
Transcription Factor AP4 Mediates Cell Fate Decisions: To Divide, Age, or Die




{kind=link}
{kind=link}
Simple Summary
Abstract
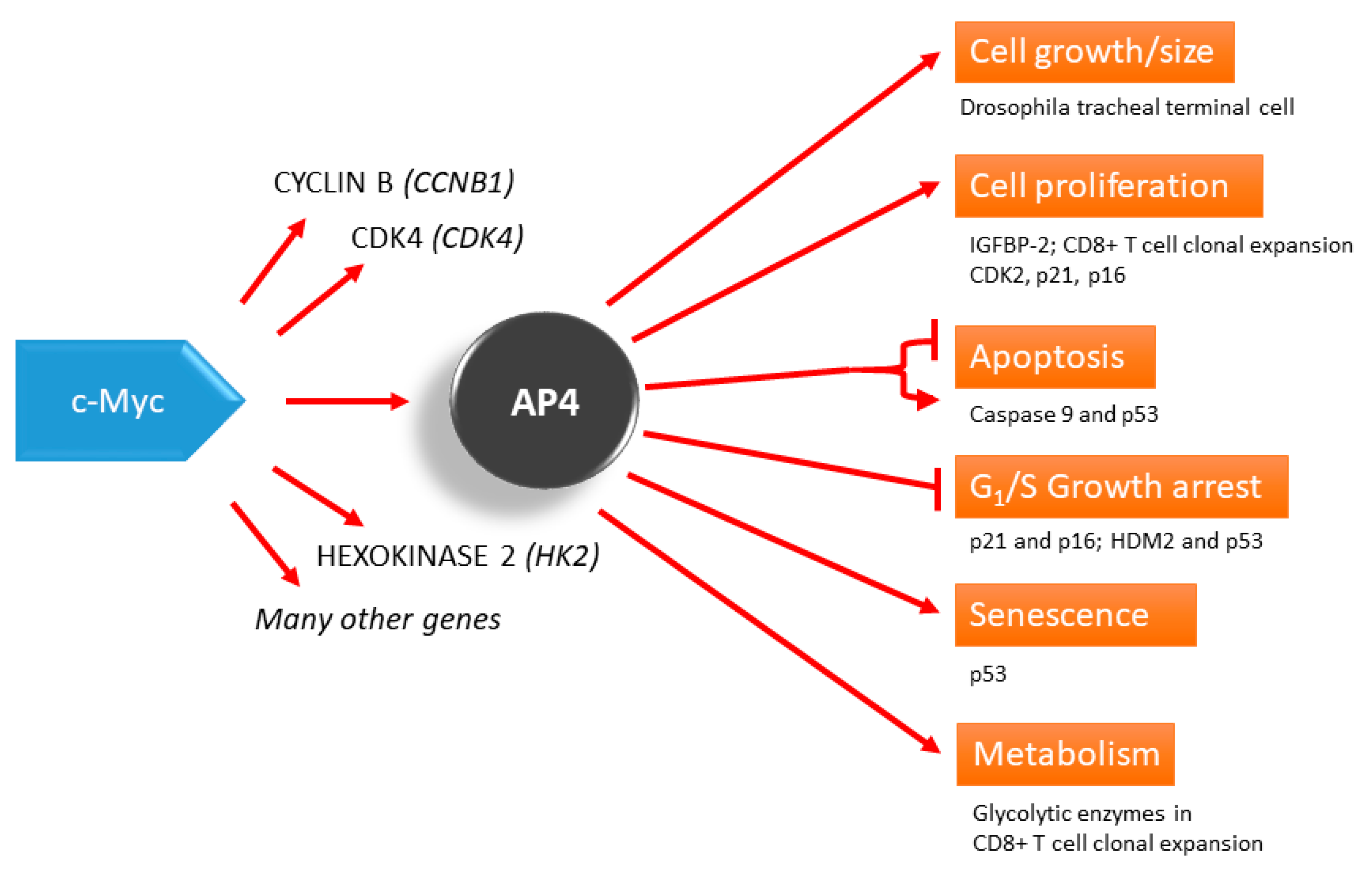
1. AP4-Mediated Repression vs. Induction of Target Genes
2. AP4 DNA Binding Sites as Targets for SNPs
3. AP4 Controls Cell Proliferation, Senescence, and Apoptosis
4. AP4 in Adaptive Immunity
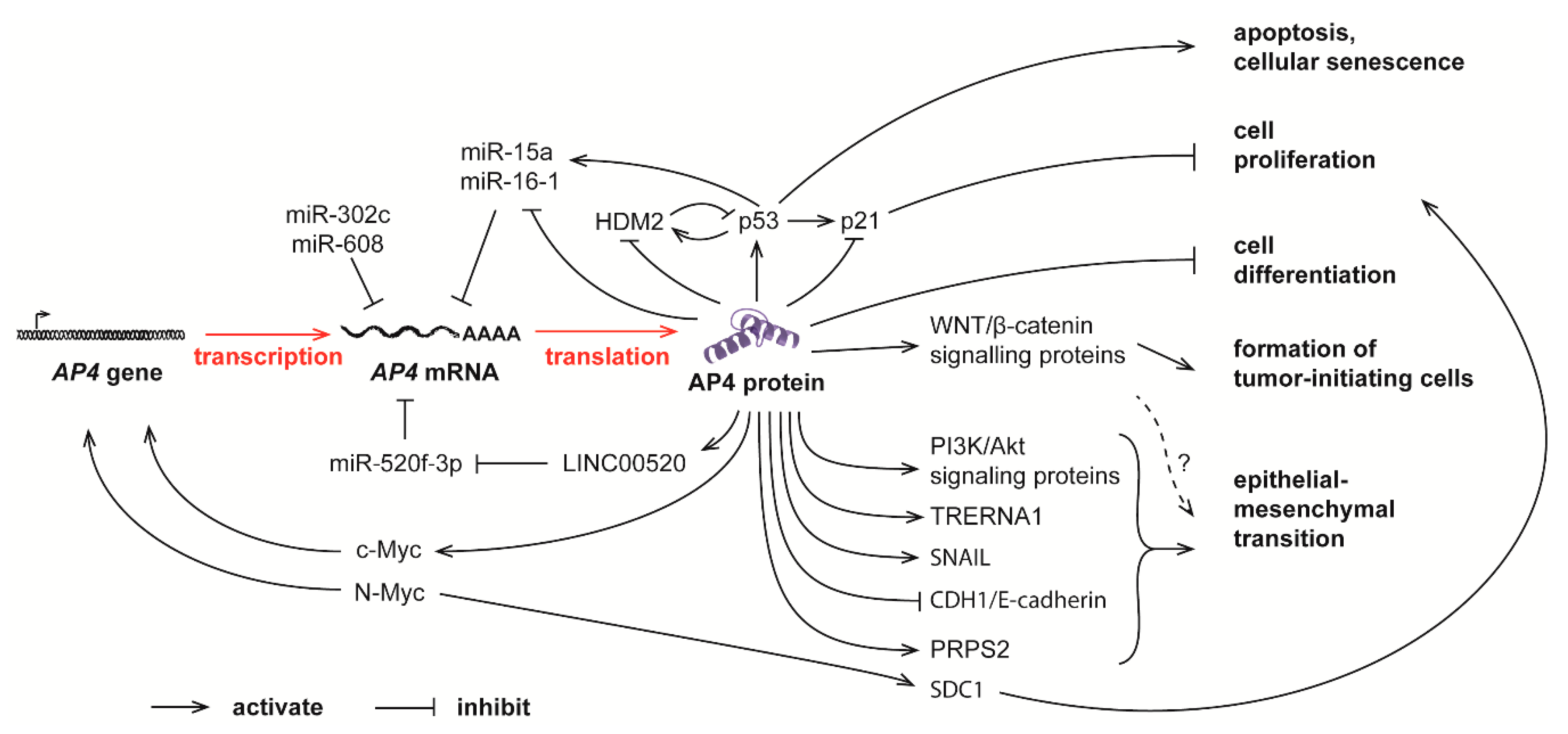
5. Pathological Functions of AP4 in Cancer
6. AP4 Regulates Epithelial-Mesenchymal Transition and Cell Migration in Cancer Cells
7. AP4 Is Essential for Cell Cycle Regulation and Cellular Stress Response in Cancer
8. Role of AP4 in Stem Cells and Tumor Initiating Cells
9. Connections between Non-Coding RNAs and AP4 in Cancer
10. Pathways Involved in AP4-Mediated Regulation of EMT and the Cell Cycle in Cancer
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Narlikar, L.; Ovcharenko, I. Identifying regulatory elements in eukaryotic genomes. Brief. Funct. Genom. Proteom. 2009, 8, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Mermod, N.; Williams, T.J.; Tjian, R. Enhancer binding factors AP-4 and AP-1 act in concert to activate SV40 late transcription in vitro. Nature 1988, 332, 557–561. [Google Scholar] [CrossRef]
- Jackstadt, R.; Roh, S.; Neumann, J.; Jung, P.; Hoffmann, R.; Horst, D.; Berens, C.; Bornkamm, G.W.; Kirchner, T.; Menssen, A.; et al. AP4 is a mediator of epithelial-mesenchymal transition and metastasis in colorectal cancer. J. Exp. Med. 2013, 210, 1331–1350. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.F.; Luscher, B.; Admon, A.; Mermod, N.; Tjian, R. Transcription factor AP-4 contains multiple dimerization domains that regulate dimer specificity. Genes Dev. 1990, 4, 1741–1752. [Google Scholar] [CrossRef]
- Badinga, L.; Song, S.; Simmen, R.C.M.; Simmen, F.A. A Distal Regulatory Region of the Insulin-Like Growth Factor Binding Protein-2 (IGFBP-2) Gene Interacts with the Basic Helix–Loop–Helix Transcription Factor, AP-4. Endocrine 1998, 8, 281–289. [Google Scholar] [CrossRef]
- Hong, S.J.; Choi, H.J.; Hong, S.; Huh, Y.; Chae, H.; Kim, K.S. Transcription factor GATA-3 regulates the transcriptional activity of dopamine beta-hydroxylase by interacting with Sp1 and AP4. Neurochem. Res. 2008, 33, 1821–1831. [Google Scholar] [CrossRef]
- Fodor, E.; Weinrich, S.L.; Meister, A.; Mermod, N.; Rutter, W.J. A pancreatic exocrine cell factor and AP4 bind overlapping sites in the amylase 2A enhancer. Biochemistry 1991, 30, 8102–8108. [Google Scholar] [CrossRef]
- Ku, W.C.; Chiu, S.K.; Chen, Y.J.; Huang, H.H.; Wu, W.G.; Chen, Y.J. Complementary quantitative proteomics reveals that transcription factor AP-4 mediates E-box-dependent complex formation for transcriptional repression of HDM2. Mol. Cell. Proteom. 2009, 8, 2034–2050. [Google Scholar] [CrossRef]
