
Quantification and Optimization of Standard-of-Care Therapy to Delay the Emergence of Resistant Bone Metastatic Prostate Cancer

 ,
, 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods and Materials
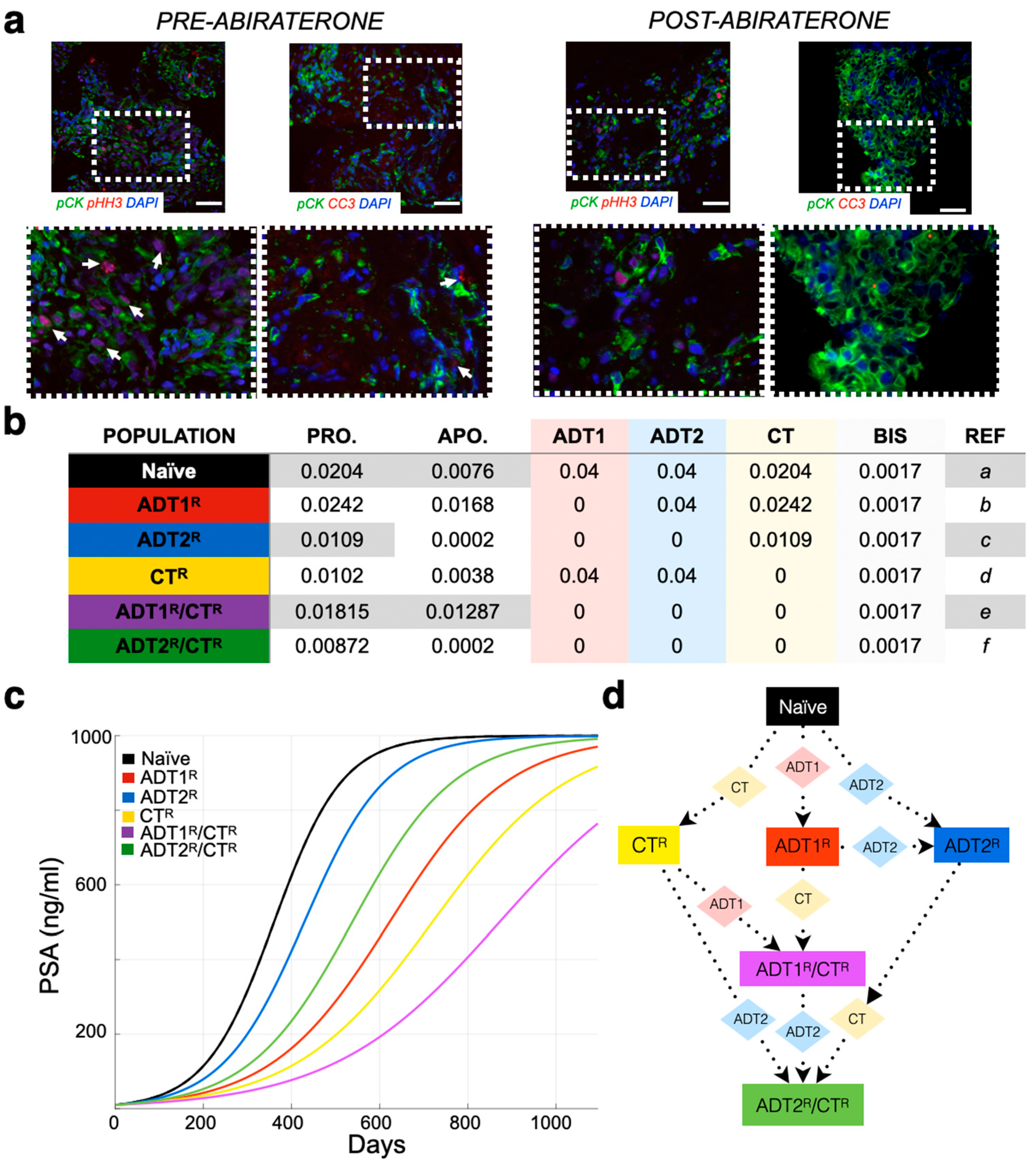
2.1. Immunofluorescent Staining and Analysis
2.2. Model and Assumptions
2.3. Resistance-Informed Intervention-Decision Algorithm
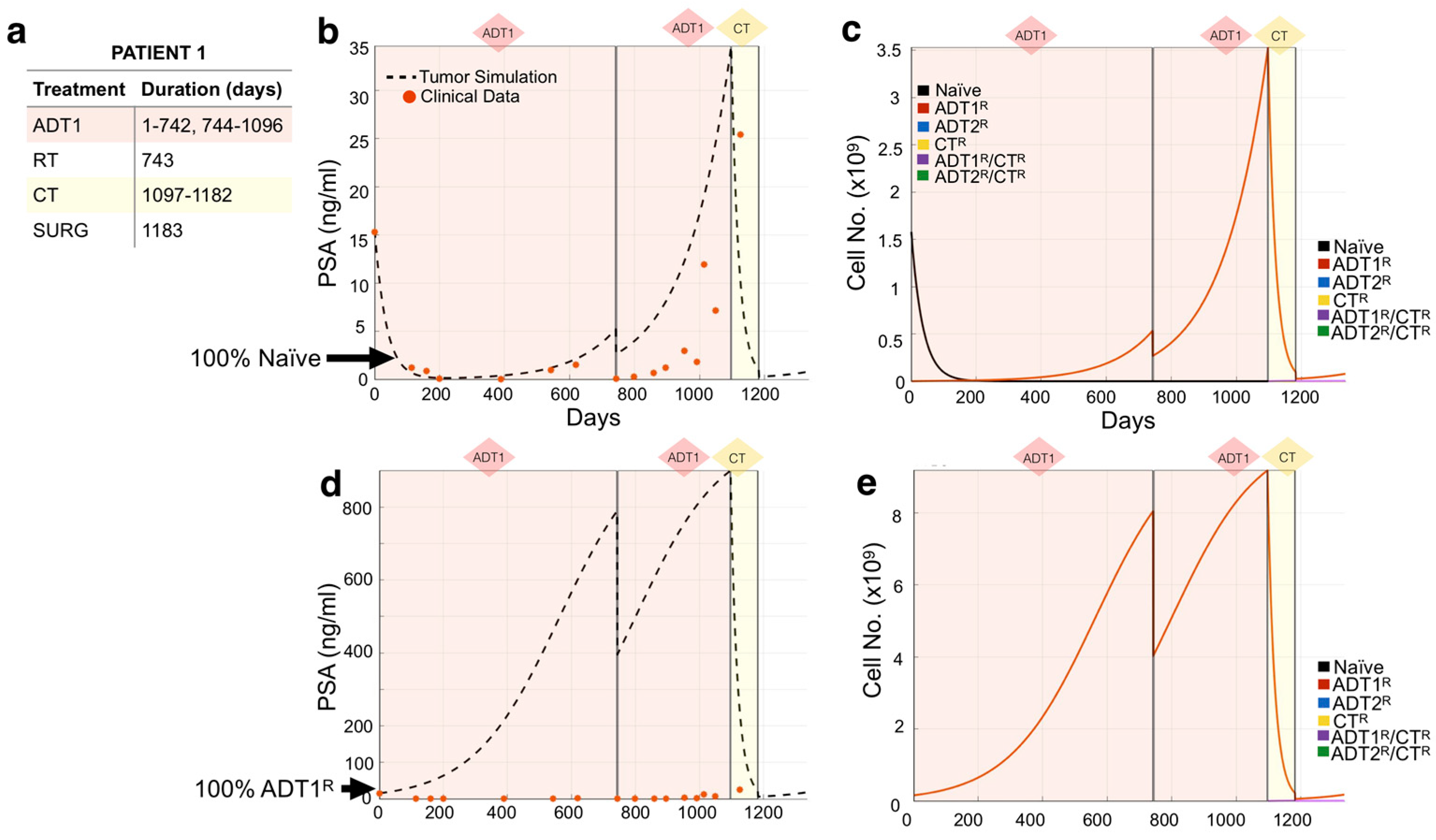
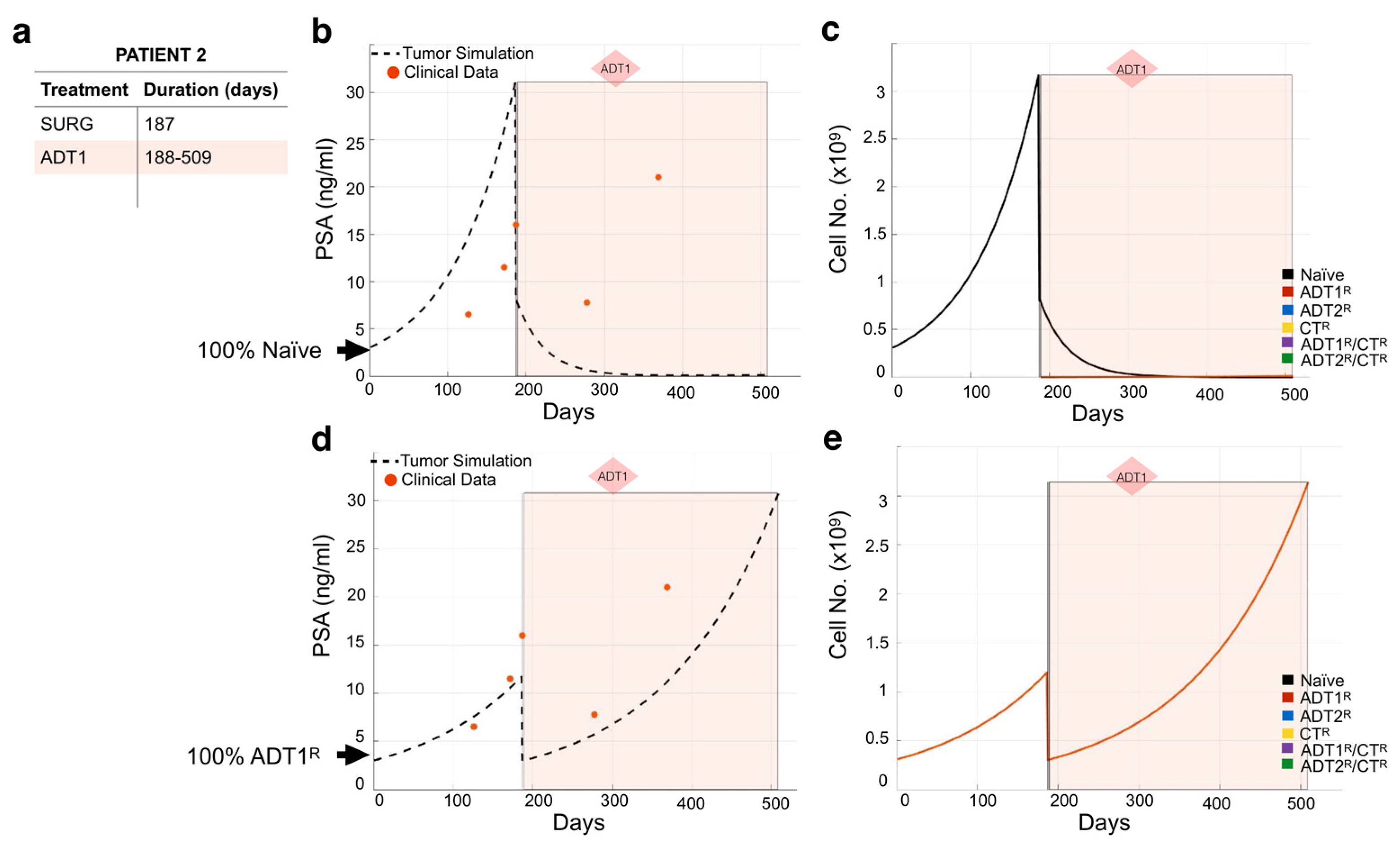
2.4. Model Validation and Limitations
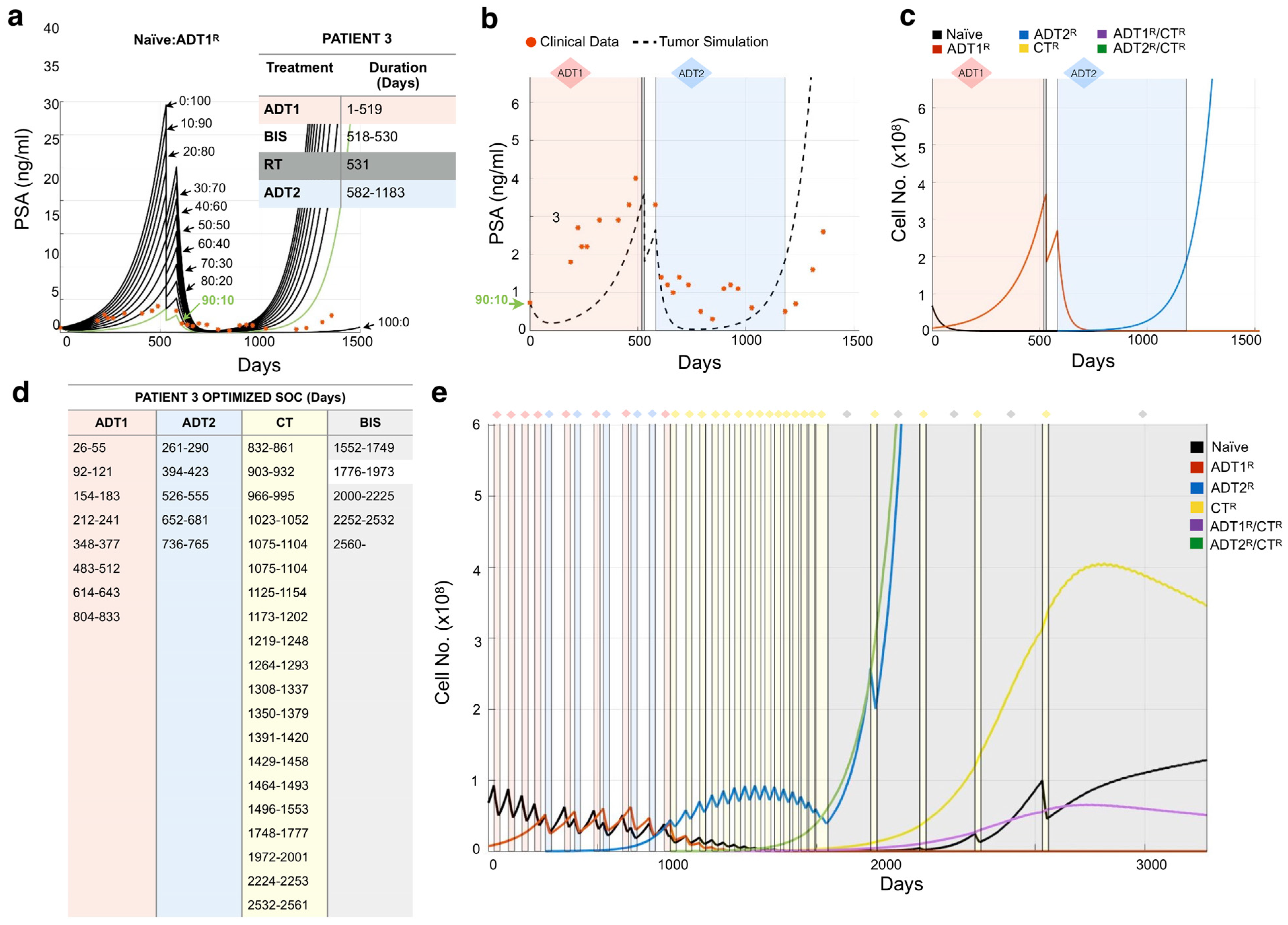
3. Results
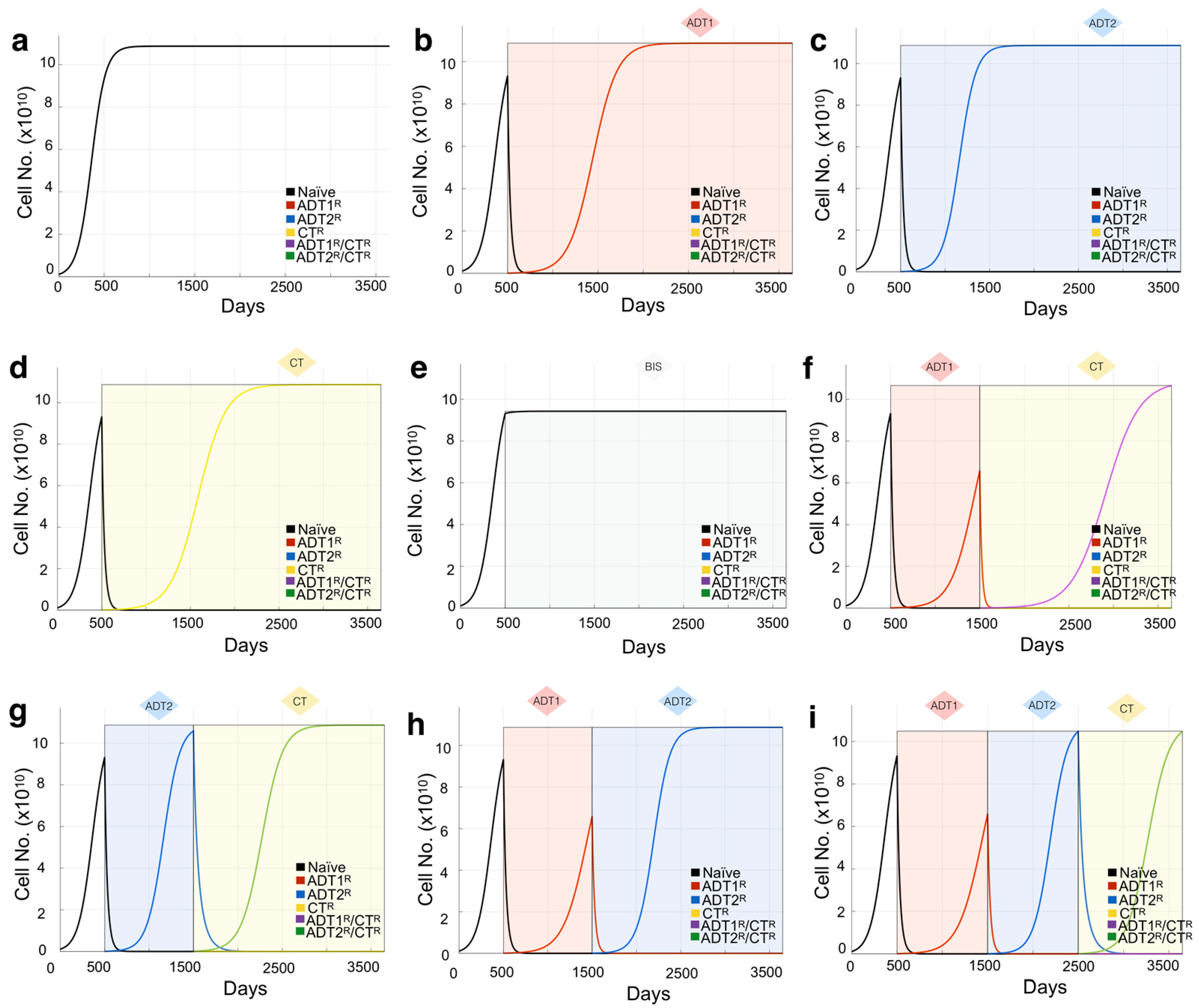
3.1. Defining the Impact of ADT on Prostate Cancer Growth Rates
3.2. Modeling Individual Patient Responses to Standard-of-Care Therapy
3.3. Design of Resistance-Informed Patient Treatment Strategy to Extend Overall Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Azzouni, F.; Mohler, J. Biology of castration-recurrent prostate cancer. Urol. Clin. N. Am. 2012, 39, 435–452. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Frieling, J.S.; Basanta, D.; Lynch, C.C. Current and emerging therapies for bone metastatic castration-resistant prostate cancer. Cancer Control 2015, 22, 109–120. [Google Scholar] [CrossRef]
