
Novel Cyclophilin Inhibitor Decreases Cell Proliferation and Tumor Growth in Models of Hepatocellular Carcinoma

 , , ,
, , ,  ,
,  ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Drugs
2.2. PPIase Activity Assay
2.3. Toxicity Studies
2.3.1. In Vitro
CellTiter Glo Studies
- Evaluation of PBMCs
- Evaluation of Human Monocytes-Macrophages
- Evaluation of Human Monocytes–Macrophages–Derived Dendritic Cells
- Evaluation of Primary Human Hepatocytes
- Evaluation of Human Bone Marrow Cells
- Evaluation of iPS Cardiomyocytes
- Evaluation of iPS Neurons
- Evaluation of RPTEC Kidney Cells
- CellTiter Glo
Primary Human Hepatocytes
2.3.2. In Vivo
2.4. Pharmacokinetic Studies
2.4.1. Single Oral Administration (PO)
2.4.2. Intravenous (IV) and Oral Administration (PO)
2.5. Cell Proliferation Assay: Acumen
2.6. Cell Proliferation and Viability Assay with Acridine Orange and DAPI
2.7. Clonogenic Assay
2.8. Fluorocytometry
2.8.1. Quantification of Mitochondrial Membrane Potential and Cell Permeability
2.8.2. Cell Cycle Analysis
2.8.3. Phospho–Histone 3 (PH3) Staining
2.8.4. Analysis
2.9. Immunofluorescence
Mitotic Quantification
2.10. In Vivo Efficacy
2.10.1. Chemicals
2.10.2. Cells and Mice
2.10.3. Treatment and Analysis
2.11. Statistical Analysis
3. Results
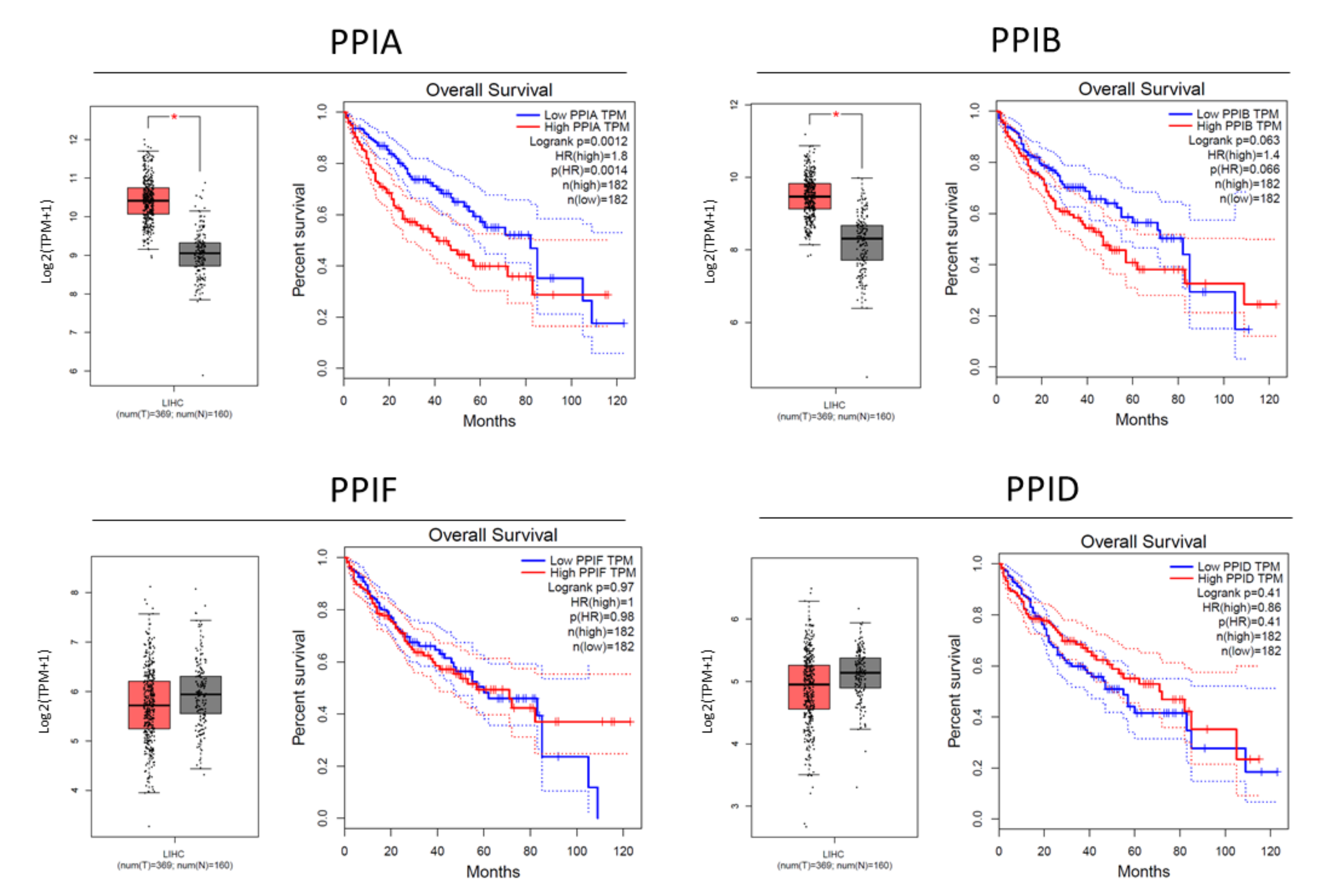
3.1. Cyclophilin Overexpression in HCC
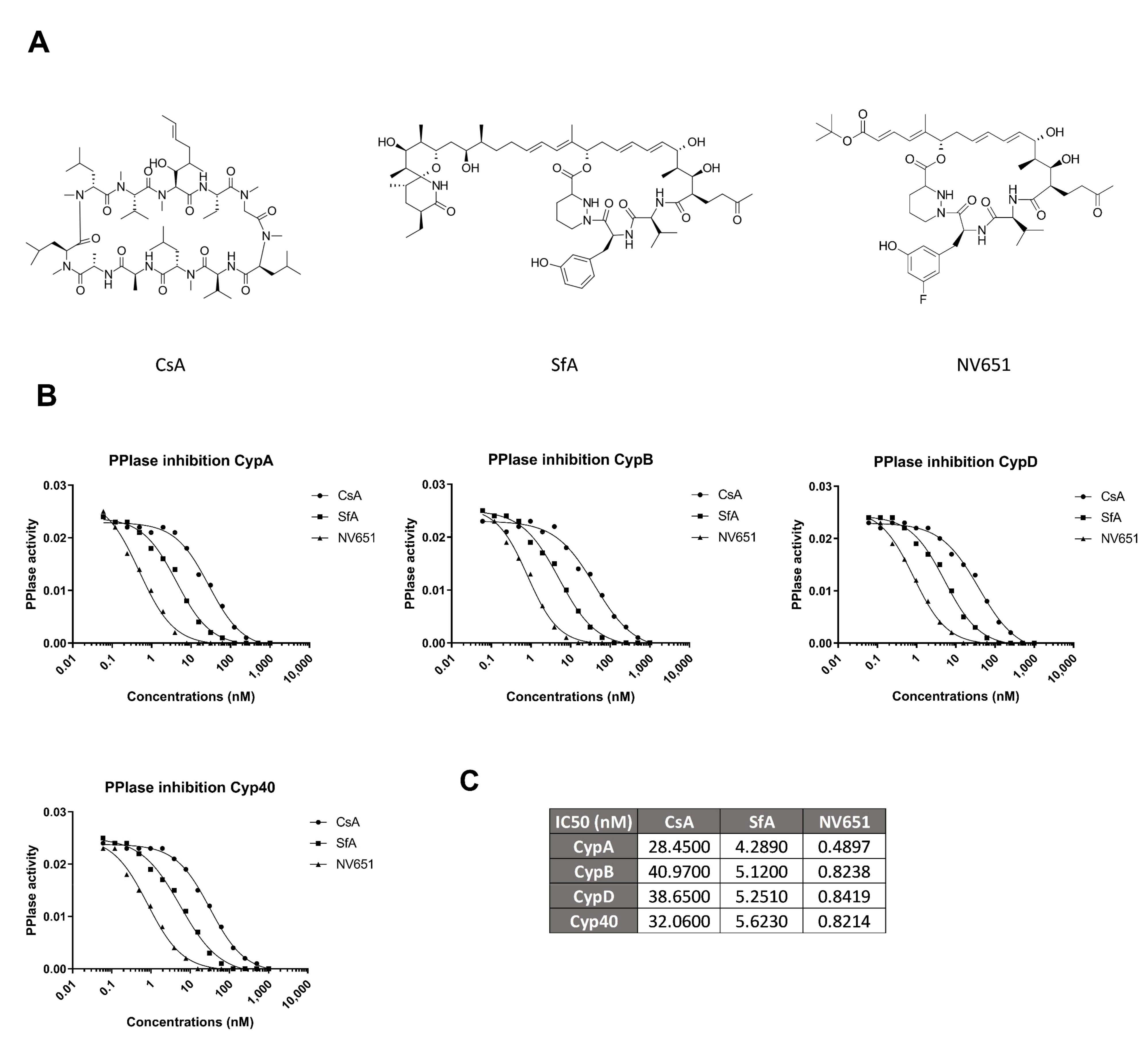
3.2. NV651 Is More Potent Than Common Cyclophilin Inhibitors at Reducing PPIase Activity of Cyclophilins
