
PRKAR1A and Thyroid Tumors




Abstract
Simple Summary
Abstract
1. Introduction
1.1. Incidence of Thyroid Cancer
1.2. Subtypes of Thyroid Cancer
1.3. Evaluation of a Thyroid Nodule
1.4. Thyroid Cancer as Part of Genetic Syndromes
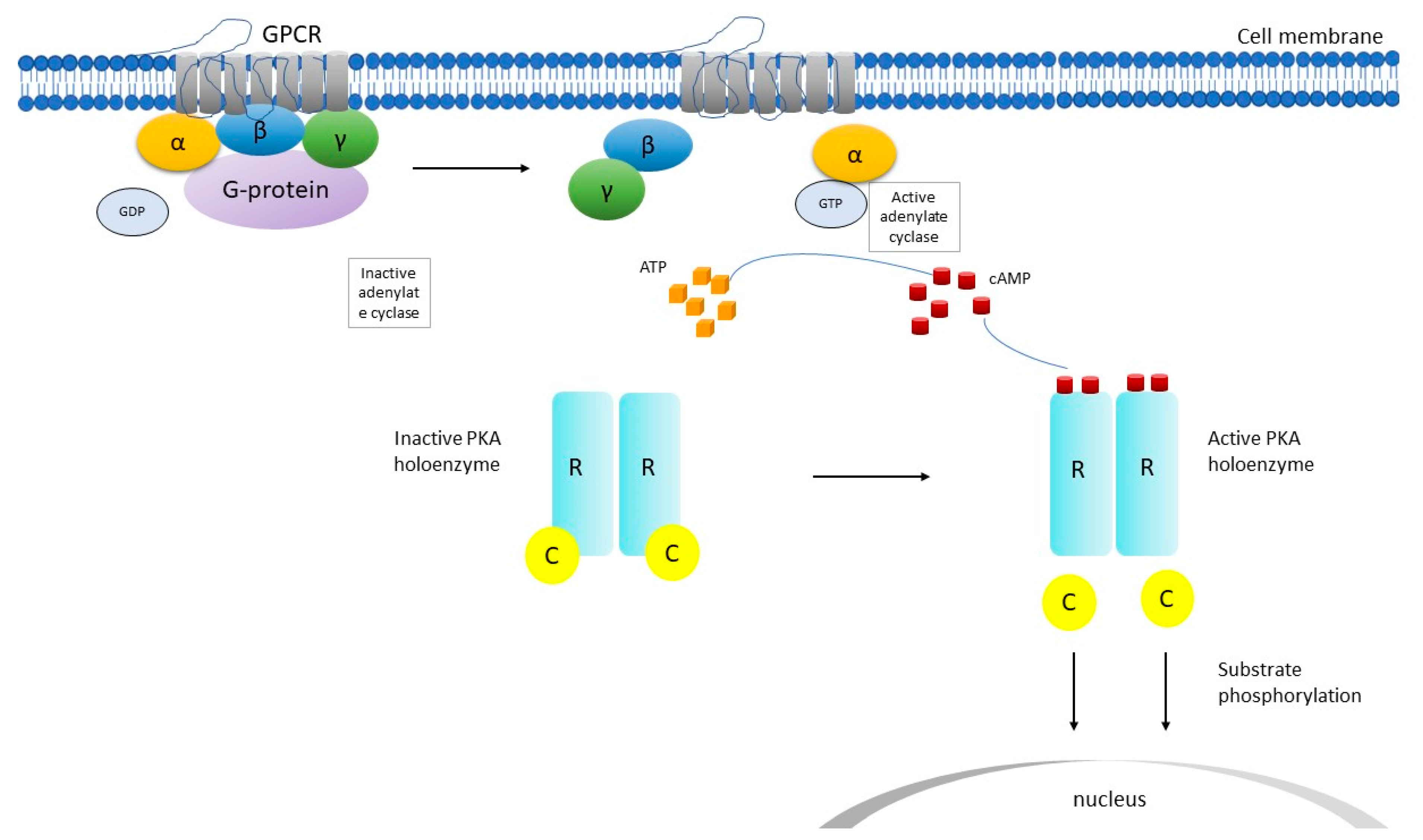
2. PRKAR1A Structure and Function
3. Role of PRKAR1A in Thyroid Cancer
3.1. Mouse Studies
3.1.1. Mouse Models
3.1.2. Activation of mTOR Pathway
3.1.3. Targeting Downstream Effectors of cAMP
3.2. Studies in Humans
4. Other Molecular Events in Thyroid Cancer
5. Medullary Thyroid Cancer as Part of MEN2 Syndromes
6. Anaplastic Thyroid Carcinoma
7. Systemic Treatments for Thyroid Cancer
7.1. Immunotherapy
7.2. Treatment for PRKAR1A-Associated Thyroid Tumors
8. Clinical Surveillance in Patients with PRKAR1A-Associated Thyroid Tumors
9. Conclusions
Funding
Conflicts of Interest
References
- Pringle, D.R.; Vasko, V.V.; Yu, L.; Manchanda, P.K.; Lee, A.A.; Zhang, X.; Kirschner, J.M.; Parlow, A.F.; Saji, M.; Jarjoura, D.; et al. Follicular thyroid cancers demonstrate dual activation of PKA and mTOR as modeled by thyroid-specific deletion of Prkar1a and Pten in mice. J. Clin. Endocrinol. Metab. 2014, 99, E804–E812. [Google Scholar] [CrossRef]
- Eng, C.; Mulligan, L.M.; Smith, D.P.; Healey, C.S.; Frilling, A.; Raue, F.; Neumann, H.P.; Ponder, M.A.; Ponder, B.A. Low frequency of germline mutations in the RET proto-oncogene in patients with apparently sporadic medullary thyroid carcinoma. Clin. Endocrinol. 1995, 43, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Raman, P.; Koenig, R.J. Pax-8-PPAR-gamma fusion protein in thyroid carcinoma. Nat. Rev. Endocrinol. 2014, 10, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Younis, E. Oncogenesis of Thyroid Cancer. Asian Pac. J. Cancer Prev. 2017, 18, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Besic, N.; Auersperg, M.; Golouh, R. Prognostic factors in follicular carcinoma of the thyroid—A multivariate survival analysis. Eur. J. Surg. Oncol. 1999, 25, 599–605. [Google Scholar] [CrossRef]
- Nguyen, X.V.; Roy Choudhury, K.; Tessler, F.N.; Hoang, J.K. Effect of Tumor Size on Risk of Metastatic Disease and Survival for Thyroid Cancer: Implications for Biopsy Guidelines. Thyroid 2018, 28, 295–300. [Google Scholar] [CrossRef]
- Sugino, K.; Ito, K.; Nagahama, M.; Kitagawa, W.; Shibuya, H.; Ohkuwa, K.; Yano, Y.; Uruno, T.; Akaishi, J.; Kameyama, K.; et al. Prognosis and prognostic factors for distant metastases and tumor mortality in follicular thyroid carcinoma. Thyroid 2011, 21, 751–757. [Google Scholar] [CrossRef]
- Burman, K.D.; Wartofsky, L. Clinical Practice. Thyroid Nodules. N. Engl. J. Med. 2015, 373, 2347–2356. [Google Scholar] [CrossRef]
- Yassa, L.; Cibas, E.S.; Benson, C.B.; Frates, M.C.; Doubilet, P.M.; Gawande, A.A.; Moore, F.D., Jr.; Kim, B.W.; Nose, V.; Marqusee, E.; et al. Long-term assessment of a multidisciplinary approach to thyroid nodule diagnostic evaluation. Cancer 2007, 111, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.S. American Thyroid Association Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2009, 19, 1167–1214. [Google Scholar] [CrossRef]
- Frates, M.C.; Benson, C.B.; Charboneau, J.W.; Cibas, E.S.; Clark, O.H.; Coleman, B.G.; Cronan, J.J.; Doubilet, P.M.; Evans, D.B.; Goellner, J.R.; et al. Management of thyroid nodules detected at US: Society of Radiologists in Ultrasound consensus conference statement. Radiology 2005, 237, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Oertel, Y.C.; Miyahara-Felipe, L.; Mendoza, M.G.; Yu, K. Value of repeated fine needle aspirations of the thyroid: An analysis of over ten thousand FNAs. Thyroid 2007, 17, 1061–1066. [Google Scholar] [CrossRef]
- Brander, A.; Viikinkoski, P.; Nickels, J.; Kivisaari, L. Thyroid gland: US screening in a random adult population. Radiology 1991, 181, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Papini, E.; Guglielmi, R.; Bianchini, A.; Crescenzi, A.; Taccogna, S.; Nardi, F.; Panunzi, C.; Rinaldi, R.; Toscano, V.; Pacella, C.M. Risk of malignancy in nonpalpable thyroid nodules: Predictive value of ultrasound and color-Doppler features. J. Clin. Endocrinol. Metab. 2002, 87, 1941–1946. [Google Scholar] [CrossRef]
- Carroll, B.A. Asymptomatic thyroid nodules: Incidental sonographic detection. AJR Am. J. Roentgenol. 1982, 138, 499–501. [Google Scholar] [CrossRef]
- Sclabas, G.M.; Staerkel, G.A.; Shapiro, S.E.; Fornage, B.D.; Sherman, S.I.; Vassillopoulou-Sellin, R.; Lee, J.E.; Evans, D.B. Fine-needle aspiration of the thyroid and correlation with histopathology in a contemporary series of 240 patients. Am. J. Surg. 2003, 186, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Greaves, T.S.; Olvera, M.; Florentine, B.D.; Raza, A.S.; Cobb, C.J.; Tsao-Wei, D.D.; Groshen, S.; Singer, P.; Lopresti, J.; Martin, S.E. Follicular lesions of thyroid: A 5-year fine-needle aspiration experience. Cancer 2000, 90, 335–341. [Google Scholar] [CrossRef]
