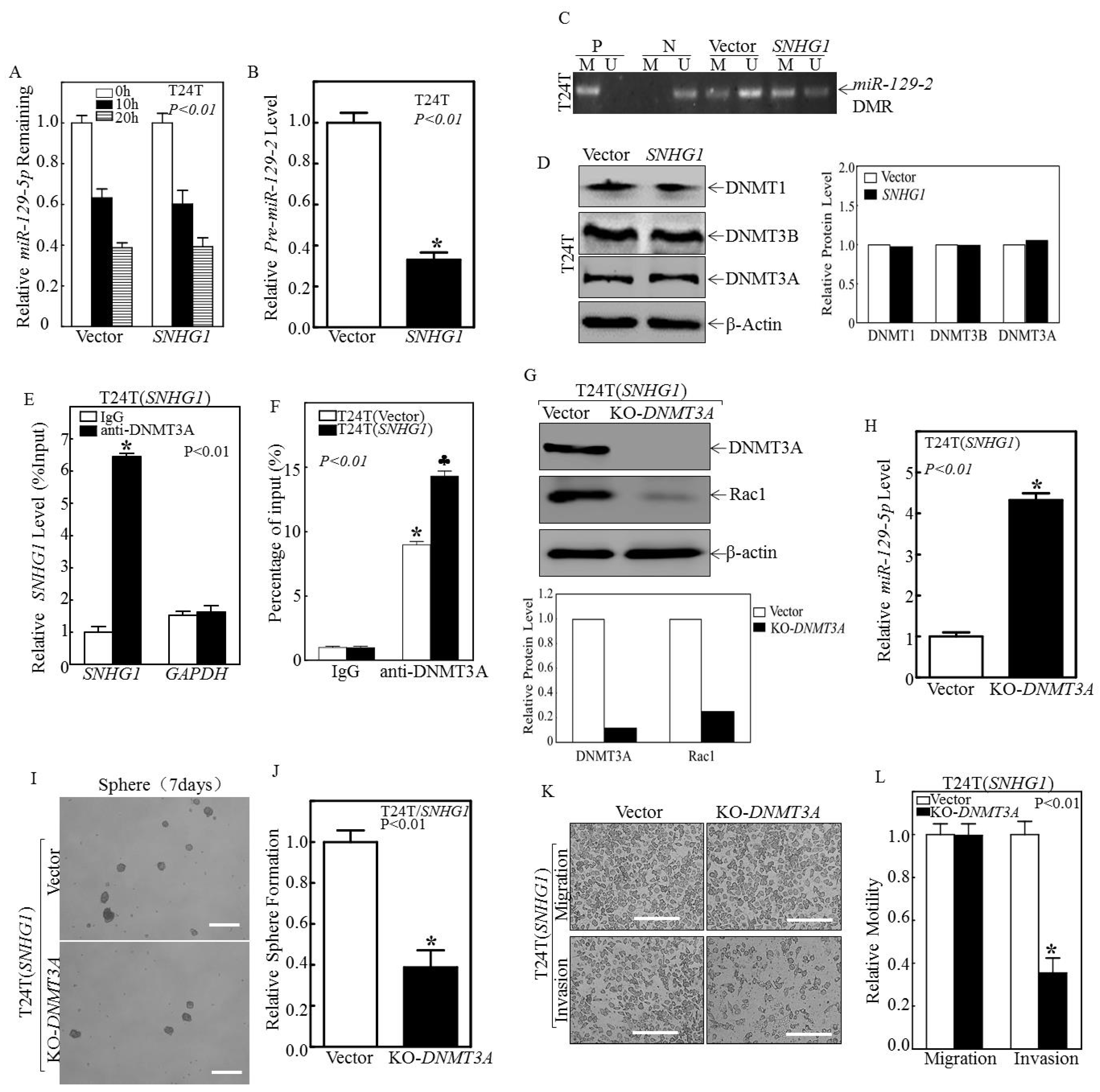
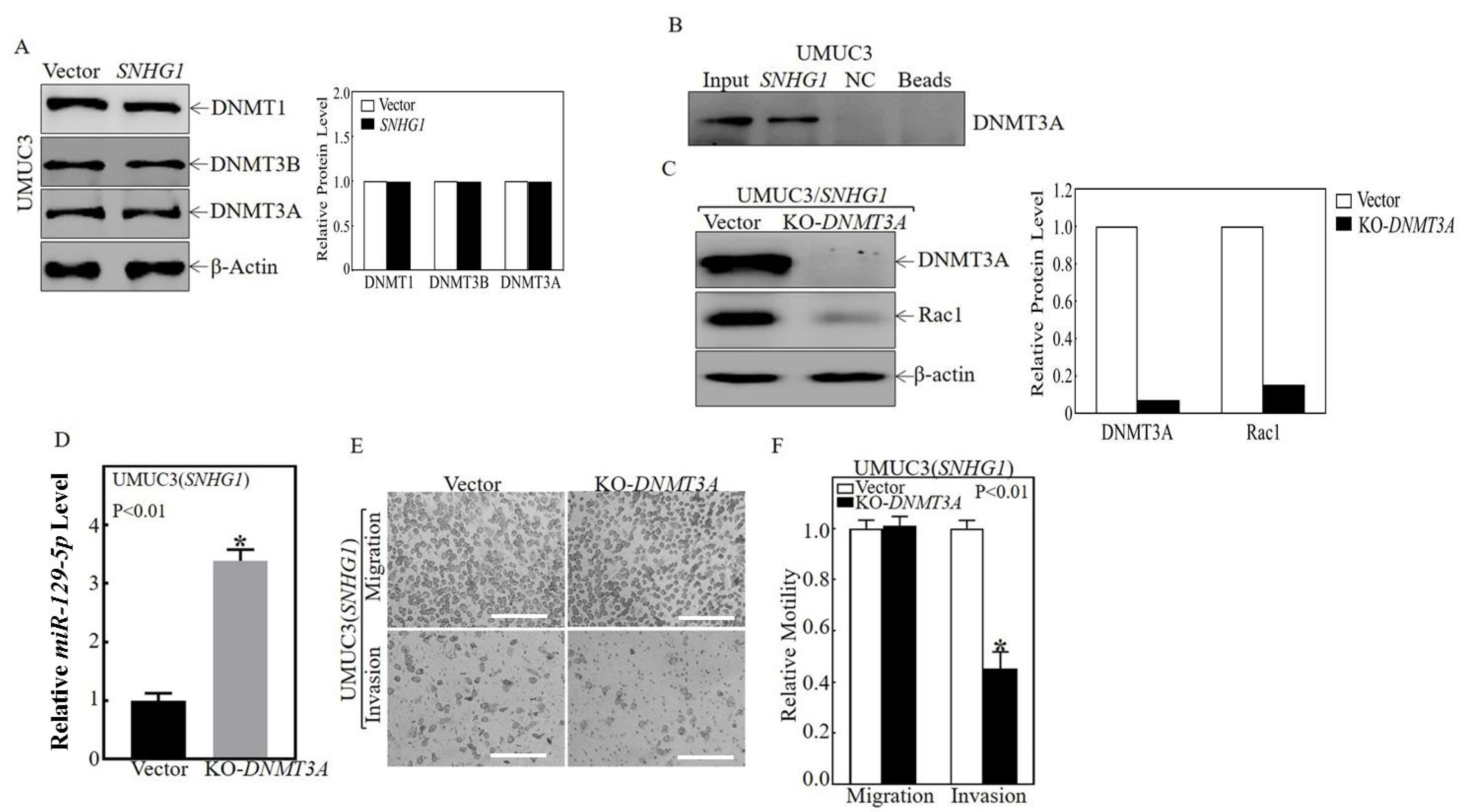
3.1. SNHG1 Over-Expression Induces the Stem-Cell-like and Invasive Behaviors of Cultured Bladder Cancer Cells by Upregulating Rac1 Expression
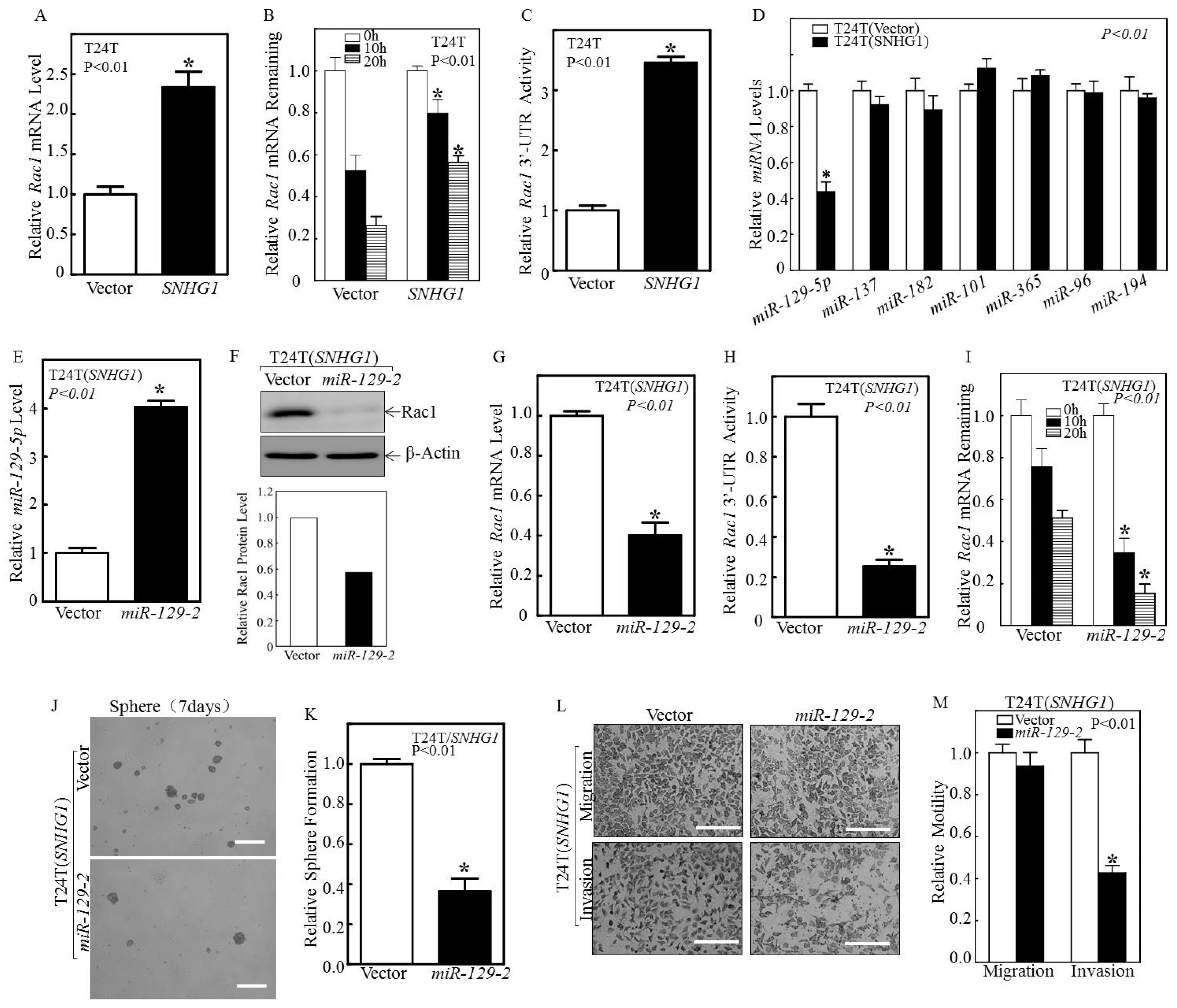
3.2. SNHG1 Stabilizes Rac1 mRNA by Suppressing miR-129-2-5p Transcription
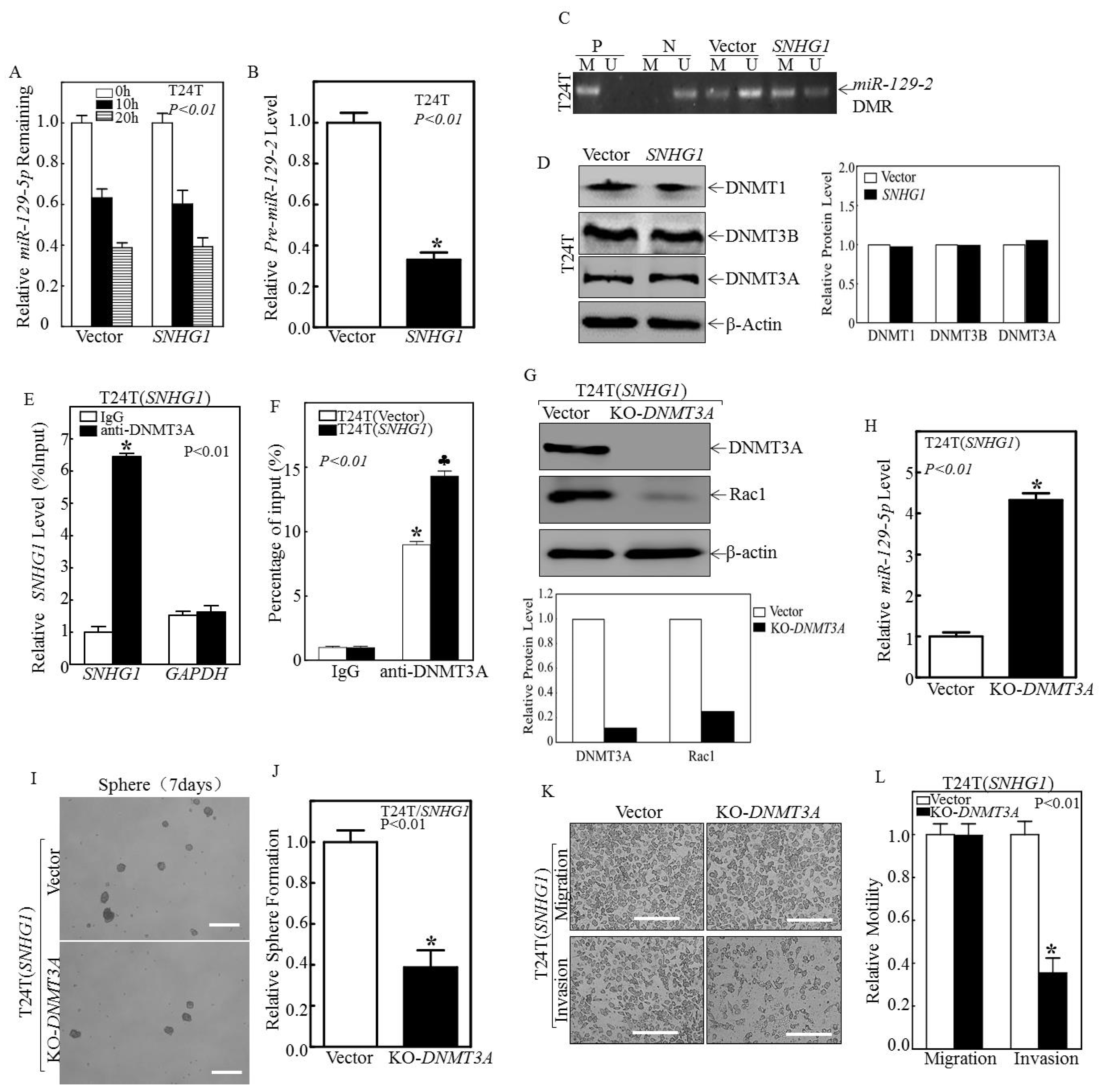
3.3. SNHG1 Binds DNMT3A, and the Binding Tethers DNMT3A to the miR-129-2 Promoter, Hyper-Methylates It, and Suppresses Its Transcription
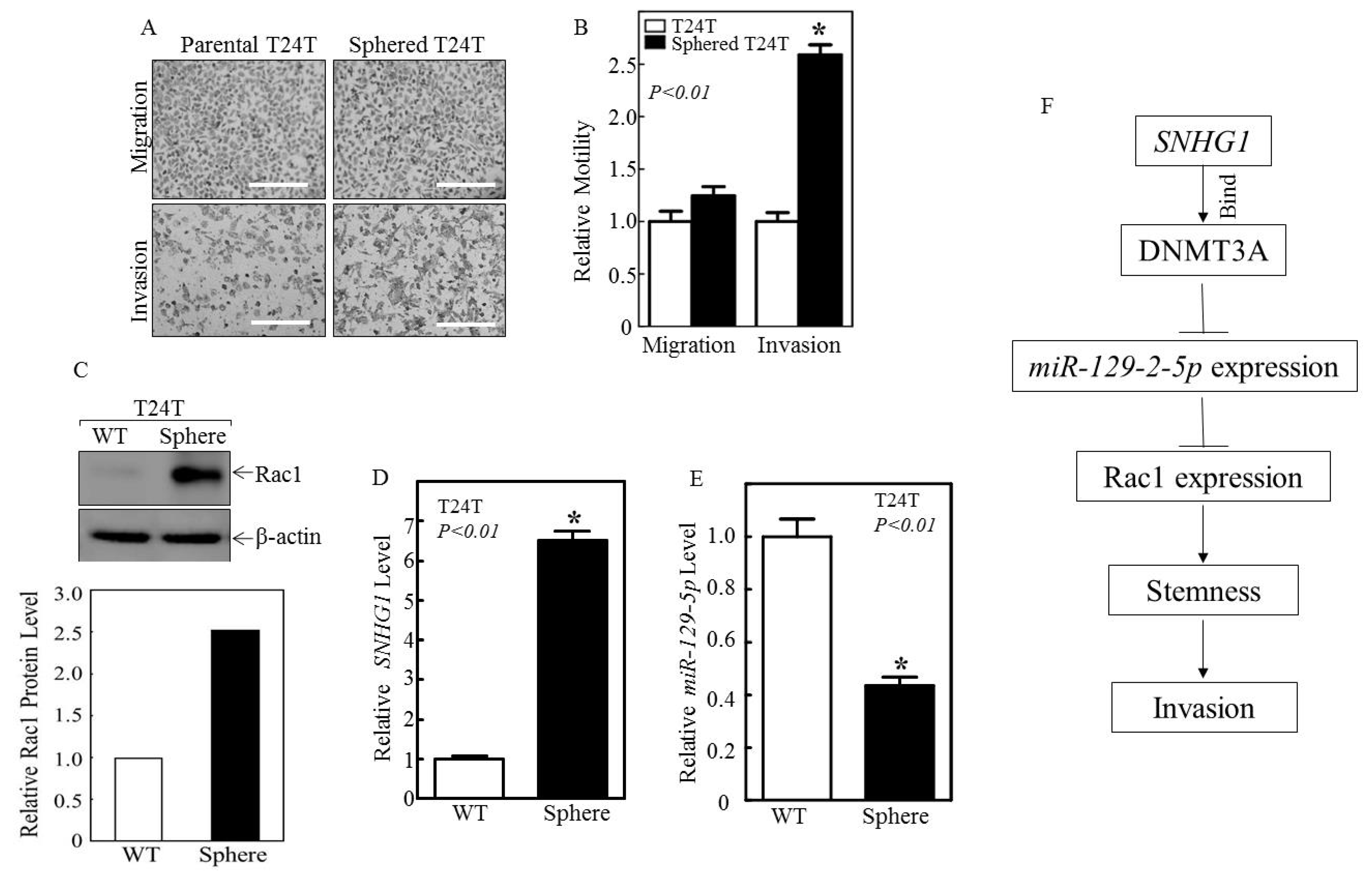
3.4. Sphered T24T Cells Are More Invasive than Non-Sphered Counterparts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Cancer Society. Cancer Facts and Figures 2022; American Cancer Society: Atlanta, GA, USA, 2022. [Google Scholar]
- Wu, X.R. Urothelial tumorigenesis: A tale of divergent pathways. Nat. Rev. Cancer 2005, 5, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Boormans, J.L.; Zwarthoff, E.C. Limited Funds for Bladder Cancer Research and What Can We Do about It. Bladder Cancer 2016, 2, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Lokeshwar, S.D.; Lopez, M.; Sarcan, S.; Aguilar, K.; Morera, D.S.; Shaheen, D.M.; Lokeshwar, B.L.; Lokeshwar, V.B. Molecular Oncology of Bladder Cancer from Inception to Modern Perspective. Cancers 2022, 14, 2578. [Google Scholar] [CrossRef] [PubMed]
- Said, N.; Theodorescu, D. Pathways of metastasis suppression in bladder cancer. Cancer Metastasis Rev. 2009, 28, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.M.; Hahn, N.M.; Efstathiou, J.A.; Lerner, S.P.; Malmstrom, P.U.; Choi, W.; Guo, C.C.; Lotan, Y.; Kassouf, W. Bladder cancer. Lancet 2016, 388, 2796–2810. [Google Scholar] [CrossRef]
- TCGA. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef]
