Cancer-Associated Fibroblasts: Versatile Players in the Tumor Microenvironment

 , ,
, ,  ,
,  and
and
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
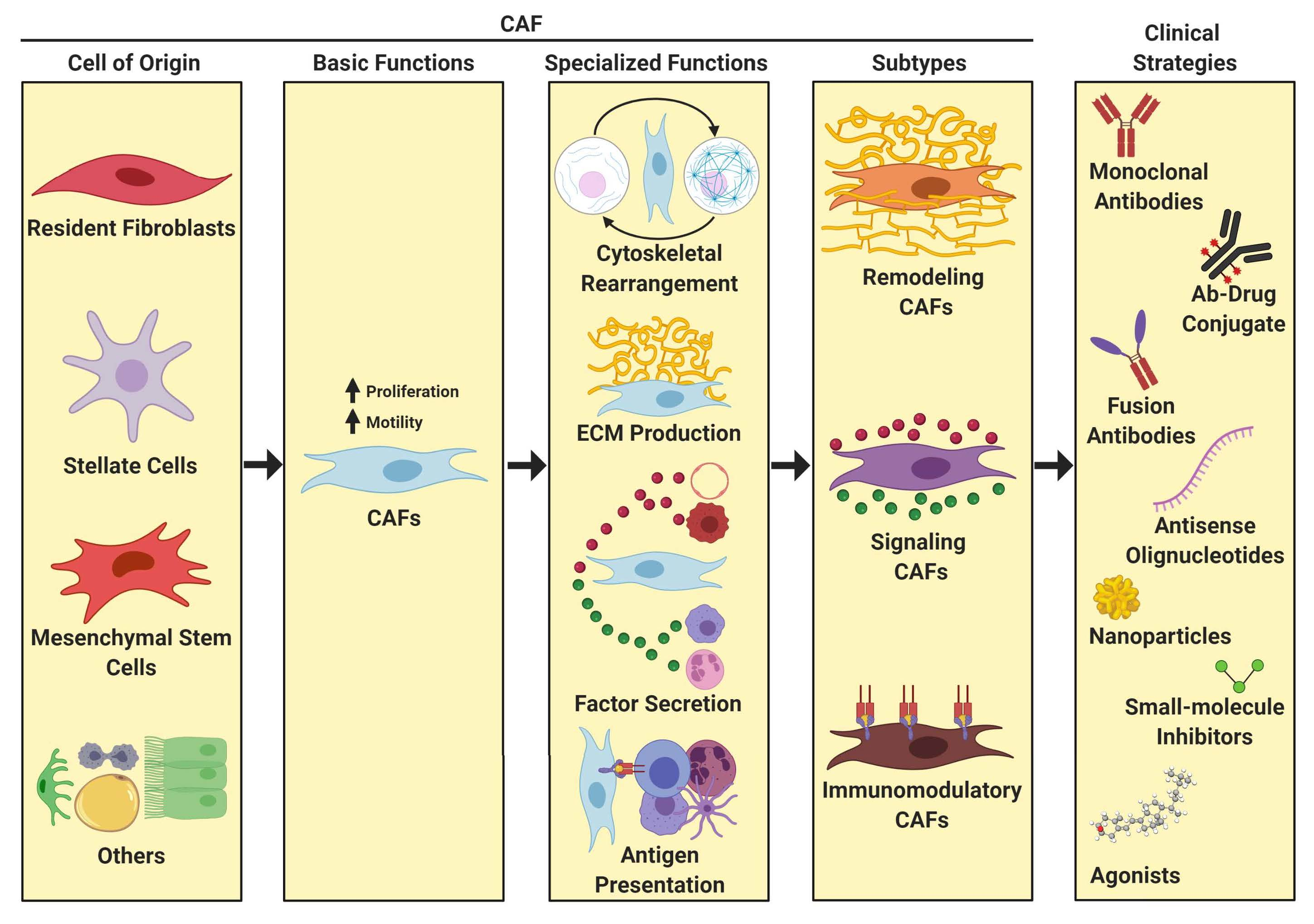
2. History of CAFs
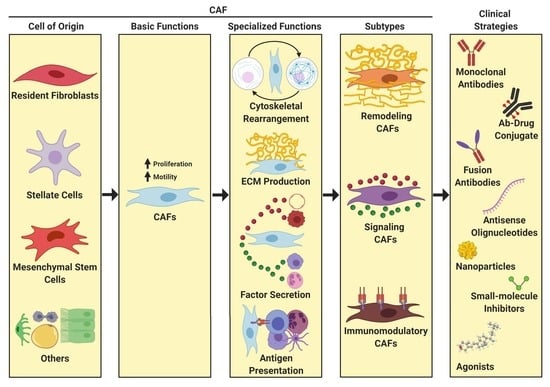
3. Definition and Properties of CAFs
3.1. Comparison of Normal Activated Fibroblasts (NAFs), Fibrosis-Associated Fibroblasts (FAFs), and CAFs