- Tomimori, E.; Pedrinola, F.; Cavaliere, H.; Knobel, M.; Medeiros-Neto, G. Prevalence of incidental thyroid disease in a relatively low iodine intake area. Thyroid 1995, 5, 273–276. [Google Scholar] [CrossRef]
- Wiest, P.W.; Hartshorne, M.F.; Inskip, P.D.; Crooks, L.A.; Vela, B.S.; Telepak, R.J.; Williamson, M.R.; Blumhardt, R.; Bauman, J.M.; Tekkel, M. Thyroid palpation versus high-resolution thyroid ultrasonography in the detection of nodules. J. Ultrasound Med. 1998, 17, 487–496. [Google Scholar] [CrossRef]
- Burman, K.D.; Wartofsky, L. Thyroid Nodules. N. Engl. J. Med. 2016, 374, 1294–1295. [Google Scholar] [CrossRef] [PubMed]
- Nagy, R.; Ganapathi, S.; Comeras, I.; Peterson, C.; Orloff, M.; Porter, K.; Eng, C.; Ringel, M.D.; Kloos, R.T. Frequency of germline PTEN mutations in differentiated thyroid cancer. Thyroid 2011, 21, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Yeager, N.; Klein-Szanto, A.; Kimura, S.; Di Cristofano, A. Pten loss in the mouse thyroid causes goiter and follicular adenomas: Insights into thyroid function and Cowden disease pathogenesis. Cancer Res. 2007, 67, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Kirschner, L.S.; Carney, J.A. Clinical and molecular features of the Carney complex: Diagnostic criteria and recommendations for patient evaluation. J. Clin. Endocrinol. Metab. 2001, 86, 4041–4046. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, L.S.; Sandrini, F.; Monbo, J.; Lin, J.P.; Carney, J.A.; Stratakis, C.A. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the carney complex. Hum. Mol. Genet. 2000, 9, 3037–3046. [Google Scholar] [CrossRef]
- Sandrini, F.; Matyakhina, L.; Sarlis, N.J.; Kirschner, L.S.; Farmakidis, C.; Gimm, O.; Stratakis, C.A. Regulatory subunit type I-alpha of protein kinase A (PRKAR1A): A tumor-suppressor gene for sporadic thyroid cancer. Genes Chromosomes Cancer 2002, 35, 182–192. [Google Scholar] [CrossRef]
- Bertherat, J.; Groussin, L.; Sandrini, F.; Matyakhina, L.; Bei, T.; Stergiopoulos, S.; Papageorgiou, T.; Bourdeau, I.; Kirschner, L.S.; Vincent-Dejean, C.; et al. Molecular and functional analysis of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res. 2003, 63, 5308–5319. [Google Scholar]
- Bossis, I.; Voutetakis, A.; Bei, T.; Sandrini, F.; Griffin, K.J.; Stratakis, C.A. Protein kinase A and its role in human neoplasia: The Carney complex paradigm. Endocr. Relat. Cancer 2004, 11, 265–280. [Google Scholar] [CrossRef]
- Kirschner, L.S.; Carney, J.A.; Pack, S.D.; Taymans, S.E.; Giatzakis, C.; Cho, Y.S.; Cho-Chung, Y.S.; Stratakis, C.A. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat. Genet. 2000, 26, 89–92. [Google Scholar] [CrossRef]
- McKnight, G.S.; Clegg, C.H.; Uhler, M.D.; Chrivia, J.C.; Cadd, G.G.; Correll, L.A.; Otten, A.D. Analysis of the cAMP-dependent protein kinase system using molecular genetic approaches. Recent Prog. Horm. Res. 1988, 44, 307–335. [Google Scholar] [CrossRef]
- Cho-Chung, Y.S.; Pepe, S.; Clair, T.; Budillon, A.; Nesterova, M. cAMP-dependent protein kinase: Role in normal and malignant growth. Crit. Rev. Oncol. Hematol. 1995, 21, 33–61. [Google Scholar] [CrossRef]
- Skalhegg, B.S.; Tasken, K. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front. Biosci. 2000, 5, D678–D693. [Google Scholar] [CrossRef] [PubMed]
- Bossis, I.; Stratakis, C.A. Minireview: PRKAR1A: Normal and abnormal functions. Endocrinology 2004, 145, 5452–5458. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.Q.; Stratakis, C.A. How does cAMP/protein kinase A signaling lead to tumors in the adrenal cortex and other tissues? Mol. Cell Endocrinol. 2011, 336, 162–168. [Google Scholar] [CrossRef]
- Amieux, P.S.; McKnight, G.S. The essential role of RI alpha in the maintenance of regulated PKA activity. Ann. N. Y. Acad. Sci. 2002, 968, 75–95. [Google Scholar] [CrossRef]
- Cho-Chung, Y.S.; Nesterova, M.; Pepe, S.; Lee, G.R.; Noguchi, K.; Srivastava, R.K.; Srivastava, A.R.; Alper, O.; Park, Y.G.; Lee, Y.N. Antisense DNA-targeting protein kinase A-RIA subunit: A novel approach to cancer treatment. Front. Biosci. 1999, 4, D898–D907. [Google Scholar] [CrossRef][Green Version]
- Scott, J.D. Cyclic nucleotide-dependent protein kinases. Pharmacol. Ther. 1991, 50, 123–145. [Google Scholar] [CrossRef]
- Griffioen, G.; Thevelein, J.M. Molecular mechanisms controlling the localisation of protein kinase A. Curr. Genet. 2002, 41, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Colledge, M.; Scott, J.D. AKAPs: From structure to function. Trends Cell Biol. 1999, 9, 216–221. [Google Scholar] [CrossRef]