- Thomsen, M.B.; Nordentoft, I.; Lamy, P.; Høyer, S.; Vang, S.; Hedegaard, J.; Borre, M.; Jensen, J.B.; Ørntoft, T.F.; Dyrskjøt, L. Spatial and temporal clonal evolution during development of metastatic urothelial carcinoma. Mol. Oncol. 2016, 10, 1450–1460. [Google Scholar] [CrossRef]
- Weiss, F.; Lauffenburger, D.; Friedl, P. Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat. Rev. Cancer 2022, 22, 157–173. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Bodgi, L.; Bahmad, H.F.; Araji, T.; Al Choboq, J.; Bou-Gharios, J.; Cheaito, K.; Zeidan, Y.H.; Eid, T.; Geara, F.; Abou-Kheir, W. Assessing Radiosensitivity of Bladder Cancer in vitro: A 2D vs. 3D Approach. Front. Oncol. 2019, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.X.; Zhang, H.; Tepper, C.G.; Lin, T.Y.; Davis, R.R.; Keck, J.; Ghosh, P.M.; Gill, P.; Airhart, S.; Bult, C.; et al. Development and Characterization of Bladder Cancer Patient-Derived Xenografts for Molecularly Guided Targeted Therapy. PLoS ONE 2015, 10, e0134346. [Google Scholar]
- Jäger, W.; Xue, H.; Hayashi, T.; Janssen, C.; Awrey, S.; Wyatt, A.W.; Anderson, S.; Moskalev, I.; Haegert, A.; Alshalalfa, M.; et al. Patient-derived bladder cancer xenografts in the preclinical development of novel targeted therapies. Oncotarget 2015, 6, 21522–21532. [Google Scholar] [CrossRef]
- Gouin, K.H., 3rd; Ing, N.; Plummer, J.T.; Rosser, C.J.; Ben Cheikh, B.; Oh, C.; Chen, S.S.; Chan, K.S.; Furuya, H.; Tourtellotte, W.G.; et al. An N-Cadherin 2 expressing epithelial cell subpopulation predicts response to surgery, chemotherapy and immunotherapy in bladder cancer. Nat. Commun. 2021, 12, 4906. [Google Scholar] [CrossRef]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.L.; et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef]