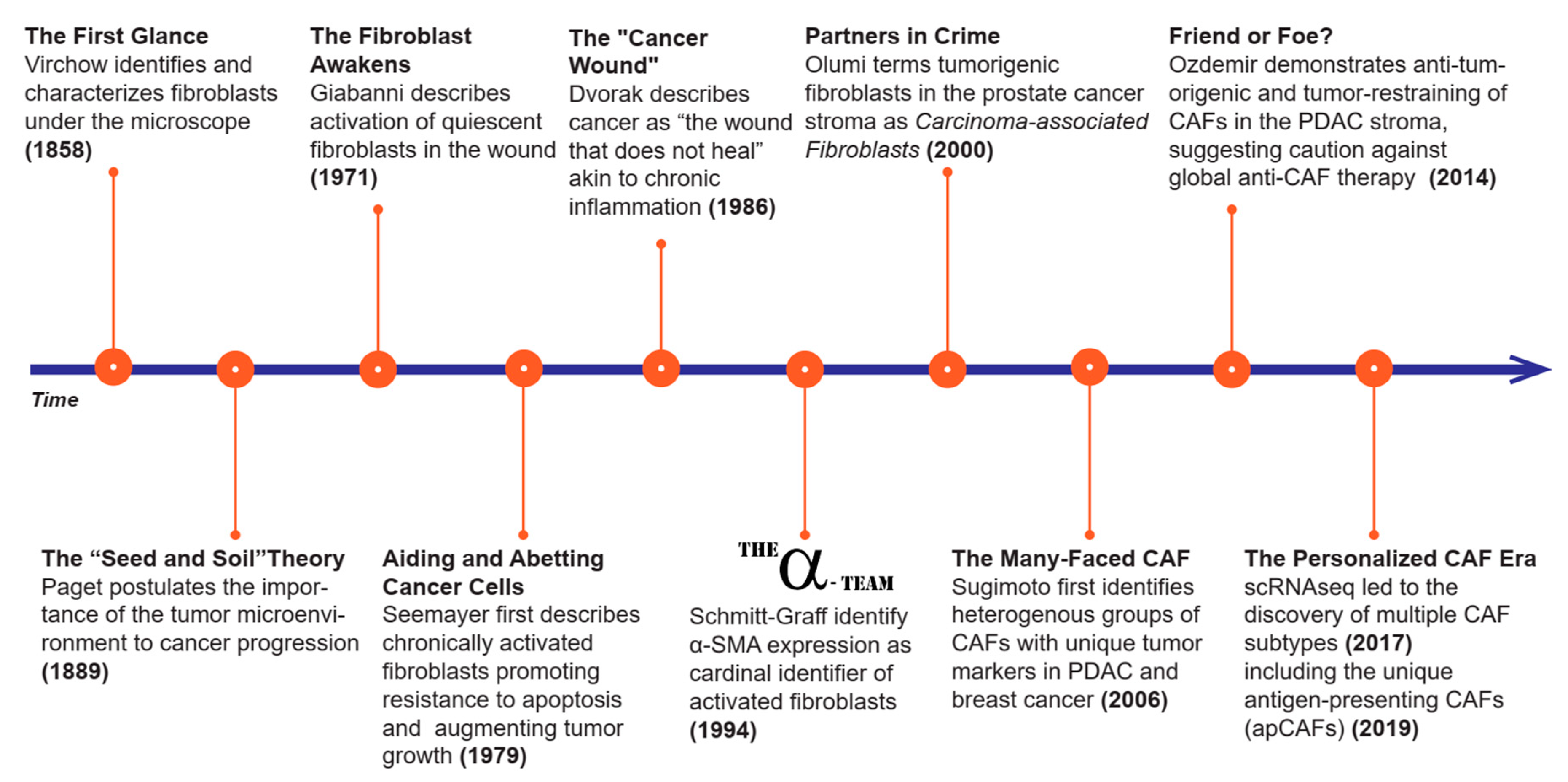
3.2. Origin of CAFs
3.2.1. Tissue Resident Fibroblasts
3.2.2. Stellate Cells
3.2.3. Mesenchymal Stem Cells (MSCs)
3.2.4. Mesothelial Cells
3.2.5. Other Sources of CAFs
3.3. Function of CAFS
3.3.1. ECM Deposition
3.3.2. Cytoskeletal Rearrangement
3.3.3. Factor Secretion
3.3.4. Antigen Presentation
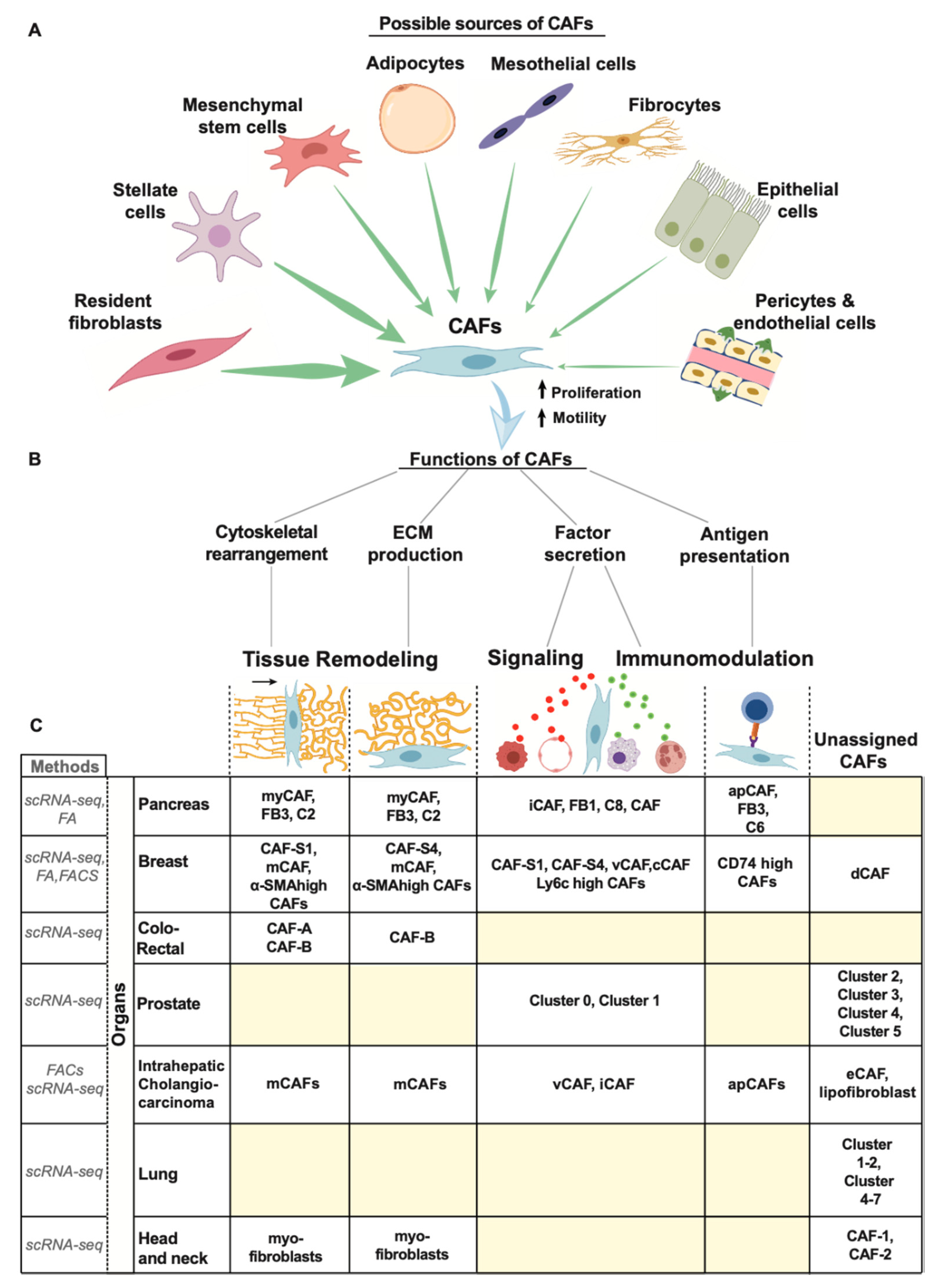
3.4. CAF Heterogeneity