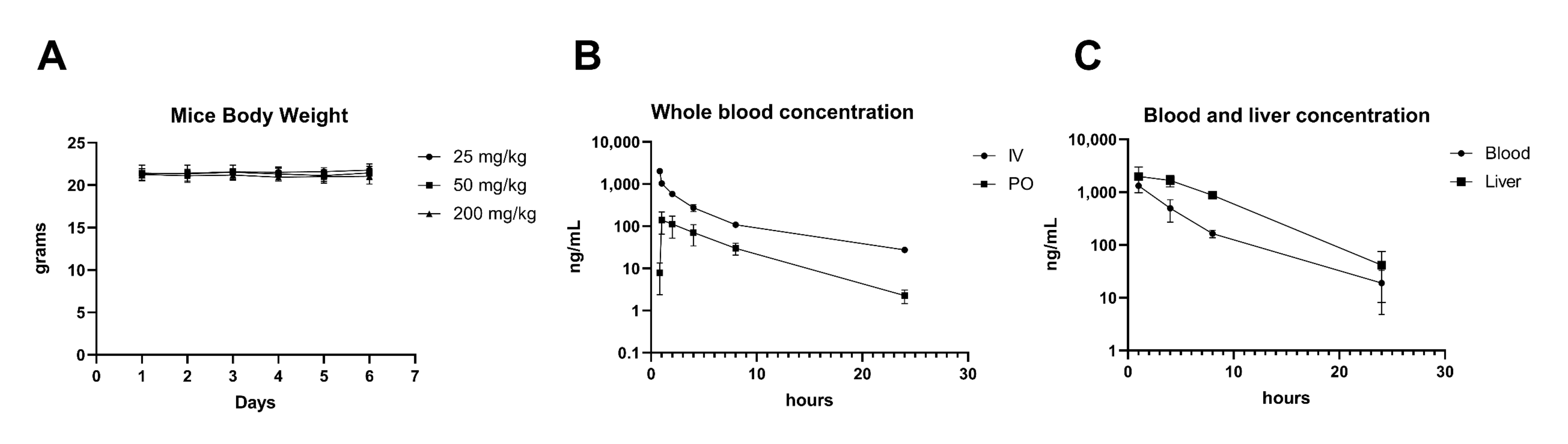
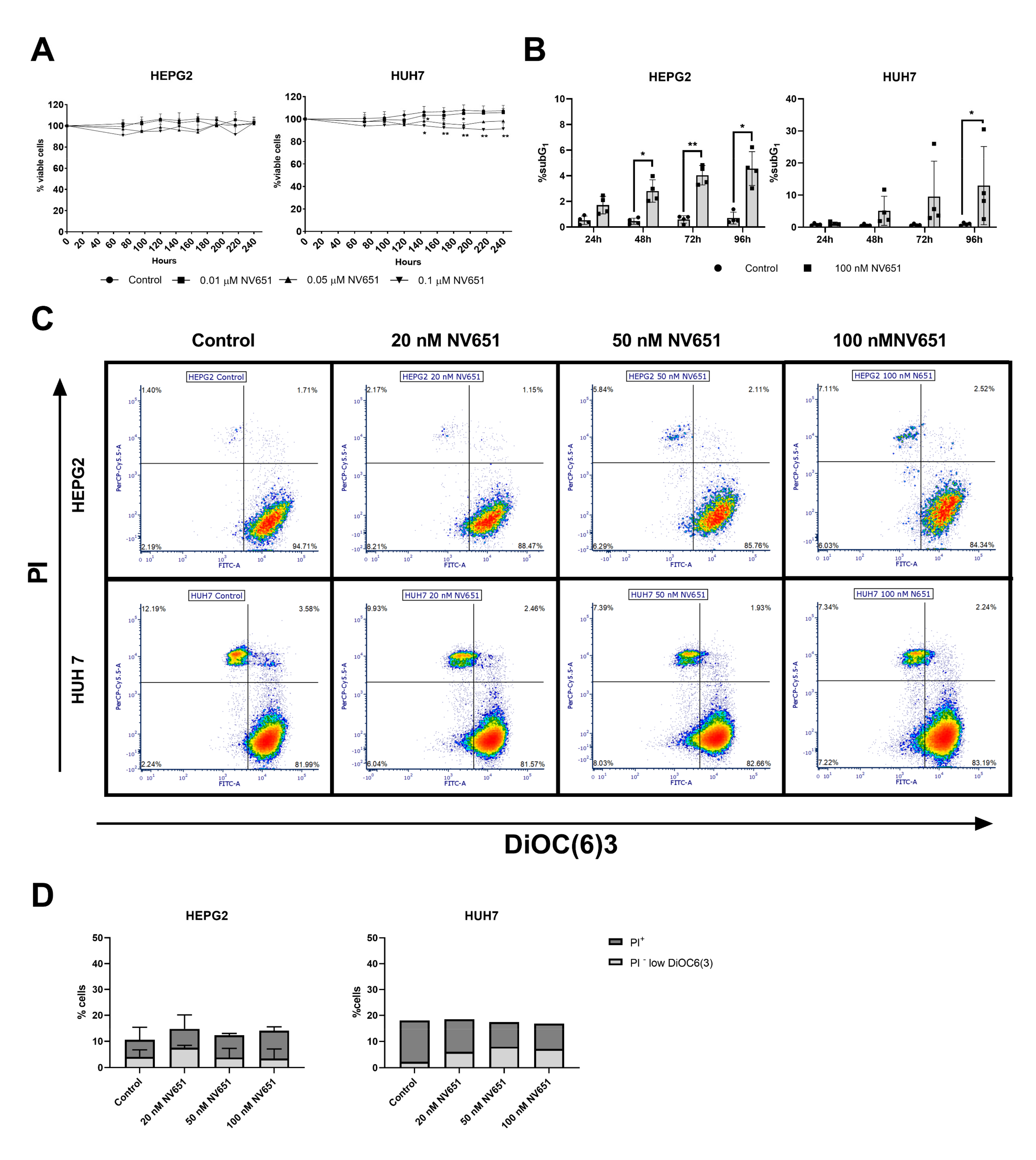
3.3. NV651 Showed Low Cytotoxic Activity against Normal Cell Types In Vitro and Promising In Vivo Tolerability
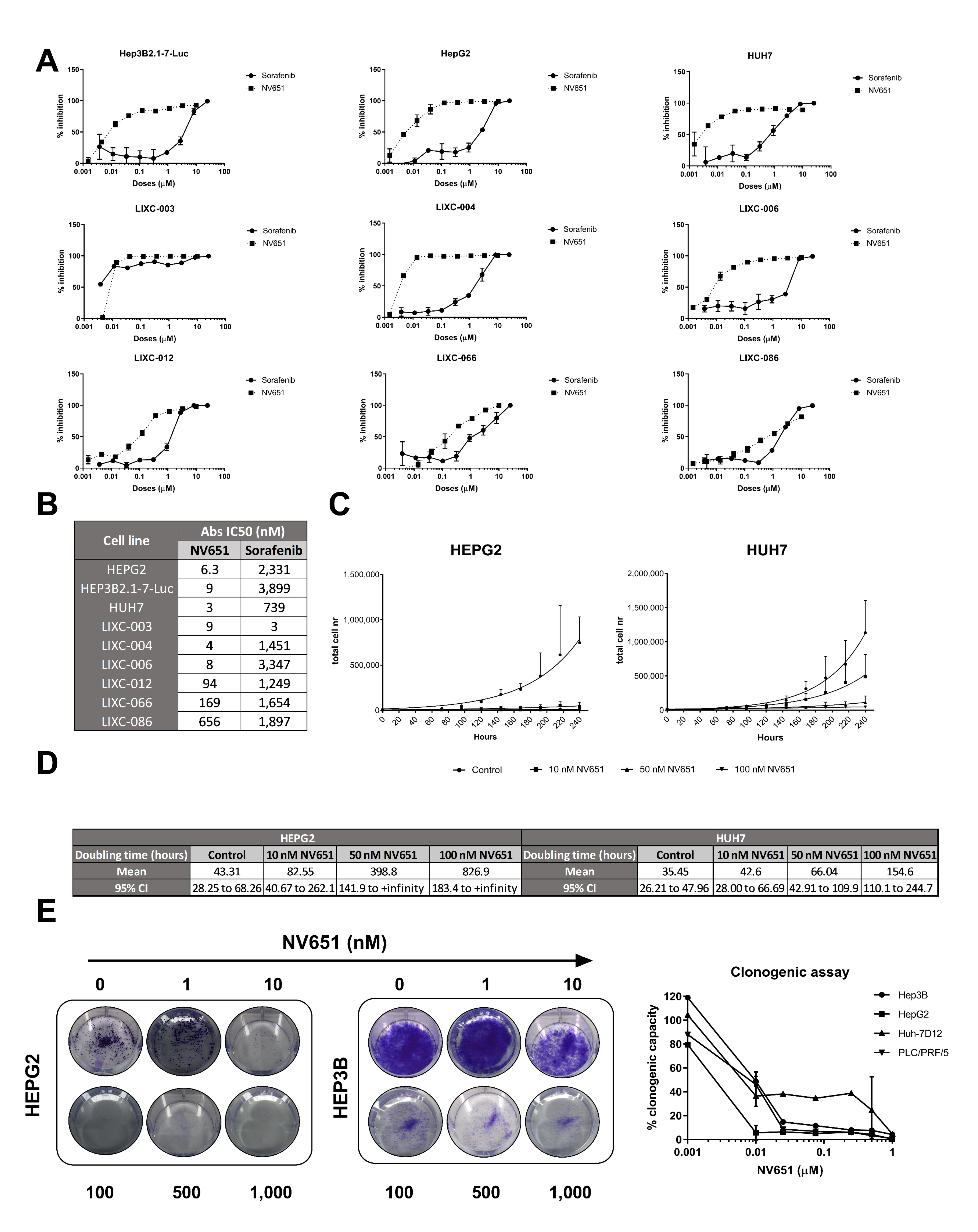
3.4. NV651 Displays a More Potent Antiproliferative Effect Than Sorafenib in HCC Cell Lines
3.5. Prolonged Treatment of NV651 at a High Concentration Causes a Slight Increase in Cell Death
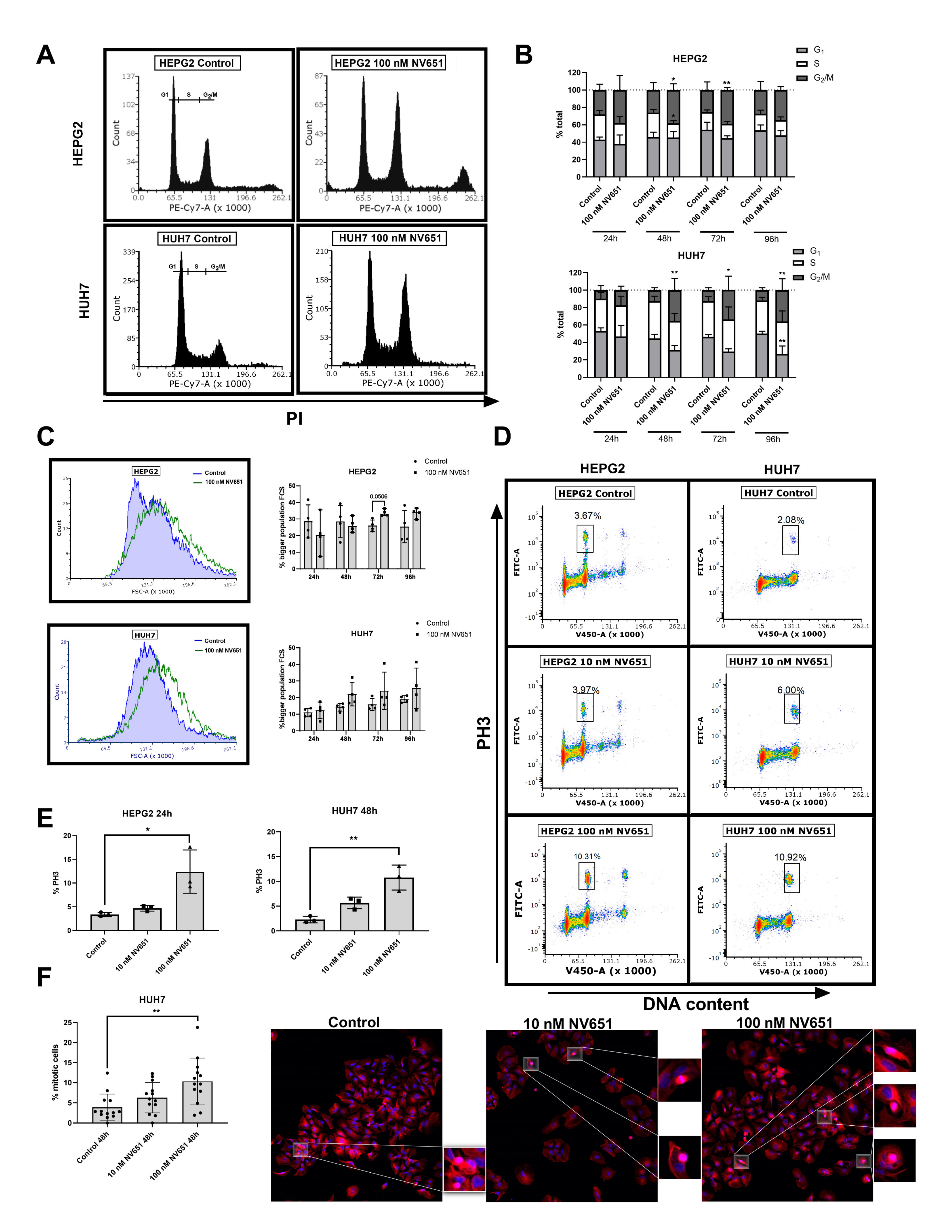
3.6. NV651 Induces Cell Cycle Perturbations
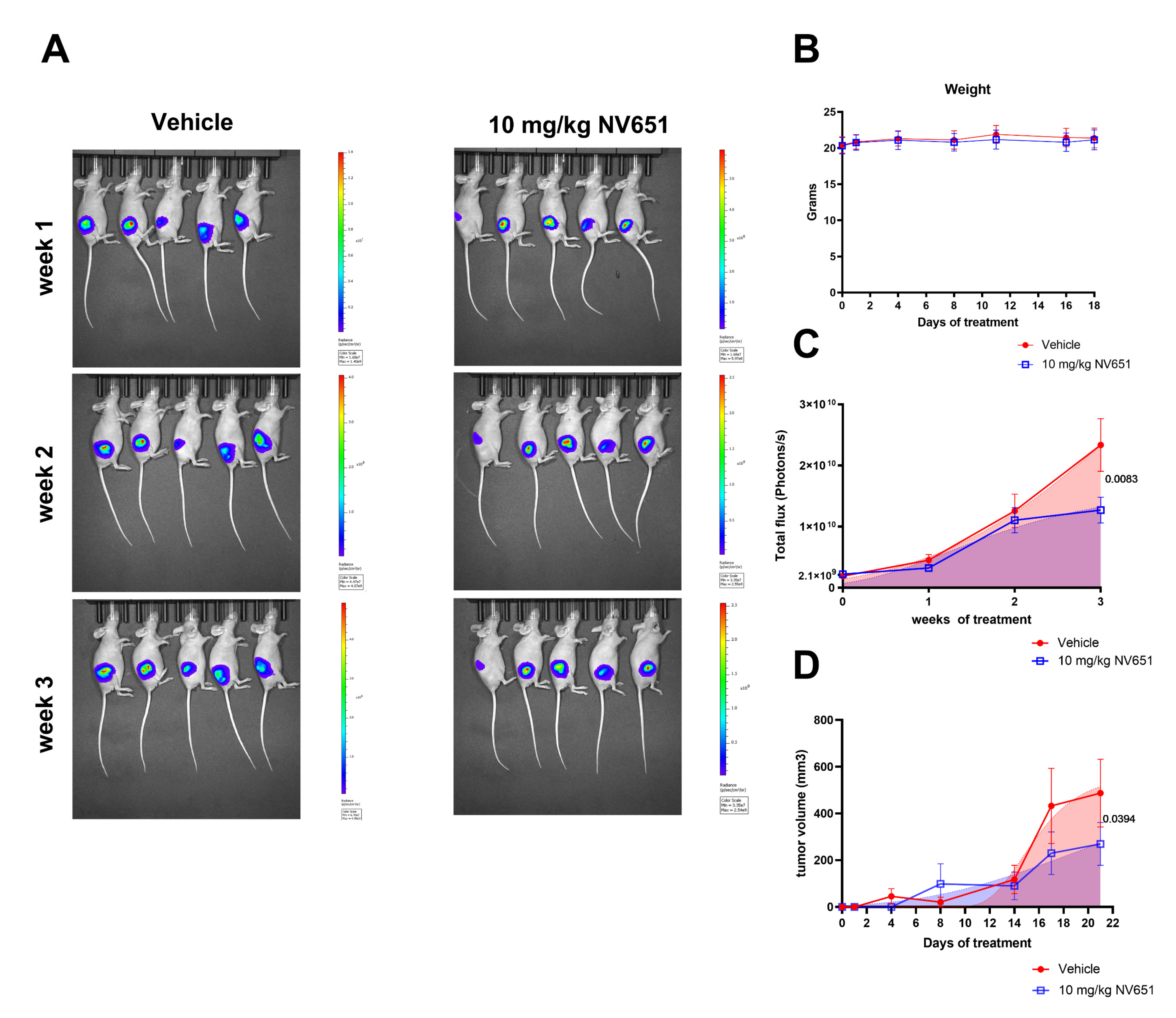
3.7. NV651 Decreases Tumor Growth In Vivo
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Liver Fact Sheet. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/11-Liver-fact-sheet.pdf (accessed on 19 May 2021).
- Janevska, D.; Chaloska-Ivanova, V.; Janevski, V. Hepatocellular Carcinoma: Risk Factors, Diagnosis and Treatment. Open Access Maced. J. Med. Sci. 2015, 3, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Raza, A. Hepatocellular carcinoma review: Current treatment, and evidence-based medicine. World J. Gastroenterol. 2014, 20, 4115–4127. [Google Scholar] [CrossRef]
- Llovet, J.M.; Di Bisceglie, A.M.; Bruix, J.; Kramer, B.S.; Lencioni, R.; Zhu, A.X.; Sherman, M.; Schwartz, M.; Lotze, M.; Talwalkar, J.; et al. Design and Endpoints of Clinical Trials in Hepatocellular Carcinoma. J. Natl. Cancer Inst. 2008, 100, 698–711. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Zhu, Y.