- Imai, K.; Okamoto, T. Transcriptional repression of human immunodeficiency virus type 1 by AP-4. J. Biol. Chem. 2006, 281, 12495–12505. [Google Scholar] [CrossRef] [PubMed]
- Glahder, J.A.; Hansen, C.N.; Vinther, J.; Madsen, B.S.; Norrild, B. A promoter within the E6 ORF of human papillomavirus type 16 contributes to the expression of the E7 oncoprotein from a monocistronic mRNA. J. Gen. Virol. 2003, 84, 3429–3441. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Raya, A.; DeJesus, P.; Chao, S.H.; Quon, K.C.; Caldwell, J.S.; Chanda, S.K.; Izpisua-Belmonte, J.C.; Schultz, P.G. Identification of p53 regulators by genome-wide functional analysis. Proc. Natl. Acad. Sci. USA 2004, 101, 3456–3461. [Google Scholar] [CrossRef]
- Kim, M.Y.; Jeong, B.C.; Lee, J.H.; Kee, H.J.; Kook, H.; Kim, N.S.; Kim, Y.H.; Kim, J.K.; Ahn, K.Y.; Kim, K.K. A repressor complex, AP4 transcription factor and geminin, negatively regulates expression of target genes in nonneuronal cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13074–13079. [Google Scholar] [CrossRef]
- Cui, Y.; Narayanan, C.S.; Zhou, J.; Kumar, A. Exon-I is involved in positive as well as negative regulation of human angiotensinogen gene expression. Gene 1998, 224, 97–107. [Google Scholar] [CrossRef]
- Fyhrquist, F.; Metsarinne, K.; Tikkanen, I. Role of angiotensin II in blood pressure regulation and in the pathophysiology of cardiovascular disorders. J. Hum. Hypertens. 1995, 9 (Suppl. 5), S19–S24. [Google Scholar]
- Arias, T.D.; Jorge, L.F.; Barrantes, R. Uses and misuses of definitions of genetic polymorphism. A perspective from population pharmacogenetics. Br. J. Clin. Pharmacol. 1991, 31, 117–119. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Butter, F.; Davison, L.; Viturawong, T.; Scheibe, M.; Vermeulen, M.; Todd, J.A.; Mann, M. Proteome-wide analysis of disease-associated SNPs that show allele-specific transcription factor binding. PLoS Genet. 2012, 8, e1002982. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Curtin, K.; Wolff, R.K.; Albertsen, H.; Sweeney, C.; Caan, B.J.; Ulrich, C.M.; Potter, J.D.; Slattery, M.L. The MLH1 -93 G>A promoter polymorphism and genetic and epigenetic alterations in colon cancer. Genes Chromosomes Cancer 2008, 47, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Haron, N.H.; Mohamad Hanif, E.A.; Abdul Manaf, M.R.; Yaakub, J.A.; Harun, R.; Mohamed, R.; Mohamed Rose, I. Microsatellite Instability and Altered Expressions of MLH1 and MSH2 in Gastric Cancer. Asian Pac. J. Cancer Prev. 2019, 20, 509–517. [Google Scholar] [CrossRef]
