
Extracellular Vesicles and Their Current Role in Cancer Immunotherapy


 , and
, and
Abstract
Simple Summary
Abstract
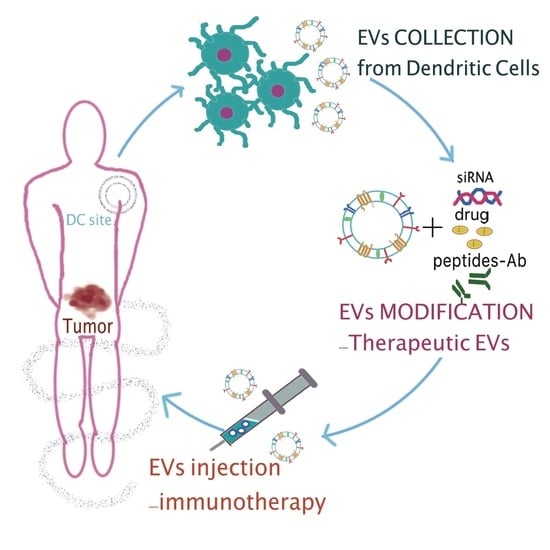
1. Extracellular Vesicles: An Introduction
2. EVs for Therapeutic and Drug Delivery Purposes
3. Cargo-Loading Methods of EVs
4. EVs Surface Functionalization: An Overview
4.1. Post-Isolation Methods
4.2. Genetic Engineering of Parental Cells for Surface Functionalization
5. Cancer Immunotherapy
6. What Is a Cancer Vaccine?
7. EVs in Anti-Tumor Immunotherapy
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Susa, F.; Limongi, T.; Dumontel, B.; Vighetto, V.; Cauda, V. Engineered extracellular vesicles as a reliable tool in cancer nanomedicine. Cancers 2019, 11, 1979. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Johansson, K.; Mossberg, M.; Kahn, R.; Karpman, D. Exosomes and microvesicles in normal physiology, pathophysiology, and renal diseases. Pediatr. Nephrol. 2019, 34, 11–30. [Google Scholar] [CrossRef]
- Sutaria, D.S.; Badawi, M.; Phelps, M.A.; Schmittgen, T.D. Achieving the Promise of Therapeutic Extracellular Vesicles: The Devil is in Details of Therapeutic Loading. Pharm. Res. 2017, 34, 1053–1066. [Google Scholar] [CrossRef]
- Kalra, H.; Drummen, G.P.; Mathivanan, S. Focus on Extracellular Vesicles: Introducing the Next Small Big Thing. Int. J. Mol. Sci. 2016, 17, 170. [Google Scholar] [CrossRef]
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef]
- Van der Pol, E.; Böing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2017, 8, 220–232. [Google Scholar] [CrossRef]
- Chen, Y.; Li, G.; Liu, M.L. Microvesicles as Emerging Biomarkers and Therapeutic Targets in Cardiometabolic Diseases. Genom. Proteom. Bioinf. 2018, 16, 50–62. [Google Scholar] [CrossRef]
- Xu, X.; Lai, Y.; Hua, Z.C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef] [PubMed]
- Jurj, A.; Zanoaga, O.; Braicu, C.; Lazar, V.; Tomuleasa, C.; Irimie, A.; Berindan-Neagoe, I. A comprehensive picture of extracellular vesicles and their contents. Molecular transfer to cancer cells. Cancers 2020, 12, 298. [Google Scholar] [CrossRef]
- Battistelli, M.; Falcieri, E. Apoptotic bodies: Particular extracellular vesicles involved in intercellular communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef]
- Van Dommelen, S.M.; Vader, P.; Lakhal, S.; Kooijmans, S.A.A.; Van Solinge, W.W.; Wood, M.J.A.; Schiffelers, R.M. Microvesicles and exosomes: Opportunities for cell-derived membrane vesicles in drug delivery. J. Control. Release 2012, 161, 635–644. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Andreu, Z.; Yáñez-Mó, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.L.; Chen, K.C.; Hsieh, J.T.; Shen, T.L. Exosomes in cancer development and clinical applications. Cancer Sci. 2018, 109, 2364–2374. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Li, W. Dendritic cell-derived exosomes for cancer immunotherapy: Hope and challenges. Ann. Transl. Med. 2017, 5, 3–5. [Google Scholar] [CrossRef]
- Kugeratski, F.G.; Kalluri, R. Exosomes as mediators of immune regulation and immunotherapy in cancer. FEBS J. 2021, 288, 10–35. [Google Scholar] [CrossRef]
- Syn, N.L.; Wang, L.; Chow, E.K.; Lim, C.T.; Goh, B.C. Exosomes in Cancer Nanomedicine and Immunotherapy: Prospects and Challenges. Trends Biotechnol. 2017, 35, 665–676. [Google Scholar] [CrossRef]
- Villata, S.; Canta, M.; Cauda, V. EVs and Bioengineering: From Cellular Products to Engineered Nanomachines. Int. J. Mol. Sci. 2020, 21, 6048. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.M.; Sawant, S.S.; Kunda, N.K. Exosomes as drug delivery systems: A brief overview and progress update. Eur. J. Pharm. Biopharm. 2020, 154, 259–269. [Google Scholar] [CrossRef]