-J.; Zheng, B.; Wang, H.-Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Perrucci, G.L.; Gowran, A.; Zanobini, M.; Capogrossi, M.C.; Pompilio, G.; Nigro, P. Peptidyl-prolyl isomerases: A full cast of critical actors in cardiovascular diseases. Cardiovasc. Res. 2015, 106, 353–364. [Google Scholar] [CrossRef]
- Davis, T.L.; Walker, J.R.; Campagna-Slater, V.; Finerty, P.J.; Paramanathan, R.; Bernstein, G.; MacKenzie, F.; Tempel, W.; Ouyang, H.; Lee, W.H.; et al. Structural and Biochemical Characterization of the Human Cyclophilin Family of Peptidyl-Prolyl Isomerases. PLoS Biol. 2010, 8, e1000439. [Google Scholar] [CrossRef]
- Nigro, P.; Pompilio, G.; Capogrossi, M.C. Cyclophilin A: A key player for human disease. Cell Death Dis. 2013, 4, e888. [Google Scholar] [CrossRef]
- Kim, J.; Choi, T.G.; Ding, Y.; Kim, Y.; Ha, K.S.; Lee, K.H.; Kang, I.; Ha, J.; Kaufman, R.J.; Lee, J.; et al. Overexpressed cyclophilin B suppresses apoptosis associated with ROS and Ca2+ homeostasis after ER stress. J. Cell Sci. 2008, 121, 3636–3648. [Google Scholar] [CrossRef]
- Elrod, J.W.; Molkentin, J.D. Physiologic Functions of Cyclophilin D and the Mitochondrial Permeability Transition Pore. Circ. J. 2013, 77, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Lawen, A. Biosynthesis of cyclosporins and other natural peptidyl prolyl cis/trans isomerase inhibitors. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Farmer, J.D.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Naoumov, N.V. Cyclophilin inhibition as potential therapy for liver diseases. J. Hepatol. 2014, 61, 1166–1174. [Google Scholar] [CrossRef]