- Savio, A.J.; Bapat, B. Modulation of transcription factor binding and epigenetic regulation of the MLH1 CpG island and shore by polymorphism rs1800734 in colorectal cancer. Epigenetics 2017, 12, 441–448. [Google Scholar] [CrossRef]
- Thomas, R.; Trapani, D.; Goodyer-Sait, L.; Tomkova, M.; Fernandez-Rozadilla, C.; Sahnane, N.; Woolley, C.; Davis, H.; Chegwidden, L.; Kriaucionis, S.; et al. The polymorphic variant rs1800734 influences methylation acquisition and allele-specific TFAP4 binding in the MLH1 promoter leading to differential mRNA expression. Sci. Rep. 2019, 9, 13463. [Google Scholar] [CrossRef]
- Liu, N.Q.; Ter Huurne, M.; Nguyen, L.N.; Peng, T.; Wang, S.Y.; Studd, J.B.; Joshi, O.; Ongen, H.; Bramsen, J.B.; Yan, J.; et al. The non-coding variant rs1800734 enhances DCLK3 expression through long-range interaction and promotes colorectal cancer progression. Nat. Commun. 2017, 8, 14418. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003, 4, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.W.; Barradas, M.; Stone, J.C.; van Aelst, L.; Serrano, M.; Lowe, S.W. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998, 12, 3008–3019. [Google Scholar] [CrossRef]
- Hurlin, P.J. Control of vertebrate development by MYC. Cold Spring Harb. Perspect. Med. 2013, 3, a014332. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Jung, P.; Menssen, A.; Mayr, D.; Hermeking, H. AP4 encodes a c-MYC-inducible repressor of p21. Proc. Natl. Acad. Sci. USA 2008, 105, 15046–15051. [Google Scholar] [CrossRef]
- Wong, M.M.; Liu, M.F.; Chiu, S.K. Cropped, Drosophila transcription factor AP-4, controls tracheal terminal branching and cell growth. BMC Dev. Biol. 2015, 15, 20. [Google Scholar] [CrossRef]
- Jackstadt, R.; Jung, P.; Hermeking, H. AP4 directly downregulates p16 and p21 to suppress senescence and mediate transformation. Cell Death Dis. 2013, 4, e775. [Google Scholar] [CrossRef]
- Jackstadt, R.; Hermeking, H. AP4 is required for mitogen- and c-MYC-induced cell cycle progression. Oncotarget 2014, 5, 7316–7327. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, B.; Guo, Y.; Liang, Q.; Wu, C.; Wu, L.; Tao, K.; Wang, G.; Chen, J. Down-regulation of AP-4 inhibits proliferation, induces cell cycle arrest and promotes apoptosis in human gastric cancer cells. PLoS ONE 2012, 7, e37096. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Guo, W.; Chen, S.; Xu, Y.; Li, P.; Wang, H.; Chu, H.; Li, J.; Du, Y.; Chen, X.; et al. Silencing of AP-4 inhibits proliferation, induces cell cycle arrest and promotes apoptosis in human lung cancer cells. Oncol. Lett. 2016, 11, 3735–3742. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Meng, Y.; Xu, J.J.; Zhang, Q.Y. The Transcription Factor AP4 Promotes Oncogenic Phenotypes and Cisplatin Resistance by Regulating LAPTM4B Expression. Mol. Cancer Res. 2018, 16, 857–868. [Google Scholar] [CrossRef]