- Choi, W.; Czerniak, B.; Ochoa, A.; Su, X.; Siefker-Radtke, A.; Dinney, C.; McConkey, D.J. Intrinsic basal and luminal subtypes of muscle-invasive bladder cancer. Nat. Rev. Urol. 2014, 11, 400–410. [Google Scholar] [CrossRef]
- Guo, C.C.; Majewski, T.; Zhang, L.; Yao, H.; Bondaruk, J.; Wang, Y.; Zhang, S.; Wang, Z.; Lee, J.G.; Lee, S.; et al. Dysregulation of EMT Drives the Progression to Clinically Aggressive Sarcomatoid Bladder Cancer. Cell Rep. 2019, 27, 1781–1793.e4. [Google Scholar] [CrossRef]
- He, F.; Melamed, J.; Tang, M.S.; Huang, C.; Wu, X.R. Oncogenic HRAS Activates Epithelial-to-Mesenchymal Transition and Confers Stemness to p53-Deficient Urothelial Cells to Drive Muscle Invasion of Basal Subtype Carcinomas. Cancer Res. 2015, 75, 2017–2028. [Google Scholar] [CrossRef]
- Schardt, J.; Roth, B.; Seiler, R. Forty years of cisplatin-based chemotherapy in muscle-invasive bladder cancer: Are we understanding how, who and when? World J. Urol. 2019, 37, 1759–1765. [Google Scholar] [CrossRef]
- Evans, J.R.; Feng, F.Y.; Chinnaiyan, A.M. The bright side of dark matter: lncRNAs in cancer. J. Clin. Investig. 2016, 126, 2775–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wu, D.; Chen, J.; Chen, S.; He, F.; Fu, H.; Wu, Q.; Shuan, L.; Wang, W.; Li, X. Long non-coding RNA HOXA-AS2 promotes the migration, invasion and stemness of bladder cancer via regulating miR-125b/Smad2 axis. Exp. Cell Res. 2019, 375, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jiang, X.; Wang, Z.; Zhong, X.; Tai, S.; Cui, Y. Small nucleolar RNA host gene 1: A new biomarker and therapeutic target for cancers. Pathol. Res. Pract. 2018, 214, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, W.; Li, T.Y.; Yu, X.D.; Zhu, Y.F.; Ding, F.; Li, D.S.; Yang, T. Long noncoding RNA SNHG1 predicts a poor prognosis and promotes hepatocellular carcinoma tumorigenesis. Biomed. Pharmacother. 2016, 80, 73–79. [Google Scholar] [CrossRef]