- Wang, L.; Sunahara, R.K.; Krumins, A.; Perkins, G.; Crochiere, M.L.; Mackey, M.; Bell, S.; Ellisman, M.H.; Taylor, S.S. Cloning and mitochondrial localization of full-length D-AKAP2, a protein kinase A anchoring protein. Proc. Natl. Acad. Sci. USA 2001, 98, 3220–3225. [Google Scholar] [CrossRef]
- Kirschner, L.S.; Kusewitt, D.F.; Matyakhina, L.; Towns, W.H., 2nd; Carney, J.A.; Westphal, H.; Stratakis, C.A. A mouse model for the Carney complex tumor syndrome develops neoplasia in cyclic AMP-responsive tissues. Cancer Res. 2005, 65, 4506–4514. [Google Scholar] [CrossRef]
- Robinson-White, A.; Hundley, T.R.; Shiferaw, M.; Bertherat, J.; Sandrini, F.; Stratakis, C.A. Protein kinase-A activity in PRKAR1A-mutant cells, and regulation of mitogen-activated protein kinases ERK1/2. Hum. Mol. Genet. 2003, 12, 1475–1484. [Google Scholar] [CrossRef]
- Haymart, M.R.; Repplinger, D.J.; Leverson, G.E.; Elson, D.F.; Sippel, R.S.; Jaume, J.C.; Chen, H. Higher serum thyroid stimulating hormone level in thyroid nodule patients is associated with greater risks of differentiated thyroid cancer and advanced tumor stage. J. Clin. Endocrinol. Metab. 2008, 93, 809–814. [Google Scholar] [CrossRef]
- Esapa, C.T.; Harris, P.E. Mutation analysis of protein kinase A catalytic subunit in thyroid adenomas and pituitary tumours. Eur. J. Endocrinol. 1999, 141, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Griffin, K.J.; Kirschner, L.S.; Matyakhina, L.; Stergiopoulos, S.G.; Robinson-White, A.; Lenherr, S.M.; Weinberg, F.D.; Claflin, E.S.; Batista, D.; Bourdeau, I.; et al. A transgenic mouse bearing an antisense construct of regulatory subunit type 1A of protein kinase A develops endocrine and other tumours: Comparison with Carney complex and other PRKAR1A induced lesions. J. Med. Genet. 2004, 41, 923–931. [Google Scholar] [CrossRef][Green Version]
- Pringle, D.R.; Yin, Z.; Lee, A.A.; Manchanda, P.K.; Yu, L.; Parlow, A.F.; Jarjoura, D.; La Perle, K.M.; Kirschner, L.S. Thyroid-specific ablation of the Carney complex gene, PRKAR1A, results in hyperthyroidism and follicular thyroid cancer. Endocr. Relat. Cancer 2012, 19, 435–446. [Google Scholar] [CrossRef][Green Version]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, M.; Lippincott-Schwartz, J.; Stratakis, C.A.; Bossis, I. mTOR kinase and the regulatory subunit of protein kinase A (PRKAR1A) spatially and functionally interact during autophagosome maturation. Autophagy 2007, 3, 151–153. [Google Scholar] [CrossRef]
- Day, M.E.; Gaietta, G.M.; Sastri, M.; Koller, A.; Mackey, M.R.; Scott, J.D.; Perkins, G.A.; Ellisman, M.H.; Taylor, S.S. Isoform-specific targeting of PKA to multivesicular bodies. J. Cell Biol. 2011, 193, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Blancquaert, S.; Wang, L.; Paternot, S.; Coulonval, K.; Dumont, J.E.; Harris, T.E.; Roger, P.P. cAMP-dependent activation of mammalian target of rapamycin (mTOR) in thyroid cells. Implication in mitogenesis and activation of CDK4. Mol. Endocrinol. 2010, 24, 1453–1468. [Google Scholar] [CrossRef][Green Version]
- Kari, S.; Vasko, V.V.; Priya, S.; Kirschner, L.S. PKA Activates AMPK Through LKB1 Signaling in Follicular Thyroid Cancer. Front. Endocrinol. 2019, 10, 769. [Google Scholar] [CrossRef] [PubMed]
- Jenne, D.E.; Reimann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Muller, O.; Back, W.; Zimmer, M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Hoglund, P.; et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef]
- Monteverde, T.; Muthalagu, N.; Port, J.; Murphy, D.J. Evidence of cancer-promoting roles for AMPK and related kinases. FEBS J. 2015, 282, 4658–4671. [Google Scholar] [CrossRef] [PubMed]
- Brunton, J.; Steele, S.; Ziehr, B.; Moorman, N.; Kawula, T. Feeding uninvited guests: mTOR and AMPK set the table for intracellular pathogens. PLoS Pathog. 2013, 9, e1003552. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.G.; Wang, L.; Li, W.; Li, J.Z.; Li, J. miR-143 inhibits oncogenic traits by degrading NUAK2 in glioblastoma. Int. J. Mol. Med. 2016, 37, 1627–1635. [Google Scholar] [CrossRef]
- Tang, L.; Tong, S.J.; Zhan, Z.; Wang, Q.; Tian, Y.; Chen, F. Expression of NUAK2 in gastric cancer tissue and its effects on the proliferation of gastric cancer cells. Exp. Ther. Med. 2017, 13, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Miranda, F.; Mannion, D.; Liu, S.; Zheng, Y.; Mangala, L.S.; Redondo, C.; Herrero-Gonzalez, S.; Xu, R.; Taylor, C.; Chedom, D.F.; et al. Salt-Inducible Kinase 2 Couples Ovarian Cancer Cell Metabolism with Survival at the Adipocyte-Rich Metastatic Niche. Cancer Cell 2016, 30, 273–289. [Google Scholar] [CrossRef]
- Hubaux, R.; Thu, K.L.; Vucic, E.A.; Pikor, L.A.; Kung, S.H.; Martinez, V.D.; Mosslemi, M.; Becker-Santos, D.D.; Gazdar, A.F.; Lam, S.; et al. Microtubule affinity-regulating kinase 2 is associated with DNA damage response and cisplatin resistance in non-small cell lung cancer. Int. J. Cancer 2015, 137, 2072–2082. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Goransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Makela, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef]