- Tsujimoto, K.; Ono, T.; Sato, M.; Nishida, T.; Oguma, T.; Tadakuma, T. Regulation of the expression of caspase-9 by the transcription factor activator protein-4 in glucocorticoid-induced apoptosis. J. Biol. Chem. 2005, 280, 27638–27644. [Google Scholar] [CrossRef]
- D’Annibale, S.; Kim, J.; Magliozzi, R.; Low, T.Y.; Mohammed, S.; Heck, A.J.; Guardavaccaro, D. Proteasome-dependent degradation of transcription factor activating enhancer-binding protein 4 (TFAP4) controls mitotic division. J. Biol. Chem. 2014, 289, 7730–7737. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, X.; Samma, M.K.; Kung, S.K.P.; Lee, C.M.; Chiu, S.K. Differential cellular responses by oncogenic levels of c-Myc expression in long-term confluent retinal pigment epithelial cells. Mol. Cell. Biochem. 2018, 443, 193–204. [Google Scholar] [CrossRef]
- Stern, J.; Temple, S. Retinal pigment epithelial cell proliferation. Exp. Biol. Med. 2015, 240, 1079–1086. [Google Scholar] [CrossRef]
- Wang, Y.; Wong, M.M.; Zhang, X.; Chiu, S.K. Ectopic AP4 expression induces cellular senescence via activation of p53 in long-term confluent retinal pigment epithelial cells. Exp. Cell Res. 2015, 339, 135–146. [Google Scholar] [CrossRef]
- Singer, A.; Adoro, S.; Park, J.H. Lineage fate and intense debate: Myths, models and mechanisms of CD4-versus CD8-lineage choice. Nat. Rev. Immunol. 2008, 8, 788–801. [Google Scholar] [CrossRef]
- Carpenter, A.C.; Bosselut, R. Decision checkpoints in the thymus. Nat. Immunol. 2010, 11, 666–673. [Google Scholar] [CrossRef]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef]
- Issuree, P.D.A.; Ng, C.P.; Littman, D.R. Heritable Gene Regulation in the CD4:CD8 T Cell Lineage Choice. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Egawa, T.; Littman, D.R. Transcription factor AP4 modulates reversible and epigenetic silencing of the Cd4 gene. Proc. Natl. Acad. Sci. USA 2011, 108, 14873–14878. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.; Verbaro, D.J.; Tonc, E.; Holmgren, M.; Cella, M.; Colonna, M.; Bhattacharya, D.; Egawa, T. The Transcription Factor AP4 Mediates Resolution of Chronic Viral Infection through Amplification of Germinal Center B Cell Responses. Immunity 2016, 45, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.; Pinto, A.K.; Curtis, J.D.; Persaud, S.P.; Cella, M.; Lin, C.C.; Edelson, B.T.; Allen, P.M.; Colonna, M.; Pearce, E.L.; et al. c-Myc-induced transcription factor AP4 is required for host protection mediated by CD8+ T cells. Nat. Immunol. 2014, 15, 884–893. [Google Scholar] [CrossRef]