- Lu, Q.; Shan, S.; Li, Y.; Zhu, D.; Jin, W.; Ren, T. Long noncoding RNA SNHG1 promotes non-small cell lung cancer progression by up-regulating MTDH via sponging miR-145-5p. FASEB J. 2018, 32, 3957–3967. [Google Scholar] [CrossRef]
- Cui, L.; Dong, Y.; Wang, X.; Zhao, X.; Kong, C.; Liu, Y.; Jiang, X.; Zhang, X. Downregulation of long noncoding RNA SNHG1 inhibits cell proliferation, metastasis, and invasion by suppressing the Notch-1 signaling pathway in pancreatic cancer. J. Cell. Biochem. 2018, 120, 6106–6112. [Google Scholar] [CrossRef]
- Guo, X.; Huang, H.; Jin, H.; Xu, J.; Risal, S.; Li, J.; Li, X.; Yan, H.; Zeng, X.; Xue, L.; et al. ISO, via Upregulating MiR-137 Transcription, Inhibits GSK3beta-HSP70-MMP-2 Axis, Resulting in Attenuating Urothelial Cancer Invasion. Mol. Ther.-Nucleic Acids 2018, 12, 337–349. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, Y.; Yang, C.; Simeone, D.M.; Sun, T.T.; DeGraff, D.J.; Tang, M.S.; Zhang, Y.; Wu, X.R. Dominant role of CDKN2B/p15INK4B of 9p21.3 tumor suppressor hub in inhibition of cell-cycle and glycolysis. Nat. Commun. 2021, 12, 2047. [Google Scholar] [CrossRef]
- Murphy, S.K.; Wylie, A.A.; Coveler, K.J.; Cotter, P.D.; Papenhausen, P.R.; Sutton, V.R.; Shaffer, L.G.; Jirtle, R.L. Epigenetic detection of human chromosome 14 uniparental disomy. Hum. Mutat. 2003, 22, 92–97. [Google Scholar] [CrossRef]
- Bagheri, V.; Memar, B.; Behzadi, R.; Aliakbarian, M.; Jangjoo, A.; Bahar, M.M.; Talebi, S.; Gholamin, M.; Abbaszadegan, M.R. Isolation and identification of chemotherapy-enriched sphere-forming cells from a patient with gastric cancer. J. Cell. Physiol. 2018, 233, 7036–7046. [Google Scholar] [CrossRef]