- Mizrak, A.; Bolukbasi, M.F.; Ozdener, G.B.; Brenner, G.J.; Madlener, S.; Erkan, E.P.; Ströbel, T.; Breakefield, X.O.; Saydam, O. Genetically engineered microvesicles carrying suicide mRNA/protein inhibit schwannoma tumor growth. Mol. Ther. 2013, 21, 101–108. [Google Scholar] [CrossRef]
- Kanada, M.; Kim, B.D.; Hardy, J.W.; Ronald, J.A.; Bachmann, M.H.; Bernard, M.P.; Perez, G.I.; Zarea, A.A.; Ge, T.J.; Withrow, A.; et al. Microvesicle-Mediated Delivery of Minicircle DNA Results in Effective Gene-Directed Enzyme Prodrug Cancer Therapy. Mol. Cancer Ther. 2019, 18, 2331–2342. [Google Scholar] [CrossRef] [PubMed]
- Yeo, R.W.Y.; Lai, R.C.; Zhang, B.; Tan, S.S.; Yin, Y.; Teh, B.J.; Lim, S.K. Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Markov, O.; Oshchepkova, A.; Mironova, N. Immunotherapy Based on Dendritic Cell-Targeted/-Derived Extracellular Vesicles-A Novel Strategy for Enhancement of the Anti-tumor Immune Response. Front. Pharmacol. 2019, 10, 1152. [Google Scholar] [CrossRef]
- American Cancer Society. How Immunotherapy Is Used to Treat Cancer. Available online: https://www.cancer.org/treatment/treatments-and-side-effects/treatment-types/immunotherapy/what-is-immunotherapy.html (accessed on 10 September 2020).
- Benecke, L.; Coray, M.; Umbricht, S.; Chiang, D.; Figueiró, F.; Muller, L. Exosomes: Small EVs with Large Immunomodulatory Effect in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 3600. [Google Scholar] [CrossRef]
- Batista, I.A.; Melo, S.A. Exosomes and the Future of Immunotherapy in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 567. [Google Scholar] [CrossRef]
- Nawaz, M.; Fatima, F.; Nazarenko, I.; Ekström, K.; Murtaza, I.; Anees, M.; Sultan, A.; Neder, L.; Camussi, G.; Valadi, H.; et al. Extracellular vesicles in ovarian cancer: Applications to tumor biology, immunotherapy and biomarker discovery. Expert Rev. Proteomics. 2016, 13, 395–409. [Google Scholar] [CrossRef]
- Bae, S.; Brumbaugh, J.; Bonavida, B. Exosomes derived from cancerous and non-cancerous cells regulate the anti-tumor response in the tumor microenvironment. Genes Cancer 2018, 9, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Gercel-Taylor, C. Exosomes/microvesicles: Mediators of cancer-associated immunosuppressive microenvironments. Semin. Immunopathol. 2011, 33, 441–454. [Google Scholar] [CrossRef]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef]
- Shenoda, B.B.; Ajit, S.K. Modulation of Immune Responses by Exosomes Derived from Antigen-Presenting Cells. Clin. Med. Insights Pathol. 2016, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Martin-Gayo, E.; Yu, X.G. Role of Dendritic Cells in Natural Immune Control of HIV-1 Infection. Front. Immunol. 2019, 10, 1306. [Google Scholar] [CrossRef] [PubMed]
- Turnis, M.E.; Rooney, C.M. Enhancement of dendritic cells as vaccines for cancer. Immunotherapy 2010, 2, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.A.; Yang, X.; Giannoudis, P.; McGonagle, D. Mesenchymal Stem Cells: Discovery in Bone Marrow and Beyond. In Mesenchymal Stem Cells and Skeletal Regeneration; Jones, E.A., Yang, X., Giannoudis, P., McGonagle, D., Eds.; Academic Press: Boston, MA, USA, 2013; Chapter 2; pp. 7–13. [Google Scholar]
- Marigo, I.; Dazzi, F. The immunomodulatory properties of mesenchymal stem cells. Semin. Immunopathol. 2011, 33, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.S.; Arslan, F.; Yin, Y.; Tan, S.S.; Lai, R.C.; Choo, A.B.H.; Padmanabhan, J.; Lee, C.N.; de Kleijn, D.P.; Lim, S.K. Enabling a robust scalable manufacturing process for therapeutic exosomes through oncogenic immortalization of human ESC-derived MSCs. J. Transl. Med. 2011, 9, 47. [Google Scholar] [CrossRef]