- Morrissey, C.; Roudier, M.P.; Dowell, A.; True, L.D.; Ketchanji, M.; Welty, C.; Corey, E.; Lange, P.H.; Higano, C.S.; Vessella, R.L. Effects of androgen deprivation therapy and bisphosphonate treatment on bone in patients with metastatic castration-resistant prostate cancer: Results from the University of Washington Rapid Autopsy Series. J. Bone Miner. Res. 2013, 28, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.J.; Torre, T.; Grob, M.; Yu, J.; Avital, I.; Brücher, B.L.D.M.; Stojadinovic, A.; Man, Y.G. Current approaches, challenges and future directions for monitoring treatment response in prostate cancer. J. Cancer 2014, 5, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Fenton, J.J.; Weyrich, M.S.; Durbin, S.; Liu, Y.; Bang, H.; Melnikow, J. Prostate-Specific Antigen-Based Screening for Prostate Cancer: Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2018, 319, 1914–1931. [Google Scholar] [CrossRef]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R1896. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Hu, Q. CYP17 inhibitors—abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat. Rev. Urol. 2014, 11, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Gomes, A.J.P.D.S.; Given, R.; Soto, Á.J.; Merseburger, A.S.; Özgüroğlu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [PubMed]
- McKeage, K. Docetaxel: A review of its use for the first-line treatment of advanced castration-resistant prostate cancer. Drugs 2012, 72, 1559–1577. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.; Chen, Y.H.; Carduccie, M.A.; Liu, G.; Jarrard, D.F.; Eisenberger, M.A.; Wong, Y.