- Bak, M.J.; Furmanski, P.; Shan, N.L.; Lee, H.J.; Bao, C.; Lin, Y.; Shih, W.J.; Yang, C.S.; Suh, N. Tocopherols inhibit estrogen-induced cancer stemness and OCT4 signaling in breast cancer. Carcinogenesis 2018, 39, 1045–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.M.; Yang, S.L.; Zheng, X.M.; Chen, G.G.; Chen, J.; Zhang, T. CD133 expression and alpha-fetoprotein levels define novel prognostic subtypes of HBV-associated hepatocellular carcinoma: A long-term follow-up analysis. Oncol. Lett. 2018, 15, 2985–2991. [Google Scholar] [PubMed]
- Yoon, C.H.; Hyun, K.H.; Kim, R.K.; Lee, H.; Lim, E.J.; Chung, H.Y.; An, S.; Park, M.J.; Suh, Y.; Kim, M.J.; et al. The small GTPase Rac1 is involved in the maintenance of stemness and malignancies in glioma stem-like cells. FEBS Lett. 2011, 585, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Steri, M.; Idda, M.L.; Whalen, M.B.; Orru, V. Genetic variants in mRNA untranslated regions. Wiley Interdiscip. Rev. RNA 2018, 9, e1474. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Becquet, D.; Franc, J.L.; Francois-Bellan, A.M. Circadian processes in the RNA life cycle. Wiley Interdiscip. Rev. RNA 2018, 9, e1467. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Feng, B.; Han, S.; Lu, L.; Chen, Y.; Chu, X.; Wang, R.; Chen, L. MicroRNA-129 in Human Cancers: From Tumorigenesis to Clinical Treatment. Cell. Physiol. Biochem. 2016, 39, 2186–2202. [Google Scholar] [CrossRef]
- Subramaniam, D.; Thombre, R.; Dhar, A.; Anant, S. DNA methyltransferases: A novel target for prevention and therapy. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef]