- Irfan Maqsood, M.; Matin, M.M.; Bahrami, A.R.; Ghasroldasht, M.M. Immortality of cell lines: Challenges and advantages of establishment. Cell Biol. Int. 2013, 37, 1038–1045. [Google Scholar] [CrossRef]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat. Commun. 2019, 10, 4355. [Google Scholar] [CrossRef]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.J.; Lee, C.K.; Zou, S.; Woon, E.C.; Czarny, B.; Pastorin, G. Doxorubicin-loaded cell-derived nanovesicles: An alternative targeted approach for anti-tumor therapy. Int. J. Nanomedicine 2017, 12, 2759–2767. [Google Scholar] [CrossRef]
- Dumontel, B.; Susa, F.; Limongi, T.; Canta, M.; Racca, L.; Chiodoni, A.; Garno, N.; Chiabotto, G.; Centomo, M.L.; Pignochino, Y.; et al. ZnO nanocrystals shuttled by extracellular vesicles as effective Trojan nano-horses against cancer cells. Nanomedicine 2019, 14, 2815–2833. [Google Scholar] [CrossRef] [PubMed]
- Illes, B.; Hirschle, P.; Barnert, S.; Cauda, V.; Wuttke, S.; Engelke, H. Exosome-coated metal–organic framework nanoparticles: An efficient drug delivery platform. Chem. Mater. 2017, 29, 8042–8046. [Google Scholar] [CrossRef]
- Sancho-Albero, M.; Navascués, N.; Mendoza, G.; Sebastián, V.; Arruebo, M.; Martín-Duque, P.; Santamaria, J. Exosome origin determines cell targeting and the transfer of therapeutic nanoparticles towards target cells. J. Nanobiotechnology 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Piffoux, M.; Silva, A.K.A.; Lugagne, J.-B.; Hersen, P.; Wilhelm, C.; Gazeau, F. Extracellular Vesicle Production Loaded with Nanoparticles and Drugs in a Trade-off between Loading, Yield and Purity: Towards a Personalized Drug Delivery System. Adv. Biosyst. 2017, 1, 1700044. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.P.K.; Holme, M.N.; Stevens, M.M. Re-Engineering Extracellular Vesicles as Smart Nanoscale Therapeutics. ACS Nano 2017, 11, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Futaki, S. Combined treatment with a pH-sensitive fusogenic peptide and cationic lipids achieves enhanced cytosolic delivery of exosomes. Sci. Rep. 2015, 5, 10112. [Google Scholar] [CrossRef]
- Nakase, I.; Kogure, K.; Harashima, H.; Futaki, S. Application of a fusiogenic peptide GALA for intracellular delivery. Methods Mol. Biol. 2011, 683, 525–533. [Google Scholar] [CrossRef]
- Hein, C.D.; Liu, X.M.; Wang, D. Click Chemistry, A Powerful Tool for Pharmaceutical Sciences. Pharm. Res. 2008, 25, 2216–2230. [Google Scholar] [CrossRef]
- Smyth, T.; Petrova, K.; Payton, N.M.; Persaud, I.; Redzic, J.S.; Graner, M.W.; Smith-Jones, P.; Anchordoquy, T.J. Surface Functionalization of Exosomes Using Click Chemistry. Bioconjug. Chem. 2014, 25, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Kooijamans, S.A.A.; Fliervoet, L.A.L.; van der Meel, R.; Fens, M.H.A.M.; Heijnen, H.F.G.; van Bergen en Henegouwen, P.M.P.; Vader, P.; Schiffelers, R.M. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85. [Google Scholar] [CrossRef]
- Li, S.-D.; Huang, L. Targeted Delivery of Antisense Oligodeoxynucleotide and Small Interference RNA into Lung Cancer Cells. Mol. Pharm. 2006, 3, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Yuan, D.; Deygen, I.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomedicine: Nanotechnol. Biol. Med. 2018, 14, 195–204. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Flintoft, L. Getting RNAi therapies to the brain. Nat. Rev. Genet. 2011, 12, 296. [Google Scholar] [CrossRef]