- Hamilton, G. Cyclophilin A as a target of Cisplatin chemosensitizers. Curr. Cancer Drug Targets 2014, 14, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, S.; Wang, J.; Zhang, M.; Gong, Z.; Wei, Y.; Li, L.; Zhang, Y.; Zhao, X.; Jiang, S.; et al. Cyclophilin J Is a Novel Peptidyl-Prolyl Isomerase and Target for Repressing the Growth of Hepatocellular Carcinoma. PLoS ONE 2015, 10, e0127668. [Google Scholar] [CrossRef]
- Ren, Y.-X.; Wang, S.-J.; Fan, J.-H.; Sun, S.-J.; Li, X.; Padhiar, A.; Zhang, J.-N. CD147 stimulates hepatoma cells escaping from immune surveillance of T cells by interaction with Cyclophilin A. Biomed. Pharmacother. 2016, 80, 289–297. [Google Scholar] [CrossRef]
- Gong, Z.; Chi, C.; Huang, X.; Chu, H.; Wang, J.; Du, F.; Jiang, L.; Chen, J. Cyclophilin A Is Overexpressed in Hepatocellular Carcinoma and Is Associated with the Cell Cycle. Anticancer Res. 2017, 37, 4443–4447. [Google Scholar] [CrossRef][Green Version]
- Feng, W.; Xin, Y.; Xiao, Y.; Li, W.; Sun, D. Cyclophilin A Enhances Cell Proliferation and Xenografted Tumor Growth of Early Gastric Cancer. Dig. Dis. Sci. 2015, 60, 2700–2711. [Google Scholar] [CrossRef]
- Kim, E.-Y.; Choi, Y.H.; Choi, C.G.; Nam, T.-J. Effects of the cyclophilin-type peptidylprolyl cis-trans isomerase from Pyropia yezoensis against hydrogen peroxide-induced oxidative stress in HepG2 cells. Mol. Med. Rep. 2017, 15, 4132–4138. [Google Scholar] [CrossRef]