- Guo, C.; Li, X.; Xie, J.; Liu, D.; Geng, J.; Ye, L.; Yan, Y.; Yao, X.; Luo, M. Long Noncoding RNA SNHG1 Activates Autophagy and Promotes Cell Invasion in Bladder Cancer. Front. Oncol. 2021, 11, 660551. [Google Scholar] [CrossRef]
- Feldman, A.S.; Lokeshwar, V.; Lin, D.W. A 25-year perspective on evaluation and understanding of biomarkers in urologic cancers. Urol. Oncol. Semin. Orig. Investig. 2021, 39, 602–617. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef]
- Warrick, J.I.; Walter, V.; Yamashita, H.; Chung, E.; Shuman, L.; Amponsa, V.O.; Zheng, Z.; Chan, W.; Whitcomb, T.L.; Yue, F.; et al. FOXA1, GATA3 and PPARɣ Cooperate to Drive Luminal Subtype in Bladder Cancer: A Molecular Analysis of Established Human Cell Lines. Sci. Rep. 2016, 6, 38531. [Google Scholar] [CrossRef] [PubMed]
- Aspenstrom, P.; Fransson, A.; Saras, J. Rho GTPases have diverse effects on the organization of the actin filament system. Biochem. J. 2004, 377, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Chianale, F.; Cutrupi, S.; Rainero, E.; Baldanzi, G.; Porporato, P.E.; Traini, S.; Filigheddu, N.; Gnocchi, V.F.; Santoro, M.M.; Parolini, O.; et al. Diacylglycerol kinase-alpha mediates hepatocyte growth factor-induced epithelial cell scatter by regulating Rac activation and membrane ruffling. Mol. Biol. Cell 2007, 18, 4859–4871. [Google Scholar] [CrossRef] [PubMed]
- Castilho, R.M.; Squarize, C.H.; Leelahavanichkul, K.; Zheng, Y.; Bugge, T.; Gutkind, J.S. Rac1 is required for epithelial stem cell function during dermal and oral mucosal wound healing but not for tissue homeostasis in mice. PLoS ONE 2010, 5, e10503. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.; Cho, S.J.; Chang, K.K.; Park, D.J.; Ryeom, S.W.; Yoon, S.S. Role of Rac1 Pathway in Epithelial-to-Mesenchymal Transition and Cancer Stem-like Cell Phenotypes in Gastric Adenocarcinoma. Mol. Cancer Res. 2017, 15, 1106–1116. [Google Scholar] [CrossRef]