- Luo, Z.; Zang, M.; Guo, W. AMPK as a metabolic tumor suppressor: Control of metabolism and cell growth. Future Oncol. 2010, 6, 457–470. [Google Scholar] [CrossRef]
- Dasgupta, B.; Chhipa, R.R. Evolving Lessons on the Complex Role of AMPK in Normal Physiology and Cancer. Trends Pharmacol. Sci. 2016, 37, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M.; Hay, N. The dark face of AMPK as an essential tumor promoter. Cell Logist. 2012, 2, 197–202. [Google Scholar] [CrossRef]
- Saunders, M.P.; Salisbury, A.J.; O’Byrne, K.J.; Long, L.; Whitehouse, R.M.; Talbot, D.C.; Mawer, E.B.; Harris, A.L. A novel cyclic adenosine monophosphate analog induces hypercalcemia via production of 1,25-dihydroxyvitamin D in patients with solid tumors. J. Clin. Endocrinol. Metab. 1997, 82, 4044–4048. [Google Scholar] [CrossRef] [PubMed]
- Propper, D.J.; Saunders, M.P.; Salisbury, A.J.; Long, L.; O’Byrne, K.J.; Braybrooke, J.P.; Dowsett, M.; Taylor, M.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I study of the novel cyclic AMP (cAMP) analogue 8-chloro-cAMP in patients with cancer: Toxicity, hormonal, and immunological effects. Clin. Cancer Res. 1999, 5, 1682–1689. [Google Scholar]
- Huk, D.J.; Ashtekar, A.; Magner, A.; La Perle, K.; Kirschner, L.S. Deletion of Rap1b, but not Rap1a or Epac1, Reduces Protein Kinase A-Mediated Thyroid Cancer. Thyroid 2018, 28, 1153–1161. [Google Scholar] [CrossRef]
- Tsygankova, O.M.; Saavedra, A.; Rebhun, J.F.; Quilliam, L.A.; Meinkoth, J.L. Coordinated regulation of Rap1 and thyroid differentiation by cyclic AMP and protein kinase A. Mol. Cell. Biol. 2001, 21, 1921–1929. [Google Scholar] [CrossRef]
- Ribeiro-Neto, F.; Leon, A.; Urbani-Brocard, J.; Lou, L.; Nyska, A.; Altschuler, D.L. cAMP-dependent oncogenic action of Rap1b in the thyroid gland. J. Biol. Chem. 2004, 279, 46868–46875. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Saporito-Irwin, S.; DeClue, J.E.; Wienecke, R.; Guha, A. Alterations in the rap1 signaling pathway are common in human gliomas. Oncogene 1997, 15, 1611–1616. [Google Scholar] [CrossRef]
- Chevillard, S.; Ugolin, N.; Vielh, P.; Ory, K.; Levalois, C.; Elliott, D.; Clayman, G.L.; El-Naggar, A.K. Gene expression profiling of differentiated thyroid neoplasms: Diagnostic and clinical implications. Clin. Cancer Res. 2004, 10, 6586–6597. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Minato, N. Rap1 GTPase: Functions, regulation, and malignancy. J. Biochem. 2003, 134, 479–484. [Google Scholar] [CrossRef]
- Chen, C.H.; Chuang, H.C.; Huang, C.C.; Fang, F.M.; Huang, H.Y.; Tsai, H.T.; Su, L.J.; Shiu, L.Y.; Leu, S.; Chien, C.Y. Overexpression of Rap-1A indicates a poor prognosis for oral cavity squamous cell carcinoma and promotes tumor cell invasion via Aurora-A modulation. Am. J. Pathol. 2013, 182, 516–528. [Google Scholar] [CrossRef]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Dremier, S.; Milenkovic, M.; Blancquaert, S.; Dumont, J.E.; Doskeland, S.O.; Maenhaut, C.; Roger, P.P. Cyclic adenosine 3’,5’-monophosphate (cAMP)-dependent protein kinases, but not exchange proteins directly activated by cAMP (Epac), mediate thyrotropin/cAMP-dependent regulation of thyroid cells. Endocrinology 2007, 148, 4612–4622. [Google Scholar] [CrossRef][Green Version]
- Almahariq, M.; Tsalkova, T.; Mei, F.C.; Chen, H.; Zhou, J.; Sastry, S.K.; Schwede, F.; Cheng, X. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol. Pharmacol. 2013, 83, 122–128. [Google Scholar] [CrossRef]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. 2008, 40, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Luo, C.; Cheng, X.; Lu, M. Lithium and an EPAC-specific inhibitor ESI-09 synergistically suppress pancreatic cancer cell proliferation and survival. Acta Biochim. Biophys. Sin. 2017, 49, 573–580. [Google Scholar] [CrossRef]
- Takahashi, M.; Li, Y.; Dillon, T.J.; Stork, P.J. Phosphorylation of Rap1 by cAMP-dependent Protein Kinase (PKA) Creates a Binding Site for KSR to Sustain ERK Activation by cAMP. J. Biol. Chem. 2017, 292, 1449–1461. [Google Scholar] [CrossRef]
- Wang, Z.; Dillon, T.J.; Pokala, V.; Mishra, S.; Labudda, K.; Hunter, B.; Stork, P.J. Rap1-mediated activation of extracellular signal-regulated kinases by cyclic AMP is dependent on the mode of Rap1 activation. Mol. Cell. Biol. 2006, 26, 2130–2145. [Google Scholar] [CrossRef]
- Takahashi, M.; Dillon, T.J.; Liu, C.; Kariya, Y.; Wang, Z.; Stork, P.J. Protein kinase A-dependent phosphorylation of Rap1 regulates its membrane localization and cell migration. J. Biol. Chem. 2013, 288, 27712–27723. [Google Scholar] [CrossRef]