- Yang, J.; Wu, S.; Hou, L.; Zhu, D.; Yin, S.; Yang, G.; Wang, Y. Therapeutic Effects of Simultaneous Delivery of Nerve Growth Factor mRNA and Protein via Exosomes on Cerebral Ischemia. Mol. Ther. Nucleic Acids 2020, 21, 512–522. [Google Scholar] [CrossRef]
- Aloe, L.; Rocco, M.L.; Balzamino, B.O.; Micera, A. Nerve growth factor: A focus on neuroscience and therapy. Curr. Neuropharmacol. 2015, 13, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.; Kaur, K.; Pauli, N.; Wilson, P.C. Harnessing the immune system’s arsenal: Producing human monoclonal antibodies for therapeutics and investigating immune responses. F1000 Biol. Rep. 2011, 3, 1–8. [Google Scholar] [CrossRef]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front. Immunol. 2018, 9, 26866. [Google Scholar] [CrossRef]
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Redman, J.M.; Collins, J.M.; Bilusic, M. Cancer vaccines: Enhanced immunogenic modulation through therapeutic combinations. Hum. Vaccin. Immunother. 2017, 13, 2561–2574. [Google Scholar] [CrossRef]
- Beatty, P.L.; Finn, O.J. Therapeutic and Prophylactic Cancer Vaccines. In Encyclopedia of Immunobiology; Ratcliffe, M.J.H., Ed.; Academic Press: Oxford, UK, 2016; pp. 542–549. [Google Scholar]
- Esmatabadi, M.J.D.; Bakhshinejad, B.; Motlagh, F.M.; Babashah, S.; Sadeghizadeh, M. Therapeutic resistance and cancer recurrence mechanisms: Unfolding the story of tumour coming back. J. Biosci. 2016, 41, 497–506. [Google Scholar] [CrossRef]
- Terraneo, N.; Jacob, F.; Dubrovska, A.; Grünberg, J. Novel Therapeutic Strategies for Ovarian Cancer Stem Cells. Front. Oncol. 2020, 10, 319. [Google Scholar] [CrossRef]
- Civenni, G.; Albino, D.; Shinde, D.; Vázquez, R.; Merulla, J.; Kokanovic, A.; Mapelli, S.; Carbone, G.M.; Catapano, C.V. Transcriptional Reprogramming and Novel Therapeutic Approaches for Targeting Prostate Cancer Stem Cells. Front. Oncol. 2019, 9, 385. [Google Scholar] [CrossRef] [PubMed]
- Coventry, B.J. Therapeutic vaccination immunomodulation: Forming the basis of all cancer immunotherapy. Ther. Adv. Vaccines Immunother. 2019, 7, 1–28. [Google Scholar] [CrossRef]
- Ye, Z.L.; Qian, Q.; Jin, H.J.; Qian, Q.J. Cancer vaccine: Learning lessons from immune checkpoint inhibitors. J. Cancer 2018, 9, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, A.; Grünebach, F.; Patrone, F.; Brossart, P. Anticancer vaccination strategies. Ann. Oncol. 2004, 15, iv153–iv160. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 1–10. [Google Scholar] [CrossRef]
- Nurieva, R.; Wang, J.; Sahoo, A. T-cell tolerance in cancer. Immunotherapy 2013, 5, 513–531. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Zhang, Y.; Cai, H.; Zhou, F.; Gao, H.; Deng, L.; Li, R. A PD-L1-Based Cancer Vaccine Elicits Antitumor Immunity in a Mouse Melanoma Model. Mol. Ther. Oncolytics 2019, 14, 222–232. [Google Scholar] [CrossRef]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinicalapplications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef]
- Kimiz-Gebologlu, I.; Gulce-Iz, S.; Biray-Avci, C. Monoclonal antibodies in cancer immunotherapy. Mol. Biol. Rep. 2018, 45, 2935–2940. [Google Scholar] [CrossRef]
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 3210–3229. [Google Scholar] [CrossRef]