- Wheeler, A.P.; Wells, C.M.; Smith, S.D.; Vega, F.M.; Henderson, R.B.; Tybulewicz, V.L.; Ridley, A.J. Rac1 and Rac2 regulate macrophage morphology but are not essential for migration. J. Cell Sci. 2006, 119, 2749–2757. [Google Scholar] [CrossRef]
- Lozano, E.; Betson, M.; Braga, V.M. Tumor progression: Small GTPases and loss of cell-cell adhesion. Bioessays 2003, 25, 452–463. [Google Scholar] [CrossRef]
- Nakagawa, T.; Kanai, Y.; Saito, Y.; Kitamura, T.; Kakizoe, T.; Hirohashi, S. Increased DNA methyltransferase 1 protein expression in human transitional cell carcinoma of the bladder. J. Urol. 2003, 170, 2463–2466. [Google Scholar] [CrossRef]
- Peres, R.; Furuya, H.; Pagano, I.; Shimizu, Y.; Hokutan, K.; Rosser, C.J. Angiogenin contributes to bladder cancer tumorigenesis by DNMT3b-mediated MMP2 activation. Oncotarget 2016, 7, 43109–43123. [Google Scholar] [CrossRef]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef]
- Ciarlantini, M.S.; Barquero, A.; Bayo, J.; Wetzler, D.; Dodes Traian, M.M.; Bucci, H.A.; Fiore, E.J.; Gandolfi Donadío, L.; Defelipe, L.; Turjanski, A.; et al. Development of an Improved Guanidine-Based Rac1 Inhibitor with in vivo Activity against Non-Small Cell Lung Cancer. ChemMedChem 2021, 16, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Collazo, A.; Ruiz-Calderon, J.F.; Picon, H.; Borrero-Garcia, L.D.; Lopez, I.; Castillo-Pichardo, L.; Del Mar, M.M.; Duconge, J.; Medina, J.I.; Bayro, M.J.; et al. Efficacy of Rac and Cdc42 Inhibitor MBQ-167 in Triple-negative breast cancer. Mol. Cancer Ther. 2021, 20, 2420–2432. [Google Scholar] [CrossRef] [PubMed]