- Kim, Y.; Jang, M.; Lim, S.; Won, H.; Yoon, K.-S.; Park, J.-H.; Kim, H.J.; Kim, B.-H.; Park, W.-S.; Ha, J.; et al. Role of cyclophilin B in tumorigenesis and cisplatin resistance in hepatocellular carcinoma in humans. Hepatology 2011, 54, 1661–1678. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, M.; Ma, H.; Saiyin, H.; Shen, S.; Xi, J.; Wan, B.; Yu, L. Oligo-microarray analysis reveals the role of cyclophilin A in drug resistance. Cancer Chemother. Pharmacol. 2008, 61, 459–469. [Google Scholar] [CrossRef]
- Lee, J. Novel combinational treatment of cisplatin with cyclophilin a inhibitors in human heptocellular carcinomas. Arch. Pharmacal Res. 2010, 33, 1401–1409. [Google Scholar] [CrossRef]
- Scanlon, K.J.; Wang, W.; Han, H. Cyclosporin A suppresses cisplatin-induced oncogene expression in human cancer cells. Cancer Treat. Rev. 1990, 17, 27–35. [Google Scholar] [CrossRef]
- Xin, H.; Wang, K.; Hu, G.; Xie, F.; Ouyang, K.; Tang, X.; Wang, M.; Wen, D.; Zhu, Y.; Qin, X. Establishment and Characterization of 7 Novel Hepatocellular Carcinoma Cell Lines from Patient-Derived Tumor Xenografts. PLoS ONE 2014, 9, e85308. [Google Scholar] [CrossRef]