- Pegram, M.D.; Konecny, G.; Slamon, D.J. The molecular and cellular biology of HER2/neu gene amplification/overexpression and the clinical development of herceptin (trastuzumab) therapy for breast cancer. Cancer Treat. Res. 2000, 103, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.O.; Trappey, F.A.; Clifton, G.T.; Vreeland, T.J.; Peace, K.M.; Hale, D.F.; Litton, J.K.; Murray, J.L.; Perez, S.A.; Papamichail, M.; et al. Effects of HLA status and HER2 status on outcomes in breast cancer patients at risk for recurrence—Implications for vaccine trial design. Clin. Immunol. 2018, 195, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Brossart, P.; Stuhler, G.; Flad, T.; Stevanovic, S.; Rammensee, H.G.; Kanz, L.; Brugger, W. Her-2/neu-derived peptides are tumor-associated antigens expressed by human renal cell and colon carcinoma lines and are recognized by in vitro induced specific cytotoxic T lymphocytes. Cancer Res. 1998, 58, 732–736. [Google Scholar] [PubMed]
- Kaumaya, P.T. B-cell epitope peptide cancer vaccines: A new paradigm for combination immunotherapies with novel checkpoint peptide vaccine. Future Oncol. 2020, 16, 1767–1791. [Google Scholar] [CrossRef]
- DeMaria, P.J.; Bilusic, M. Cancer Vaccines. Hematol. Oncol. Clin. North. Am. 2019, 33, 199–214. [Google Scholar] [CrossRef]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Hammerstrom, A.E.; Cauley, D.H.; Atkinson, B.J.; Sharma, P. Cancer immunotherapy: Sipuleucel-T and beyond. Pharmacotherapy 2011, 31, 813–828. [Google Scholar] [CrossRef]
- Calmeiro, J.; Carrascal, M.A.; Tavares, A.R.; Ferreira, D.A.; Gomes, C.; Falcão, A.; Cruz, M.T.; Neves, B.M. Dendritic cell vaccines for cancer immunotherapy: The role of human conventional type 1 dendritic cells. Pharmaceutics 2020, 12, 158. [Google Scholar] [CrossRef]
- Cintolo, J.A.; Datta, J.; Mathew, S.J.; Czerniecki, B.J. Dendritic cell-based vaccines: Barriers and opportunities. Future Oncol. 2012, 8, 1273–1299. [Google Scholar] [CrossRef]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology 2016, 5, e1071008. [Google Scholar] [CrossRef]
- Pitt, J.M.; André, F.; Amigorena, S.; Soria, J.C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic cell-derived exosomes for cancer therapy. J. Clin. Investig. 2016, 126, 1224–1232. [Google Scholar] [CrossRef]
- Théry, C.; Duban, L.; Segura, E.; Véron, P.; Lantz, O.; Amigorena, S. Indirect activation of naïve CD4+ T cells by dendritic cell–derived exosomes. Nat. Immunol. 2002, 3, 1156–1162. [Google Scholar] [CrossRef]
- André, F.; Chaput, N.; Schartz, N.E.C.; Flament, C.; Aubert, N.; Bernard, J.; Lemonnier, F.; Raposo, G.; Escudier, B.; Hsu, D.H.; et al. Exosomes as Potent Cell-Free Peptide-Based Vaccine. I. Dendritic Cell-Derived Exosomes Transfer Functional MHC Class I/Peptide Complexes to Dendritic Cells. J. Immunol. 2004, 172, 2126–2136. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Amigorena, S.; Théry, C. Mature dendritic cells secrete exosomes with strong ability to induce antigen-specific effector immune responses. Blood Cells Mol. Dis. 2005, 35, 89–93. [Google Scholar] [CrossRef]
- Sprent, J. Direct stimulation of naïve T cells by antigen-presenting cell vesicles. Blood Cells Mol. Dis. 2005, 35, 17–20. [Google Scholar] [CrossRef]
- Viaud, S.; Terme, M.; Flament, C.; Taieb, J.; André, F.; Novault, S.; Escudier, B.; Robert, C.; Caillat-Zucman, S.; Tursz, T.; et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL-15Rα. PLoS ONE 2009, 4, e4942. [Google Scholar] [CrossRef]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Zhang, Y.F.; Huang, Y.; Qu, P.; Ma, B.; Si, S.Y.; Li, Z.S.; Li, W.X.; Li, X.; Ge, W.; et al. The anti-tumor immune response induced by a combination of MAGE-3/MAGE-n-derived peptides. Oncol. Rep. 2008, 20, 245–252. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of the first phase 1 clinical trial. J. Transl. Med. 2005, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, J.; Shao, C.; Liu, S.; Yu, Y.; Wang, Q.; Cao, X. Efficient induction of antitumor T cell immunity by exosomes derived from heat-shocked lymphoma cells. Eur. J. Immunol. 2006, 36, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Shen, Y.; Chen, T.; Xu, F.; Chen, X.; Zheng, S. The functions and clinical applications of tumor-derived exosomes. Oncotarget 2016, 7, 60736–60751. [Google Scholar] [CrossRef]