-N.; Hahn, N.M.; Kohli, M.; Vogelzang, N.J.; et al. Impact on overall survival (OS) with chemohormonal therapy versus hormonal therapy for hormone-sensitive newly metastatic prostate cancer (mPrCa): An ECOG-led phase III randomized trial. J. Clin. Oncol. 2014, 32. [Google Scholar] [CrossRef]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- Morken, J.D.; Packer, A.; Everett, R.A.; Nagy, J.D.; Kuang, Y. Mechanisms of resistance to intermittent androgen deprivation in patients with prostate cancer identified by a novel computational method. Cancer Res. 2014, 74, 3673–3683. [Google Scholar] [CrossRef]
- Jackson, T.L. A mathematical investigation of the multiple pathways to recurrent prostate cancer: Comparison with experimental data. Neoplasia 2004, 6, 697–704. [Google Scholar] [CrossRef]
- Zhang, J.; Cunningham, J.J.; Brown, J.S.; Gatenby, R.A. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat. Commun. 2017, 8, 1816. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Feng, Z.; Hu, Z.; Wang, G.; Yuan, X.; Wang, H.; Hu, D. Prostate cancer magnetic resonance imaging (MRI): Multidisciplinary standpoint. Quant. Imaging Med. Surg. 2013, 3, 100–112. [Google Scholar] [PubMed]
- Hirata, Y.; Akakura, K.; Higano, C.S.; Bruchovsky, N.; Aihara, K. Quantitative mathematical modeling of PSA dynamics of prostate cancer patients treated with intermittent androgen suppression. J. Mol. Cell Biol. 2012, 4, 127–132. [Google Scholar] [CrossRef]