- Nair, S.; Archer, G.E.; Tedder, T.F. Isolation and Generation of Human Dendritic Cells. Curr. Protoc. Immunol. 2012, 99, 7–32. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Serrano, S.S.; Grönberg, A.; Longato, L.; Rombouts, K.; Kuo, J.; Gregory, M.; Moss, S.; Elmér, E.; Mazza, G.; Gallay, P.; et al. Evaluation of NV556, a Novel Cyclophilin Inhibitor, as a Potential Antifibrotic Compound for Liver Fibrosis. Cells 2019, 8, 1409. [Google Scholar] [CrossRef]
- Meng, D.; Li, P.; Xie, M. Expression and role of cyclophilin B in stomach cancer. Genet. Mol. Res. 2015, 14, 5346–5354. [Google Scholar] [CrossRef] [PubMed]
- Dunyak, B.M.; Gestwicki, J.E. Peptidyl-Proline Isomerases (PPIases): Targets for Natural Products and Natural Product-Inspired Compounds. J. Med. Chem. 2016, 59, 9622–9644. [Google Scholar] [CrossRef]
- Williams, P.D.; Owens, C.R.; Dziegielewski, J.; Moskaluk, C.A.; Read, P.W.; Larner, J.M.; Story, M.D.; Brock, W.A.; Amundson, S.A.; Lee, J.K.; et al. Cyclophilin B Expression Is Associated with In Vitro Radioresistance and Clinical Outcome after Radiotherapy. Neoplasia 2011, 13, 1122–1131, IN7–IN14. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Sun, Z.; Sun, Y.; Chen, X.; Zhu, X.; Fan, C.; Liu, B.; Zhao, Y.; Zhang, W. Association of increased ligand cyclophilin A and receptor CD147 with hypoxia, angiogenesis, metastasis and prognosis of tongue squamous cell carcinoma. Histopathology 2012, 60, 793–803. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Enriquez-Navas, P.M.; Kam, Y.; Das, T.; Hassan, S.; Silva, A.; Foroutan, P.; Ruiz, E.; Martinez, G.; Minton, S.; Gillies, R.J.; et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci. Transl. Med. 2016, 8, 327ra24. [Google Scholar] [CrossRef]