- De la Torre Gomez, C.; Goreham, R.V.; Bech Serra, J.J.; Nann, T.; Kussmann, M. “Exosomics”-A Review of Biophysics, Biology and Biochemistry of Exosomes With a Focus on Human Breast Milk. Front. Genet. 2018, 9, 92. [Google Scholar] [CrossRef]
- Whiteside, T.L.; Mandapathil, M.; Szczepanski, M.; Szajnik, M. Mechanisms of tumor escape from the immune system: Adenosine-producing Treg, exosomes and tumor-associated TLRs. Bull. Cancer 2011, 98, E25–E31. [Google Scholar] [CrossRef]
- Xavier, C.P.R.; Caires, H.R.; Barbosa, M.A.G.; Bergantim, R.; Guimarães, J.E.; Vasconcelos, M.H. The Role of Extracellular Vesicles in the Hallmarks of Cancer and Drug Resistance. Cells 2020, 9, 1141. [Google Scholar] [CrossRef]
- Bu, N.; Li, Q.-L.; Feng, Q.; Sun, B.-Z. Immune protection effect of exosomes against attack of L1210 tumor cells. Leuk. Lymphoma 2006, 47, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, C.; Wei, W.; Shen, C.; Deng, X.; Chen, L.; Ma, L.; Hao, S. Dendritic Cells Pulsed with Leukemia Cell-Derived Exosomes More Efficiently Induce Antileukemic Immunities. PLoS ONE 2014, 9, e91463. [Google Scholar] [CrossRef]
- Gu, X.; Erb, U.; Büchler, M.W.; Zöller, M. Improved vaccine efficacy of tumor exosome compared to tumor lysate loaded dendritic cells in mice. Int. J. Cancer 2015, 136, E74–E84. [Google Scholar] [CrossRef]
- Pineda, B.; Sánchez García, F.J.; Olascoaga, N.K.; Pérez de la Cruz, V.; Salazar, A.; Moreno-Jiménez, S.; Pedro, N.H.; Marquez-Navarro, A.; Plata, A.O.; Sotelo, J. Malignant Glioma Therapy by Vaccination with Irradiated C6 Cell-Derived Microvesicles Promotes an Antitumoral Immune Response. Mol. Ther. 2019, 27, 1612–1620. [Google Scholar] [CrossRef]
- Phung, C.D.; Pham, T.T.; Nguyen, H.T.; Nguyen, T.T.; Ou, W.; Jeong, J.H.; Choi, H.G.; Ku, S.K.; Yong, C.S.; Kim, J.O. Anti-CTLA-4 antibody-functionalized dendritic cell-derived exosomes targeting tumor-draining lymph nodes for effective induction of antitumor T-cell responses. Acta Biomater. 2020, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Sun, J.; Li, H.; Lin, H.; Xie, W.; Li, J.; Tan, W. Antitumor efficacy of interferon-γ-modified exosomal vaccine in prostate cancer. Prostate 2020, 80, 811–823. [Google Scholar] [CrossRef]
- Hao, S.; Bai, O.; Li, F.; Yuan, J.; Laferte, S.; Xiang, J. Mature dendritic cells pulsed with exosomes stimulate efficient cytotoxic T-lymphocyte responses and antitumour immunity. Immunology 2007, 120, 90–102. [Google Scholar] [CrossRef]
- Lu, M.; Xing, H.; Xun, Z.; Yang, T.; Ding, P.; Cai, C.; Wang, D.; Zhao, X. Exosome-based small RNA delivery: Progress and prospects. Asian J. Pharm. Sci. 2018, 13, 1–11. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, H.; Jin, Y. Delivery of Functional Small RNAs via Extracellular Vesicles In Vitro and In Vivo. Methods Mol. Biol. 2020, 2115, 107–117. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 Distribution and Perspective for Cancer Immunotherapy-Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrands, F.; Marcellin, M.; Burlet-Schiltz, O.; Gonzalez de Peredo, A.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef]
- Wieder, T.; Eigentler, T.; Brenner, E.; Röcken, M. Immune checkpoint blockade therapy. J. Allergy Clin. Immunol. 2018, 142, 1403–1414. [Google Scholar] [CrossRef]
- Matsuda, A.; Ishiguro, K.; Yan, I.K.; Patel, T. Extracellular Vesicle-Based Therapeutic Targeting of β-Catenin to Modulate Anticancer Immune Responses in Hepatocellular Cancer. Hepatol. Commun. 2019, 3, 525–541. [Google Scholar] [CrossRef] [PubMed]