- Hirata, Y.; Morino, K.; Akakura, K.; Higano, C.S.; Aihara, K. Personalizing Androgen Suppression for Prostate Cancer Using Mathematical Modeling. Sci. Rep. 2018, 8, 2673. [Google Scholar] [CrossRef]
- Jain, H.V.; Clinton, S.K.; Bhinder, A.; Friedman, A. Mathematical modeling of prostate cancer progression in response to androgen ablation therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 19701–19706. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Parker, C.; Eeles, R.A.; Schröder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate cancer. Lancet 2015. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Wang, L.; Dehm, S.M.; Hillman, D.W.; Sicotte, H.; Tan, W.; Gormley, M.; Bhargava, V.; Jimenez, R.; Xie, F.; Yin, P.; et al. A Prospective Genome-Wide Study of Prostate Cancer Metastases Reveals Association of Wnt Pathway Activation and Increased Cell Cycle Proliferation with Primary Resistance to Abiraterone Acetate-Prednisone. Ann. Oncol. 2017, 29, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Network NCC. NCCN Guidelines for Treatment of Prostate Cancer; Network NCC: Brindisi, Italy, 2019. [Google Scholar]
- Weir, E.G.; Partin, A.W.; Epstein, J.I. Correlation of serum prostate specific antigen and quantitative immunohistochemistry. J. Urol. 2000, 163, 1739–1742. [Google Scholar] [CrossRef]