- Vogel, K.S.; Klesse, L.J.; Velasco-Miguel, S.; Meyers, K.; Rushing, E.J.; Parada, L.F. Mouse tumor model for neurofibromatosis type 1. Science 1999, 286, 2176–2179. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Miyoshi, H.; Kojima, Y.; Oshima, M.; Taketo, M.M. Accelerated onsets of gastric hamartomas and hepatic adenomas/carcinomas in Lkb1+/-p53-/- compound mutant mice. Oncogene 2006, 25, 1816–1820. [Google Scholar] [CrossRef][Green Version]
- Cichowski, K.; Shih, T.S.; Schmitt, E.; Santiago, S.; Reilly, K.; McLaughlin, M.E.; Bronson, R.T.; Jacks, T. Mouse models of tumor development in neurofibromatosis type 1. Science 1999, 286, 2172–2176. [Google Scholar] [CrossRef]
- Almeida, M.Q.; Muchow, M.; Boikos, S.; Bauer, A.J.; Griffin, K.J.; Tsang, K.M.; Cheadle, C.; Watkins, T.; Wen, F.; Starost, M.F.; et al. Mouse Prkar1a haploinsufficiency leads to an increase in tumors in the Trp53+/− or Rb1+/− backgrounds and chemically induced skin papillomas by dysregulation of the cell cycle and Wnt signaling. Hum. Mol. Genet. 2010, 19, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.A.; Gordon, H.; Carpenter, P.C.; Shenoy, B.V.; Go, V.L. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine 1985, 64, 270–283. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Courcoutsakis, N.A.; Abati, A.; Filie, A.; Doppman, J.L.; Carney, J.A.; Shawker, T. Thyroid gland abnormalities in patients with the syndrome of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas (Carney complex). J. Clin. Endocrinol. Metab. 1997, 82, 2037–2043. [Google Scholar] [CrossRef]
- Casey, M.; Vaughan, C.J.; He, J.; Hatcher, C.J.; Winter, J.M.; Weremowicz, S.; Montgomery, K.; Kucherlapati, R.; Morton, C.C.; Basson, C.T. Mutations in the protein kinase A R1alpha regulatory subunit cause familial cardiac myxomas and Carney complex. J. Clin. Investig. 2000, 106, R31–R38. [Google Scholar] [CrossRef] [PubMed]
- Dumont, J.E.; Jauniaux, J.C.; Roger, P.P. The cyclic AMP-mediated stimulation of cell proliferation. Trends Biochem. Sci. 1989, 14, 67–71. [Google Scholar] [CrossRef]
- Roger, P.; Taton, M.; Van Sande, J.; Dumont, J.E. Mitogenic effects of thyrotropin and adenosine 3’,5’-monophosphate in differentiated normal human thyroid cells in vitro. J. Clin. Endocrinol. Metab. 1988, 66, 1158–1165. [Google Scholar] [CrossRef]
- Hishinuma, A.; Yamanaka, T.; Kasai, K.; So, S.; Tseng, C.C.; Bamba, N.; Ohtake, H.; Shimoda, S. Different growth control of the two human thyroid cell lines of adenomatous goiter and papillary carcinoma. Thyroid 1995, 5, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Sarlis, N.J. Expression patterns of cellular growth-controlling genes in non-medullary thyroid cancer: Basic aspects. Rev. Endocr. Metab. Disord. 2000, 1, 183–196. [Google Scholar] [CrossRef]
- Eng, C. Familial papillary thyroid cancer--many syndromes, too many genes? J. Clin. Endocrinol. Metab. 2000, 85, 1755–1757. [Google Scholar] [CrossRef] [PubMed]
- van der Laan, B.F.; Freeman, J.L.; Asa, S.L. Expression of growth factors and growth factor receptors in normal and tumorous human thyroid tissues. Thyroid 1995, 5, 67–73. [Google Scholar] [CrossRef]
- Farid, N.R.; Zou, M.; Shi, Y. Genetics of follicular thyroid cancer. Endocrinol. Metab. Clin. N. Am. 1995, 24, 865–883. [Google Scholar] [CrossRef]
- Maenhaut, C.; Roger, P.P.; Reuse, S.; Dumont, J.E. Activation of the cyclic AMP cascade as an oncogenic mechanism: The thyroid example. Biochimie 1991, 73, 29–36. [Google Scholar] [CrossRef]
- Bertherat, J.; Horvath, A.; Groussin, L.; Grabar, S.; Boikos, S.; Cazabat, L.; Libe, R.; Rene-Corail, F.; Stergiopoulos, S.; Bourdeau, I.; et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5’-monophosphate-dependent protein kinase (PRKAR1A): Phenotype analysis in 353 patients and 80 different genotypes. J. Clin. Endocrinol. Metab. 2009, 94, 2085–2091. [Google Scholar] [CrossRef]
- Carney, J.A.; Lyssikatos, C.; Seethala, R.R.; Lakatos, P.; Perez-Atayde, A.; Lahner, H.; Stratakis, C.A. The Spectrum of Thyroid Gland Pathology in Carney Complex: The Importance of Follicular Carcinoma. Am. J. Surg. Pathol. 2018, 42, 587–594. [Google Scholar] [CrossRef]
- Adeniran, A.J.; Zhu, Z.; Gandhi, M.; Steward, D.L.; Fidler, J.P.; Giordano, T.J.; Biddinger, P.W.; Nikiforov, Y.E. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am. J. Surg. Pathol. 2006, 30, 216–222. [Google Scholar] [CrossRef]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer: Genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar] [PubMed]
- Soares, P.; Trovisco, V.; Rocha, A.S.; Lima, J.; Castro, P.; Preto, A.; Maximo, V.; Botelho, T.; Seruca, R.; Sobrinho-Simoes, M. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 2003, 22, 4578–4580. [Google Scholar] [CrossRef] [PubMed]