- De Galarreta, M.R.; Bresnahan, E.; Molina-Sánchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. β-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T.F. A new paradigm for tumor immune escape: β-catenin-driven immune exclusion. J. Immunother. Cancer 2015, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; Meylan, M.; de Reyniès, A.; Sautès-Fridman, C.; Fridman, W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020, 11, 784. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Ochoa, M.C.; Olivera, I.; Melero, I. Immune Desertic Landscapes in Hepatocellular Carcinoma Shaped by β-Catenin Activation. Cancer Discov. 2019, 9, 1003–1005. [Google Scholar] [CrossRef]
- Bell, J.B.; Podetz-Pedersen, K.M.; Aronovich, E.L.; Belur, L.R.; McIvor, R.S.; Hackett, P.B. Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat. Protoc. 2007, 2, 3153–3165. [Google Scholar] [CrossRef]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Gupta, R.C. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 2016, 371, 48–61. [Google Scholar] [CrossRef]
- Subleski, J.J.; Scarzello, A.J.; Alvord, W.G.; Jiang, Q.; Stauffer, J.K.; Kronfli, A.; Saleh, B.; Back, T.; Weiss, J.M.; Wiltrout, R.H. Serum-based tracking of de novo initiated liver cancer progression reveals early immunoregulation and response to therapy. J. Hepatol. 2015, 63, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Condition | Treatment | Clinical Phase | NCT Identifier |
|---|---|---|---|
| Breast cancer | HER-2 pulsed DC Vaccine | Phase I | NCT02063724 |
| Brain tumors | Autologous DCs pulsed with CSC Lysate | Phase I | NCT02010606 |
| Prostate cancer | Autologous DCs loaded with mRNA from Primary prostate cancer tissue + hTERT + survivin | Phase I/II | NCT01197625 |
| Sarcoma/soft tissue Sarcoma/bone sarcoma | DC vaccine + tumor lysate + imiquimod | Phase I | NCT01803152 |
| Brain Metastases | Personalized cellular vaccine: tumor antigen mRNA-pulsed autologous DCs | Phase I | NCT02808416 |
| Newly diagnosed glioblastoma | AV-GBM-1: autologous DCs loaded with autologous tumor antigens derived from self-renewing TICs | Phase II | NCT03400917 |
| Multiple myeloma | ASCT + DC myeloma fusion vaccine + MAb CT-011 (pidilizumab) | Phase II | NCT01067287 |
| AML | DC AML fusion vaccine | Phase II | NCT01096602 |
| Advanced breast cancer | DCs co-cultured with CIK cells + capecitabine monotherapy | Phase II | NCT02491697 |
| Authors | Method | Main Outcomes | Refs |
|---|---|---|---|
| Théry C. et al. | In vitro | Dex can transfer functional peptide-loaded MHC class I and II complexes to DCs. | [91] |
| André F. et al. | In vitro and in vivo | Dex harbouring MHC class I/peptide complexes require DC for efficient priming of CTLs. | [92] |
| In vivo | Dex mimic the capacity of mature DCs to initiate peptide-specific CD8+ T cell responses. | ||
| Segura E. et al. | In vitro | Dex from immature DCs (imDC) and mature DCs (mDC) have different protein composition due to maturation signals. MHC class I molecules are up-regulated in mDC and reduced in mature exosomes. Molecules stimulating CD4 T cells are up-regulated in mDC and mature exosomes. | [93] |
| Sprent J. | In vitro | Peptide-pulsed Dex are immunogenic for CD8+ T cells also in the absence of APCs. | [94] |
| In vivo | Peptide-loaded Dex induce high proliferative responses and CTLs induction, so priming CD8+ T cells. | ||
| Viaud S. et al. | In vivo | Dex administration promotes proliferation, activation and cytotoxicity of NK cells. | [95] |
| In vitro | Human Dex harbouring IL-15Rα lead to NK cell proliferation and IFNγ production |