- Cyster, J.G.; Allen, C.D.C. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 2019, 177, 524–540. [Google Scholar] [CrossRef]
- De Silva, N.S.; Klein, U. Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 2015, 15, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.D.; Okada, T.; Cyster, J.G. Germinal-center organization and cellular dynamics. Immunity 2007, 27, 190–202. [Google Scholar] [CrossRef]
- Cao, J.; Tang, M.; Li, W.L.; Xie, J.; Du, H.; Tang, W.B.; Wang, H.; Chen, X.W.; Xiao, H.; Li, Y. Upregulation of activator protein-4 in human colorectal cancer with metastasis. Int. J. Surg. Pathol. 2009, 17, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Friess, H.; Ding, J.; Kleeff, J.; Fenkell, L.; Rosinski, J.A.; Guweidhi, A.; Reidhaar-Olson, J.F.; Korc, M.; Hammer, J.; Buchler, M.W. Microarray-based identification of differentially expressed growth- and metastasis-associated genes in pancreatic cancer. Cell. Mol. Life Sci. 2003, 60, 1180–1199. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cai, Q.; He, W.; Lam, T.B.; Lin, J.; Zhao, Y.; Chen, X.; Gu, P.; Huang, H.; Xue, M.; et al. AP4 modulated by the PI3K/AKT pathway promotes prostate cancer proliferation and metastasis of prostate cancer via upregulating L-plastin. Cell Death Dis. 2017, 8, e3060. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Ao, X.; Liu, Y.; Ding, D.; Jiao, W.J.; Yu, Z.; Zhai, W.X.; Dong, S.H.; He, Y.Q.; Guo, H.; et al. MicroRNA-608 Promotes Apoptosis in Non-Small Cell Lung Cancer Cells Treated With Doxorubicin Through the Inhibition of TFAP4. Front. Genet. 2019, 10, 809. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Xie, C.; Jiang, L.; Wu, G.; Zhu, J.; Zhang, S.; Tang, M.; Song, L.; Li, J. Transcription factor AP-4 promotes tumorigenic capability and activates the Wnt/beta-catenin pathway in hepatocellular carcinoma. Theranostics 2018, 8, 3571–3583. [Google Scholar] [CrossRef]
- Yang, J.; Ma, J.P.; Xiao, S.; Zhang, X.H.; Xu, J.B.; Chen, C.Q.; Cai, S.R.; He, Y.L. Evaluating the prognostic value and functional roles of transcription factor AP4 in colorectal cancer. Oncol. Lett. 2018, 15, 7545–7554. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Chen, Q.F.; Chang, B.Y.; Shen, L.J.; Li, W.; Wu, P.H.; Fan, W.J. TFAP4 Promotes Hepatocellular Carcinoma Invasion and Metastasis via Activating the PI3K/AKT Signaling Pathway. Dis. Markers 2019, 2019, 7129214. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Liu, B.; Li, J.; Jiang, J.; Zhou, R.; Huang, L.; Li, X.; He, X.; Zhou, Q. MicroRNA-302c represses epithelial-mesenchymal transition and metastasis by targeting transcription factor AP-4 in colorectal cancer. Biomed. Pharmacother. 