- Klement, G.L. Eco-evolution of cancer resistance. Sci. Transl. Med. 2016, 8, 327fs5. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, Q.-L.; Tian, Y.-H.; Huang, J.-C.; Ma, G.-L. RNA interference-mediated gene silencing of cyclophilin A enhances the radiosensitivity of PAa human lung adenocarcinoma cells in vitro. Oncol. Lett. 2017, 13, 1619–1624. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Table 1(A) IC50 values for NV651 and positive controls in various normal human cell lines | |||
| Positive Control (µM) a | NV651 (µM) | Days of Treatment | |
| Unstimulated PBMCs | 0.01 | >30 | 3 |
| PHA-stimulated PBMCs | 0.1 | >30 | 3 |
| Monocytes/Macrophages | 0.02 | 24.3 | 7 |
| Dendritic | 0.009 | 24.2 | 7 |
| Bone marrow progenitors | 0.89 | >30 | 14 |
| Hepatocytes | 1.02 | >30 | 2 |
| iPS cardiomyocytes | 39.8 | >30 | 3 |
| iPS neurons | 1.65 | >30 | 3 |
| RPTECs | 0.009 | 20.9 | 3 |
| Table 1(B) NV651 MEC and AC50 values in primary human hepatocytes evaluated for several health parameters | |||
| Cell health Parameter (Hepatocytes) | Time (h) | MEC (µM) | AC50 (µM) |
| Nuclear intensity | 24 | 8.90 | 8.94 |
| Mitochondrial Membrane potential | 24 | 12.40 | 14.20 |
| Glutathione content | 24 | 2.62 | 10.90 |
| ROS | 24 | 6.43 | 8.17 |
| Cell count (PI corrected) | 48 | 6.14 | 13.90 |
| Nuclear size | 48 | 18.10 | 21.50 |