- Lodish, H.F. Molecular Cell Biology, 4th ed.; W.H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Diamandis, E.P. Prostate-specific Antigen: Its Usefulness in Clinical Medicine. Trends Endocrinol. Metab. 1998, 9, 310–316. [Google Scholar] [CrossRef]
- Justesen, J.; Stenderup, K.; Ebbesen, E.N.; Mosekilde, L.; Steiniche, T.; Kassem, M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology 2001, 2, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.; Cook, L.M.; Lynch, C.C.; Basanta, D. An integrated computational model of the bone microenvironment in bone-metastatic prostate cancer. Cancer Res. 2014, 74, 2391–2401. [Google Scholar] [CrossRef] [PubMed]
- Jorfi, S.; Ansa-Addo, E.A.; Kholia, S.; Stratton, D.; Valley, S.; Lange, S.; Inal, J. Inhibition of microvesiculation sensitizes prostate cancer cells to chemotherapy and reduces docetaxel dose required to limit tumor growth in vivo. Sci. Rep. 2015, 5, 13006. [Google Scholar] [CrossRef]
- Wodarz, D.; Komarova, N.L. Computational Biology of Cancer: Lecture Notes and Mathematical Modeling; World Scientific: Hackensack, NJ, USA, 2008. [Google Scholar]
- Berges, R.R.; Vukanovic, J.; Epstein, J.I.; Carmichel, M.; Cisek, L.; Johnson, D.E.; Veltri, R.W.; Walsh, P.C.; Isaacs, J.T. Implication of cell kinetic changes during the progression of human prostatic cancer. Clin. Cancer Res. 1995, 1, 473–480. [Google Scholar] [PubMed]