2018, 105, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Jaeckel, S.; Kaller, M.; Jackstadt, R.; Gotz, U.; Muller, S.; Boos, S.; Horst, D.; Jung, P.; Hermeking, H. Ap4 is rate limiting for intestinal tumor formation by controlling the homeostasis of intestinal stem cells. Nat. Commun. 2018, 9, 3573. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chiu, S.K. AP4 activates cell migration and EMT mediated by p53 in MDA-MB-231 breast carcinoma cells. Mol. Cell. Biochem. 2015, 407, 57–68. [Google Scholar] [CrossRef]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef]
- Xue, C.; Yu, D.M.; Gherardi, S.; Koach, J.; Milazzo, G.; Gamble, L.; Liu, B.; Valli, E.; Russell, A.J.; London, W.B.; et al. MYCN promotes neuroblastoma malignancy by establishing a regulatory circuit with transcription factor AP4. Oncotarget 2016, 7, 54937–54951. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fears, C.Y.; Woods, A. The role of syndecans in disease and wound healing. Matrix Biol. 2006, 25, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yaccoby, S.; Liu, W.; Langford, J.K.; Pumphrey, C.Y.; Theus, A.; Epstein, J.; Sanderson, R.D. Soluble syndecan-1 promotes growth of myeloma tumors in vivo. Blood 2002, 100, 610–617. [Google Scholar] [CrossRef]
- Shi, S.; Zhong, D.; Xiao, Y.; Wang, B.; Wang, W.; Zhang, F.; Huang, H. Syndecan-1 knockdown inhibits glioma cell proliferation and invasion by deregulating a c-src/FAK-associated signaling pathway. Oncotarget 2017, 8, 40922–40934. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Moreno, M.V.; Lodi, A.; Ronen, S.M.; Ruggero, D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell 2014, 157, 1088–1103. [Google Scholar] [CrossRef]
- Miao, W.; Wang, Y. Targeted Quantitative Kinome Analysis Identifies PRPS2 as a Promoter for Colorectal Cancer Metastasis. J. Proteome Res. 2019, 18, 2279–2286. [Google Scholar] [CrossRef]
- Qiao, H.; Tan, X.; Lv, D.J.; Xing, R.W.; Shu, F.P.; Zhong, C.F.; Li, C.; Zou, Y.G.; Mao, X.M. Phosphoribosyl pyrophosphate synthetases 2 knockdown inhibits prostate cancer progression by suppressing cell cycle and inducing cell apoptosis. J. Cancer 2020, 11, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, C.; Liu, X.; Zheng, J.; Zhang, F.; Wang, D.; Xue, Y.; Li, X.; Shen, S.; Shao, L.; et al. Transcription factor AP-4 (TFAP4)-upstream ORF coding 66 aa inhibits the malignant behaviors of glioma cells by suppressing the TFAP4/long noncoding RNA 00520/microRNA-520f-3p feedback loop. Cancer Sci. 2020, 111, 891–906. [Google Scholar] [CrossRef]