| Therapeutic Agent | Condition | Outcome | Refs |
|---|---|---|---|
| Irradiated C6 glioma cell-derived MVs (IR-MVs) | Malignant C6 glioma | In vivo vaccination with IR-MVs promotes antitumor immune response leading to the apoptosis of glioblastoma cells and increases Th cells and CTL infiltration into the tumor. | [108] |
| DC-derived-exosomes functionalized with costimulatory molecules, MHCs, antigenic Ovalbumin peptide and anti-CTLA-4 antibody (EXO-OVA-mAb) | B16-OVA melanoma tumor model | Exosomes are targeted to T cells in vivo. EXO-OVA-mAb are able to effectively prime T-cell activation and proliferation, In vitro and in vivo. The fraction of memory T cells is increased in mice treated with vaccination. The antitumor efficacy is confirmed by the infiltration of both CD4 + and CD8 + cells and the CTLs/Treg ratio within the tumor site of vaccinated mice. | [109] |
| Interferon-γ-modified prostate cancer cell- derived exosomes | RM-1 prostate cancer | Vaccine induces macrophages differentiation and the production of antibodies, reduces tumor angiogenesis and metastasis rate, inhibits tumor growth and prolongs survival time of mice with metastatic prostate cancer. | [110] |
| Interferon-γ-modified prostate cancer cell- derived exosomes + IFN-γ-modified RM-1 cell vaccine | RM-1 prostate cancer | Exosomal vaccine improves the T cell response generated by the tumor cell vaccine and downregulates in the expression of IDO1 and PD-L1 immune checkpoints. Combination therapy show the highest tumor-specific cytotoxic activities compared to vaccine monotherapies and tumor growth is significantly suppressed. | [110] |
| Mature DCs pulsed with ovalbumin protein-pulsed DC-derived exosomes (EXO-pulsed DCs) | B16-OVA melanoma tumor model | EXO-pulsed DCs stimulate CD8+ T-cell proliferation and differentiation into CTL effectors In vitro and in vivo. EXO-pulsed DCs induce stronger immunity against lung tumor metastases and can eradicate established tumors. They also induce strong long-term OVA-specific CD8+ T-cell memory | [111] |
| Condition | Treatment | Year | Clinical Phase | NCT Identifier and References |
|---|---|---|---|---|
| Advanced NSCLC | Dex loaded with the MAGE tumor antigens | 2005 | Phase I | [96] |
| Metastatic melanoma | Autologous exosomes pulsed with MAGE 3 peptides | 2005 | Phase I | [99] |
| Colorectal cancer | Ascites-derived exosomes (Aex) in combination with GM-CSF | 2008 | Phase I | [125] |
| Melanoma | Human Dex bearing NKG2D ligands | 2009 | Phase I | [95] |
| NSCLC | Tumor Antigen-loaded Dex | 2010 | Phase II | NCT01159288 |
| Unresectable NSCLC | IFN-γ-Dex loaded with MHC class I- and class II-restricted cancer antigens | 2015 | Phase II | [89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacobino, C.; Canta, M.; Fornaguera, C.; Borrós, S.; Cauda, V. Extracellular Vesicles and Their Current Role in Cancer Immunotherapy. Cancers 2021, 13, 2280. https://doi.org/10.3390/cancers13092280
Giacobino C, Canta M, Fornaguera C, Borrós S, Cauda V. Extracellular Vesicles and Their Current Role in Cancer Immunotherapy. Cancers. 2021; 13(9):2280. https://doi.org/10.3390/cancers13092280
Chicago/Turabian StyleGiacobino, Carla, Marta Canta, Cristina Fornaguera, Salvador Borrós, and Valentina Cauda. 2021. "Extracellular Vesicles and Their Current Role in Cancer Immunotherapy" Cancers 13, no. 9: 2280. https://doi.org/10.3390/cancers13092280
APA StyleGiacobino, C., Canta, M., Fornaguera, C., Borrós, S., & Cauda, V. (2021). Extracellular Vesicles and Their Current Role in Cancer Immunotherapy. Cancers, 13(9), 2280. https://doi.org/10.3390/cancers13092280