- Tanaka, H.; Kono, E.; Tran, C.P.; Miyazaki, H.; Yamashiro, J.; Shimomura, T.; Fazli, L.; Wada, R.; Huang, J.; Vessella, R.L.; et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat. Med. 2010, 16, 1414–1420. [Google Scholar] [CrossRef]
- Lipton, A.; Fizazi, K.; Stopeck, A.T.; Henry, D.H.; Brown, J.E.; Yardley, D.A.; Richardson, G.E.; Siena, S.; Maroto, P.; Clemens, M.; et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: A combined analysis of 3 pivotal, randomised, phase 3 trials. Eur. J. Cancer 2012, 48, 3082–3092. [Google Scholar] [CrossRef]
- West, J.B.; Dinh, M.N.; Brown, J.S.; Zhang, J.; Anderson, A.R.; Gatenby, R.A. Multidrug Cancer Therapy in Metastatic Castrate-Resistant Prostate Cancer: An Evolution-Based Strategy. Clin. Cancer Res. 2019, 25, 4413–4421. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Nichol, D.; Kinose, F.; Abazeed, M.E.; Marusyk, A.; Haura, E.B.; Scott, J.G. Collateral sensitivity networks reveal evolutionary instability and novel treatment strategies in ALK mutated non-small cell lung cancer. Sci. Rep. 2017, 7, 1232. [Google Scholar] [CrossRef]
- Chen, W.S.; Aggarwal, R.; Zhang, L.; Zhao, S.G.; Thomas, G.V.; Beer, T.M.; Quigley, D.A.; Foye, A.; Playdle, D.; Huang, J.; et al. Genomic Drivers of Poor Prognosis and Enzalutamide Resistance in Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2019, 76, 562–571. [Google Scholar] [CrossRef]