- Shi, L.; Jackstadt, R.; Siemens, H.; Li, H.; Kirchner, T.; Hermeking, H. p53-induced miR-15a/16-1 and AP4 form a double-negative feedback loop to regulate epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer Res. 2014, 74, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Xinghua, L.; Bo, Z.; Yan, G.; Lei, W.; Changyao, W.; Qi, L.; Lin, Y.; Kaixiong, T.; Guobin, W.; Jianying, C. The overexpression of AP-4 as a prognostic indicator for gastric carcinoma. Med. Oncol. 2012, 29, 871–877. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wicha, M.S. Targeting breast cancer stem cells. J. Clin. Oncol. 2010, 28, 4006–4012. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Lewis, M.T. Identifying and targeting tumor-initiating cells in the treatment of breast cancer. Endocr. Relat. Cancer 2015, 22, R135–R155. [Google Scholar] [CrossRef] [PubMed]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef]
- Li, S.; Li, Q. Cancer stem cells and tumor metastasis (Review). Int. J. Oncol. 2014, 44, 1806–1812. [Google Scholar] [CrossRef]
- Suraneni, M.V.; Badeaux, M.D. Tumor-Initiating Cells, Cancer Metastasis and Therapeutic Implications. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Boboila, S.; Lopez, G.; Yu, J.; Banerjee, D.; Kadenhe-Chiweshe, A.; Connolly, E.P.; Kandel, J.J.; Rajbhandari, P.; Silva, J.M.; Califano, A.; et al. Transcription factor activating protein 4 is synthetically lethal and a master regulator of MYCN-amplified neuroblastoma. Oncogene 2018, 37, 5451–5465. [Google Scholar] [CrossRef]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, M.; Tsiftsoglou, S.A.; Polyzos, A.P.; Papadopoulou, D.; Valakos, D.; Klagkou, E.; Karagianni, P.; Pliatska, M.; Talianidis, I.; Agelopoulos, M.; et al. Identification of a dynamic gene regulatory network required for pluripotency factor-induced reprogramming of mouse fibroblasts and hepatocytes. EMBO J. 2021, 40, e102236. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Lasda, E.; Parker, R. Circular RNAs: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Bottoni, A.; Shimizu, M.; Spizzo, R.; Nicoloso, M.S.; Rossi, S.; Barbarotto, E.; Cimmino, A.; Adair, B.; Wojcik, S.E.; et al. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA 2011, 305, 59–67. [Google Scholar] [CrossRef]
- Wu, H.; Liu, X.; Gong, P.; Song, W.; Zhou, M.; Li, Y.; Zhao, Z.; Fan, H. Elevated TFAP4 regulates lncRNA TRERNA1 to promote cell migration and invasion in gastric cancer. Oncol. Rep. 2018, 40, 923–931. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Wang, R.; Sun, Q.; Wang, P.; Liu, M.; Xiong, S.; Luo, J.; Huang, H.; Du, Q.; Geller, D.A.; Cheng, B. Notch and Wnt/beta-catenin signaling pathway play important roles in activating liver cancer stem cells. Oncotarget 2016, 7, 5754–5768. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Li, X.Y.; Hu, C.Y.; Ford, M.; Kleer, C.G.; Weiss, S.J. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc. Natl. Acad. Sci. USA 2012, 109, 16654–16659. [Google Scholar] [CrossRef]