- Goka, E.T.; Chaturvedi, P.; Lopez, D.T.M.; Lippman, M.E. Rac signaling drives clear cell renal carcinoma tumor growth by priming the tumor microenvironment for an angiogenic switch. Mol. Cancer Ther. 2020, 19, 1462–1473. [Google Scholar] [CrossRef]
- Nunes, S.P.; Henrique, R.; Jerónimo, C.; Paramio, J.M. DNA Methylation as a Therapeutic Target for Bladder Cancer. Cells 2020, 9, 1850. [Google Scholar] [CrossRef] [PubMed]
- Zuiverloon, T.C.; Theodorescu, D. Pharmacogenomic considerations in the treatment of muscle-invasive bladder cancer. Pharmacogenomics 2017, 18, 1167–1178. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Yang, R.; Li, J.; Wang, L.; Cohen, M.; Simeone, D.M.; Costa, M.; Wu, X.-R. DNMT3A/miR-129-2-5p/Rac1 Is an Effector Pathway for SNHG1 to Drive Stem-Cell-like and Invasive Behaviors of Advanced Bladder Cancer Cells. Cancers 2022, 14, 4159. https://doi.org/10.3390/cancers14174159
Xu J, Yang R, Li J, Wang L, Cohen M, Simeone DM, Costa M, Wu X-R. DNMT3A/miR-129-2-5p/Rac1 Is an Effector Pathway for SNHG1 to Drive Stem-Cell-like and Invasive Behaviors of Advanced Bladder Cancer Cells. Cancers. 2022; 14(17):4159. https://doi.org/10.3390/cancers14174159
Chicago/Turabian StyleXu, Jiheng, Rui Yang, Jingxia Li, Lidong Wang, Mitchell Cohen, Diane M. Simeone, Max Costa, and Xue-Ru Wu. 2022. "DNMT3A/miR-129-2-5p/Rac1 Is an Effector Pathway for SNHG1 to Drive Stem-Cell-like and Invasive Behaviors of Advanced Bladder Cancer Cells" Cancers 14, no. 17: 4159. https://doi.org/10.3390/cancers14174159
APA StyleXu, J., Yang, R., Li, J., Wang, L., Cohen, M., Simeone, D. M., Costa, M., & Wu, X.-R. (2022). DNMT3A/miR-129-2-5p/Rac1 Is an Effector Pathway for SNHG1 to Drive Stem-Cell-like and Invasive Behaviors of Advanced Bladder Cancer Cells. Cancers, 14(17), 4159. https://doi.org/10.3390/cancers14174159