- Hussain, M.; Tangen, C.M.; Berry, D.L.; Higano, C.S.; Crawford, E.D.; Liu, G.; Wilding, G.; Prescott, S.; Sundaram, S.K.; Small, E.J.; et al. Intermittent versus continuous androgen deprivation in prostate cancer. N. Engl. J. Med. 2013, 368, 1314–1325. [Google Scholar] [CrossRef]
- Crook, J.M.; O’Callaghan, C.J.; Duncan, G.; Dearnaley, D.P.; Higano, C.S.; Horwitz, E.M.; Frymire, E.; Malone, S.; Chin, J.; Nabid, A.; et al. Intermittent androgen suppression for rising PSA level after radiotherapy. N. Engl. J. Med. 2012, 367, 895–903. [Google Scholar] [CrossRef]
- Oliver, R.; Williams, G.; Paris, A.; Blandy, J. Intermittent androgen deprivation after PSA-complete response as a strategy to reduce induction of hormone-resistant prostate cancer. Urology 1997, 49, 79–82. [Google Scholar] [CrossRef]
- Cosma, G.; McArdle, S.E.; Reeder, S.; Foulds, G.A.; Hood, S.; Khan, M.; Pockley, A.G. Identifying the Presence of Prostate Cancer in Individuals with PSA Levels <20 ng ml (−1) Using Computational Data Extraction Analysis of High Dimensional Peripheral Blood Flow Cytometric Phenotyping Data. Front. Immunol. 2017, 8, 1771. [Google Scholar] [PubMed]
- Huang, X.; Yuan, T.; Liang, M.; Du, M.; Xia, S.; Dittmar, R.; Wang, D.; See, W.; Costello, B.A.; Quevedo, F.; et al. Exosomal miR-1290 and miR-375 as prognostic markers in castration-resistant prostate cancer. Eur. Urol. 2015, 67, 33–41. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araujo, A.; Cook, L.M.; Frieling, J.S.; Tan, W.; Copland, J.A., II; Kohli, M.; Gupta, S.; Dhillon, J.; Pow-Sang, J.; Lynch, C.C.; et al. Quantification and Optimization of Standard-of-Care Therapy to Delay the Emergence of Resistant Bone Metastatic Prostate Cancer. Cancers 2021, 13, 677. https://doi.org/10.3390/cancers13040677
Araujo A, Cook LM, Frieling JS, Tan W, Copland JA II, Kohli M, Gupta S, Dhillon J, Pow-Sang J, Lynch CC, et al. Quantification and Optimization of Standard-of-Care Therapy to Delay the Emergence of Resistant Bone Metastatic Prostate Cancer. Cancers. 2021; 13(4):677. https://doi.org/10.3390/cancers13040677
Chicago/Turabian StyleAraujo, Arturo, Leah M. Cook, Jeremy S. Frieling, Winston Tan, John A. Copland, II, Manish Kohli, Shilpa Gupta, Jasreman Dhillon, Julio Pow-Sang, Conor C. Lynch, and et al. 2021. "Quantification and Optimization of Standard-of-Care Therapy to Delay the Emergence of Resistant Bone Metastatic Prostate Cancer" Cancers 13, no. 4: 677. https://doi.org/10.3390/cancers13040677
APA StyleAraujo, A., Cook, L. M., Frieling, J. S., Tan, W., Copland, J. A., II, Kohli, M., Gupta, S., Dhillon, J., Pow-Sang, J., Lynch, C. C., & Basanta, D. (2021). Quantification and Optimization of Standard-of-Care Therapy to Delay the Emergence of Resistant Bone Metastatic Prostate Cancer. Cancers, 13(4), 677. https://doi.org/10.3390/cancers13040677