- Wang, Y.; Bu, F.; Royer, C.; Serres, S.; Larkin, J.R.; Soto, M.S.; Sibson, N.R.; Salter, V.; Fritzsche, F.; Turnquist, C.; et al. ASPP2 controls epithelial plasticity and inhibits metastasis through beta-catenin-dependent regulation of ZEB1. Nat. Cell Biol. 2014, 16, 1092–1104. [Google Scholar] [CrossRef]
- Zirkel, A.; Lederer, M.; Stohr, N.; Pazaitis, N.; Huttelmaier, S. IGF2BP1 promotes mesenchymal cell properties and migration of tumor-derived cells by enhancing the expression of LEF1 and SNAI2 (SLUG). Nucleic Acids Res. 2013, 41, 6618–6636. [Google Scholar] [CrossRef]
- Liang, J.; Li, X.; Li, Y.; Wei, J.; Daniels, G.; Zhong, X.; Wang, J.; Sfanos, K.; Melamed, J.; Zhao, J.; et al. LEF1 targeting EMT in prostate cancer invasion is mediated by miR-181a. Am. J. Cancer Res. 2015, 5, 1124–1132. [Google Scholar] [PubMed]
- Xu, W.; Yang, Z.; Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Picksley, S.M.; Lane, D.P. The p53-mdm2 autoregulatory feedback loop: A paradigm for the regulation of growth control by p53? Bioessays 1993, 15, 689–690. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Hong, S.H.; Kim, H.S.; Shin, C.Y.; Nam, S.W.; Choi, W.S.; Han, J.W.; You, J.S. JQ1, an inhibitor of the epigenetic reader BRD4, suppresses the bidirectional MYC-AP4 axis via multiple mechanisms. Oncol. Rep. 2016, 35, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.; Potier, D.; Romanelli, L.; Jacobs, J.; Mach, J.; Hamaratoglu, F.; Aerts, S.; Halder, G. An Ectopic Network of Transcription Factors Regulated by Hippo Signaling Drives Growth and Invasion of a Malignant Tumor Model. Curr. Biol. 2016, 26, 2101–2113. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, M.M.-K.; Joyson, S.M.; Hermeking, H.; Chiu, S.K. Transcription Factor AP4 Mediates Cell Fate Decisions: To Divide, Age, or Die. Cancers 2021, 13, 676. https://doi.org/10.3390/cancers13040676
Wong MM-K, Joyson SM, Hermeking H, Chiu SK. Transcription Factor AP4 Mediates Cell Fate Decisions: To Divide, Age, or Die. Cancers. 2021; 13(4):676. https://doi.org/10.3390/cancers13040676
Chicago/Turabian StyleWong, Matthew Man-Kin, Sancy Mary Joyson, Heiko Hermeking, and Sung Kay Chiu. 2021. "Transcription Factor AP4 Mediates Cell Fate Decisions: To Divide, Age, or Die" Cancers 13, no. 4: 676. https://doi.org/10.3390/cancers13040676
APA StyleWong, M. M.-K., Joyson, S. M., Hermeking, H., & Chiu, S. K. (2021). Transcription Factor AP4 Mediates Cell Fate Decisions: To Divide, Age, or Die. Cancers, 13(4), 676. https://doi.org/10.3390/cancers13040676