- Frattini, M.; Ferrario, C.; Bressan, P.; Balestra, D.; De Cecco, L.; Mondellini, P.; Bongarzone, I.; Collini, P.; Gariboldi, M.; Pilotti, S.; et al. Alternative mutations of BRAF, RET and NTRK1 are associated with similar but distinct gene expression patterns in papillary thyroid cancer. Oncogene 2004, 23, 7436–7440. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.; Espadinha, C.; Catarino, A.L.; Moniz, S.; Pereira, T.; Sobrinho, L.G.; Leite, V. Expression of PAX8-PPAR gamma 1 rearrangements in both follicular thyroid carcinomas and adenomas. J. Clin. Endocrinol. Metab. 2002, 87, 3947–3952. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Biddinger, P.W.; Caudill, C.M.; Kroll, T.G.; Nikiforov, Y.E. PAX8-PPARgamma rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. Am. J. Surg. Pathol. 2002, 26, 1016–1023. [Google Scholar] [CrossRef]
- Dwight, T.; Thoppe, S.R.; Foukakis, T.; Lui, W.O.; Wallin, G.; Hoog, A.; Frisk, T.; Larsson, C.; Zedenius, J. Involvement of the PAX8/peroxisome proliferator-activated receptor gamma rearrangement in follicular thyroid tumors. J. Clin. Endocrinol. Metab. 2003, 88, 4440–4445. [Google Scholar] [CrossRef] [PubMed]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W., 2nd; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: Evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef]
- Ezzat, S.; Zheng, L.; Kolenda, J.; Safarian, A.; Freeman, J.L.; Asa, S.L. Prevalence of activating ras mutations in morphologically characterized thyroid nodules. Thyroid 1996, 6, 409–416. [Google Scholar] [CrossRef]
- Karga, H.; Lee, J.K.; Vickery, A.L., Jr.; Thor, A.; Gaz, R.D.; Jameson, J.L. Ras oncogene mutations in benign and malignant thyroid neoplasms. J. Clin. Endocrinol. Metab. 1991, 73, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Namba, H.; Rubin, S.A.; Fagin, J.A. Point mutations of ras oncogenes are an early event in thyroid tumorigenesis. Mol. Endocrinol. 1990, 4, 1474–1479. [Google Scholar] [CrossRef]
- Manenti, G.; Pilotti, S.; Re, F.C.; Della Porta, G.; Pierotti, M.A. Selective activation of ras oncogenes in follicular and undifferentiated thyroid carcinomas. Eur. J. Cancer 1994, 30A, 987–993. [Google Scholar] [CrossRef]
- Motoi, N.; Sakamoto, A.; Yamochi, T.; Horiuchi, H.; Motoi, T.; Machinami, R. Role of ras mutation in the progression of thyroid carcinoma of follicular epithelial origin. Pathol. Res. Pract. 2000, 196, 1–7. [Google Scholar] [CrossRef]
- Esapa, C.T.; Johnson, S.J.; Kendall-Taylor, P.; Lennard, T.W.; Harris, P.E. Prevalence of Ras mutations in thyroid neoplasia. Clin. Endocrinol. 1999, 50, 529–535. [Google Scholar] [CrossRef]
- Suarez, H.G.; du Villard, J.A.; Severino, M.; Caillou, B.; Schlumberger, M.; Tubiana, M.; Parmentier, C.; Monier, R. Presence of mutations in all three ras genes in human thyroid tumors. Oncogene 1990, 5, 565–570. [Google Scholar]
- Zhu, Z.; Gandhi, M.; Nikiforova, M.N.; Fischer, A.H.; Nikiforov, Y.E. Molecular profile and clinical-pathologic features of the follicular variant of papillary thyroid carcinoma. An unusually high prevalence of ras mutations. Am. J. Clin. Pathol. 2003, 120, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rostan, G.; Zhao, H.; Camp, R.L.; Pollan, M.; Herrero, A.; Pardo, J.; Wu, R.; Carcangiu, M.L.; Costa, J.; Tallini, G. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J. Clin. Oncol. 2003, 21, 3226–3235. [Google Scholar] [CrossRef]
- Saavedra, H.I.; Knauf, J.A.; Shirokawa, J.M.; Wang, J.; Ouyang, B.; Elisei, R.; Stambrook, P.J.; Fagin, J.A. The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene 2000, 19, 3948–3954. [Google Scholar] [CrossRef]
- Basolo, F.; Pisaturo, F.; Pollina, L.E.; Fontanini, G.; Elisei, R.; Molinaro, E.; Iacconi, P.; Miccoli, P.; Pacini, F. N-ras mutation in poorly differentiated thyroid carcinomas: Correlation with bone metastases and inverse correlation to thyroglobulin expression. Thyroid 2000, 10, 19–23. [Google Scholar] [CrossRef]
- Fagin, J.A. Minireview: Branded from the start-distinct oncogenic initiating events may determine tumor fate in the thyroid. Mol. Endocrinol. 2002, 16, 903–911. [Google Scholar] [CrossRef][Green Version]
- Hou, P.; Liu, D.; Xing, M. Functional characterization of the T1799-1801del and A1799-1816ins BRAF mutations in papillary thyroid cancer. Cell Cycle 2007, 6, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Carta, C.; Moretti, S.; Passeri, L.; Barbi, F.; Avenia, N.; Cavaliere, A.; Monacelli, M.; Macchiarulo, A.; Santeusanio, F.; Tartaglia, M.; et al. Genotyping of an Italian papillary thyroid carcinoma cohort revealed high prevalence of BRAF mutations, absence of RAS mutations and allowed the detection of a new mutation of BRAF oncoprotein (BRAF(V599lns)). Clin. Endocrinol. 2006, 64, 105–109. [Google Scholar] [CrossRef]