| DNA structure | 48 | 10.90 | 34.38 |
| Steatosis | 48 | 6.81 | 39.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simón Serrano, S.; Tavecchio, M.; Grönberg, A.; Sime, W.; Jemaà, M.; Moss, S.; Gregory, M.A.; Gallay, P.; Elmér, E.; Hansson, M.J.; et al. Novel Cyclophilin Inhibitor Decreases Cell Proliferation and Tumor Growth in Models of Hepatocellular Carcinoma. Cancers 2021, 13, 3041. https://doi.org/10.3390/cancers13123041
Simón Serrano S, Tavecchio M, Grönberg A, Sime W, Jemaà M, Moss S, Gregory MA, Gallay P, Elmér E, Hansson MJ, et al. Novel Cyclophilin Inhibitor Decreases Cell Proliferation and Tumor Growth in Models of Hepatocellular Carcinoma. Cancers. 2021; 13(12):3041. https://doi.org/10.3390/cancers13123041
Chicago/Turabian StyleSimón Serrano, Sonia, Michele Tavecchio, Alvar Grönberg, Wondossen Sime, Mohamed Jemaà, Steven Moss, Matthew Alan Gregory, Philippe Gallay, Eskil Elmér, Magnus Joakim Hansson, and et al. 2021. "Novel Cyclophilin Inhibitor Decreases Cell Proliferation and Tumor Growth in Models of Hepatocellular Carcinoma" Cancers 13, no. 12: 3041. https://doi.org/10.3390/cancers13123041
APA StyleSimón Serrano, S., Tavecchio, M., Grönberg, A., Sime, W., Jemaà, M., Moss, S., Gregory, M. A., Gallay, P., Elmér, E., Hansson, M. J., & Massoumi, R. (2021). Novel Cyclophilin Inhibitor Decreases Cell Proliferation and Tumor Growth in Models of Hepatocellular Carcinoma. Cancers, 13(12), 3041. https://doi.org/10.3390/cancers13123041