- Trovisco, V.; Vieira de Castro, I.; Soares, P.; Maximo, V.; Silva, P.; Magalhaes, J.; Abrosimov, A.; Guiu, X.M.; Sobrinho-Simoes, M. BRAF mutations are associated with some histological types of papillary thyroid carcinoma. J. Pathol. 2004, 202, 247–251. [Google Scholar] [CrossRef]
- Begum, S.; Rosenbaum, E.; Henrique, R.; Cohen, Y.; Sidransky, D.; Westra, W.H. BRAF mutations in anaplastic thyroid carcinoma: Implications for tumor origin, diagnosis and treatment. Mod. Pathol. 2004, 17, 1359–1363. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Kimura, E.T.; Gandhi, M.; Biddinger, P.W.; Knauf, J.A.; Basolo, F.; Zhu, Z.; Giannini, R.; Salvatore, G.; Fusco, A.; et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J. Clin. Endocrinol. Metab. 2003, 88, 5399–5404. [Google Scholar] [CrossRef] [PubMed]
- Namba, H.; Nakashima, M.; Hayashi, T.; Hayashida, N.; Maeda, S.; Rogounovitch, T.I.; Ohtsuru, A.; Saenko, V.A.; Kanematsu, T.; Yamashita, S. Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J. Clin. Endocrinol. Metab. 2003, 88, 4393–4397. [Google Scholar] [CrossRef]
- Xing, M. BRAF mutation in thyroid cancer. Endocr. Relat. Cancer 2005, 12, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Seyama, T.; Mizuno, T.; Tsuyama, N.; Hayashi, T.; Hayashi, Y.; Dohi, K.; Nakamura, N.; Akiyama, M. Unique association of p53 mutations with undifferentiated but not with differentiated carcinomas of the thyroid gland. Cancer Res. 1992, 52, 1369–1371. [Google Scholar]
- Dobashi, Y.; Sugimura, H.; Sakamoto, A.; Mernyei, M.; Mori, M.; Oyama, T.; Machinami, R. Stepwise participation of p53 gene mutation during dedifferentiation of human thyroid carcinomas. Diagn. Mol. Pathol. 1994, 3, 9–14. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Tallini, G.; Herrero, A.; D’Aquila, T.G.; Carcangiu, M.L.; Rimm, D.L. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999, 59, 1811–1815. [Google Scholar]
- Kurihara, T.; Ikeda, S.; Ishizaki, Y.; Fujimori, M.; Tokumoto, N.; Hirata, Y.; Ozaki, S.; Okajima, M.; Sugino, K.; Asahara, T. Immunohistochemical and sequencing analyses of the Wnt signaling components in Japanese anaplastic thyroid cancers. Thyroid 2004, 14, 1020–1029. [Google Scholar] [CrossRef]
- Block, M.A.; Jackson, C.E.; Greenawald, K.A.; Yott, J.B.; Tashjian, A.H., Jr. Clinical characteristics distinguishing hereditary from sporadic medullary thyroid carcinoma. Treatment implications. Arch. Surg. 1980, 115, 142–148. [Google Scholar] [CrossRef]
- Kouvaraki, M.A.; Shapiro, S.E.; Perrier, N.D.; Cote, G.J.; Gagel, R.F.; Hoff, A.O.; Sherman, S.I.; Lee, J.E.; Evans, D.B. RET proto-oncogene: A review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid 2005, 15, 531–544. [Google Scholar] [CrossRef]
- Brandi, M.L.; Gagel, R.F.; Angeli, A.; Bilezikian, J.P.; Beck-Peccoz, P.; Bordi, C.; Conte-Devolx, B.; Falchetti, A.; Gheri, R.G.; Libroia, A.; et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J. Clin. Endocrinol. Metab. 2001, 86, 5658–5671. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Pardi, E.; Cetani, F.; Elisei, R. Genetic and clinical features of multiple endocrine neoplasia types 1 and 2. J. Oncol. 2012, 2012, 705036. [Google Scholar] [CrossRef] [PubMed]
- Wohllk, N.; Schweizer, H.; Erlic, Z.; Schmid, K.W.; Walz, M.K.; Raue, F.; Neumann, H.P. Multiple endocrine neoplasia type 2. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.; Clayton, D.; Schuffenecker, I.; Lenoir, G.; Cote, G.; Gagel, R.F.; van Amstel, H.K.; Lips, C.J.; Nishisho, I.; Takai, S.I.; et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2: International RET mutation consortium analysis. JAMA 1996, 276, 1575–1579. [Google Scholar] [CrossRef]
- Lodish, M.B.; Stratakis, C.A. RET oncogene in MEN2, MEN2B, MTC and other forms of thyroid cancer. Expert Rev. Anticancer Ther. 2008, 8, 625–632. [Google Scholar] [CrossRef]
- Wells, S.A., Jr. Advances in the management of MEN2: From improved surgical and medical treatment to novel kinase inhibitors. Endocr. Relat. Cancer 2018, 25, T1–T13. [Google Scholar] [CrossRef]
- Romei, C.; Ciampi, R.; Elisei, R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat. Rev. Endocrinol. 2016, 12, 192–202. [Google Scholar] [CrossRef]
- Mulligan, L.M. RET revisited: Expanding the oncogenic portfolio. Nat. Rev. Cancer 2014, 14, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Tacito, A.; Ramone, T.; Ciampi, R.; Bottici, V.; Cappagli, V.; Viola, D.; Matrone, A.; Lorusso, L.; Valerio, L.; et al. Twenty-Five Years Experience on RET Genetic Screening on Hereditary MTC: An Update on The Prevalence of Germline RET Mutations. Genes 2019, 10, 698. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Ugolini, C.; Cosci, B.; Torregrossa, L.; Vivaldi, A.; Ciampi, R.; Tacito, A.; Basolo, F.; Materazzi, G.; Miccoli, P.; et al. Low prevalence of the somatic M918T RET mutation in micro-medullary thyroid cancer. Thyroid 2012, 22, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Casella, F.; Tacito, A.; Bottici, V.; Valerio, L.; Viola, D.; Cappagli, V.; Matrone, A.; Ciampi, R.; Piaggi, P.; et al. New insights in the molecular signature of advanced medullary thyroid cancer: Evidence of a bad outcome of cases with double RET mutations. J. Med. Genet. 2016, 53, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, E.; Romei, C.; Biagini, A.; Sabini, E.; Agate, L.; Mazzeo, S.; Materazzi, G.; Sellari-Franceschini, S.; Ribechini, A.; Torregrossa, L.; et al. Anaplastic thyroid carcinoma: From clinicopathology to genetics and advanced therapies. Nat. Rev. Endocrinol. 2017, 13, 644–660. [Google Scholar] [CrossRef]
- Deeken-Draisey, A.; Yang, G.Y.; Gao, J.; Alexiev, B.A. Anaplastic thyroid carcinoma: An epidemiologic, histologic, immunohistochemical, and molecular single-institution study. Hum. Pathol. 2018, 82, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Wreesmann, V.B.; Ghossein, R.A.; Patel, S.G.; Harris, C.P.; Schnaser, E.A.; Shaha, A.R.; Tuttle, R.M.; Shah, J.P.; Rao, P.H.; Singh, B. Genome-wide appraisal of thyroid cancer progression. Am. J. Pathol. 2002, 161, 1549–1556. [Google Scholar] [CrossRef]
- Charles, R.P.; Silva, J.; Iezza, G.; Phillips, W.A.; McMahon, M. Activating BRAF and PIK3CA mutations cooperate to promote anaplastic thyroid carcinogenesis. Mol. Cancer Res. 2014, 12, 979–986. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Costa, A.M.; Pereira-Castro, I.; Salvatore, G.; Hernandez, R.; Hermsem, M.J.; Herrero, A.; Fusco, A.; Cameselle-Teijeiro, J.; Santoro, M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005, 65, 10199–10207. [Google Scholar] [CrossRef]
- Gauchotte, G.; Philippe, C.; Lacomme, S.; Leotard, B.; Wissler, M.P.; Allou, L.; Toussaint, B.; Klein, M.; Vignaud, J.M.; Bressenot, A. BRAF, p53 and SOX2 in anaplastic thyroid carcinoma: Evidence for multistep carcinogenesis. Pathology 2011, 43, 447–452. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Tiedje, V.; Ting, S.; Herold, T.; Synoracki, S.; Latteyer, S.; Moeller, L.C.; Zwanziger, D.; Stuschke, M.; Fuehrer, D.; Schmid, K.W. NGS based identification of mutational hotspots for targeted therapy in anaplastic thyroid carcinoma. Oncotarget 2017, 8, 42613–42620. [Google Scholar] [CrossRef]
- Baloch, Z.W.; LiVolsi, V.A. Special types of thyroid carcinoma. Histopathology 2018, 72, 40–52. [Google Scholar] [CrossRef]
- Aschebrook-Kilfoy, B.; Ward, M.H.; Sabra, M.M.; Devesa, S.S. Thyroid cancer incidence patterns in the United States by histologic type, 1992–2006. Thyroid 2011, 21, 125–134. [Google Scholar] [CrossRef]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, M.E.; Schlumberger, M.; Jarzab, B.; Martins, R.G.; Pacini, F.; Robinson, B.; McCaffrey, J.C.; Shah, M.H.; Bodenner, D.L.; Topliss, D.; et al. A phase 2 trial of lenvatinib (E7080) in advanced, progressive, radioiodine-refractory, differentiated thyroid cancer: A clinical outcomes and biomarker assessment. Cancer 2015, 121, 2749–2756. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Schlumberger, M.J.; Muller, S.P.; Schoffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef]
- Capdevila, J.; Wirth, L.J.; Ernst, T.; Ponce Aix, S.; Lin, C.C.; Ramlau, R.; Butler, M.O.; Delord, J.P.; Gelderblom, H.; Ascierto, P.A.; et al. PD-1 Blockade in Anaplastic Thyroid Carcinoma. J. Clin. Oncol. 2020, 38, 2620–2627. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Seufert, J.; Aumann, K.; Ruf, J.; Klein, C.; Kiefer, S.; Rassner, M.; Boerries, M.; Zielke, A.; la Rosee, P.; et al. Combination of Lenvatinib and Pembrolizumab Is an Effective Treatment Option for Anaplastic and Poorly Differentiated Thyroid Carcinoma. Thyroid 2021, 31, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Bouys, L.; Bertherat, J. Management of Endocrine Disease: Carney complex: Clinical and genetic update 20 years after the identification of the CNC1 (PRKAR1A) gene. Eur. J. Endocrinol. 2021, 184, R99–R109. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Raygada, M. Carney Complex. In GeneReviews(R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]


{kind=link}
| Main Diagnostic Criteria |
|---|
|
|
|
|
|
|
|
|
|
|
|
|
| Supplementary Criteria |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitsava, G.; Stratakis, C.A.; Faucz, F.R. PRKAR1A and Thyroid Tumors. Cancers 2021, 13, 3834. https://doi.org/10.3390/cancers13153834
Pitsava G, Stratakis CA, Faucz FR. PRKAR1A and Thyroid Tumors. Cancers. 2021; 13(15):3834. https://doi.org/10.3390/cancers13153834
Chicago/Turabian StylePitsava, Georgia, Constantine A. Stratakis, and Fabio R. Faucz. 2021. "PRKAR1A and Thyroid Tumors" Cancers 13, no. 15: 3834. https://doi.org/10.3390/cancers13153834
APA StylePitsava, G., Stratakis, C. A., & Faucz, F. R. (2021). PRKAR1A and Thyroid Tumors. Cancers, 13(15), 3834. https://doi.org/10.3390/cancers13153834