3.4.1. Pancreas
3.4.2. Breast
3.4.3. Others
4. Effects of CAFs on Cancer Cell Behavior
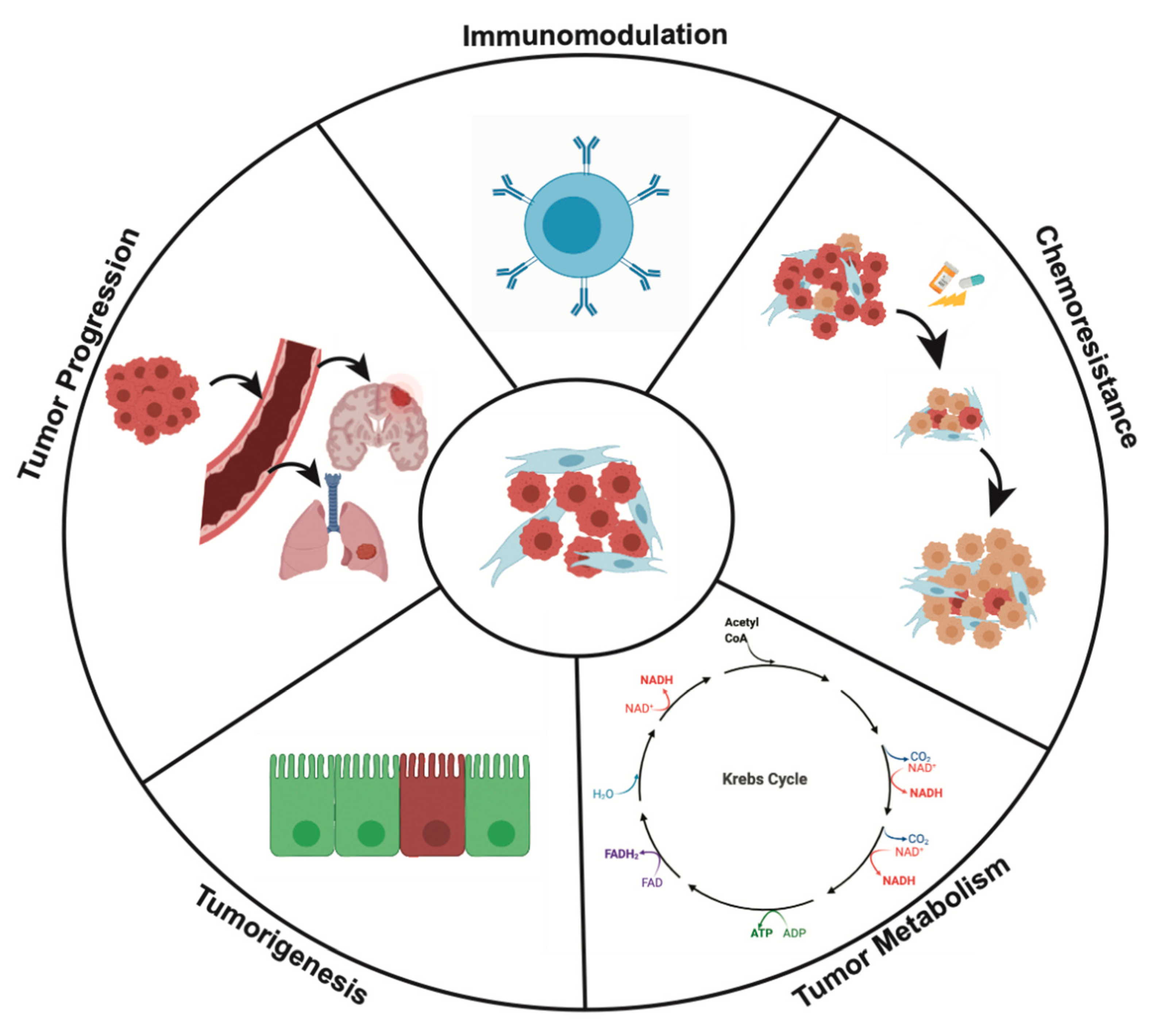
4.1. CAFs and Tumorigenesis
4.2. CAFs and Tumor Progression
4.2.1. Local Invasion and Angiogenesis
4.2.2. Epithelial-to-Mesenchymal Transition (EMT) and Distant Metastasis
4.3. CAFs and Tumor Metabolism
4.4. Immunomodulation
4.4.1. Pro-Inflammatory Milieu
4.4.2. Immunosuppression
Inhibition of Cytotoxic T Cells (CD8+)
Upregulation of Regulatory T Cells (CD4+, FOXP3+)
Modulation of Dendritic Cells
Polarization of Tumor-Associated Macrophages
Suppression of Natural Killer Cells
Recruitment of Neutrophils
Promoting Infiltration of Myeloid-Derived Suppressor Cells
4.5. CAFs and Mechanisms of Chemoresistance
4.5.1. CAF Secreted Factors
4.5.2. Impairment of Drug Delivery
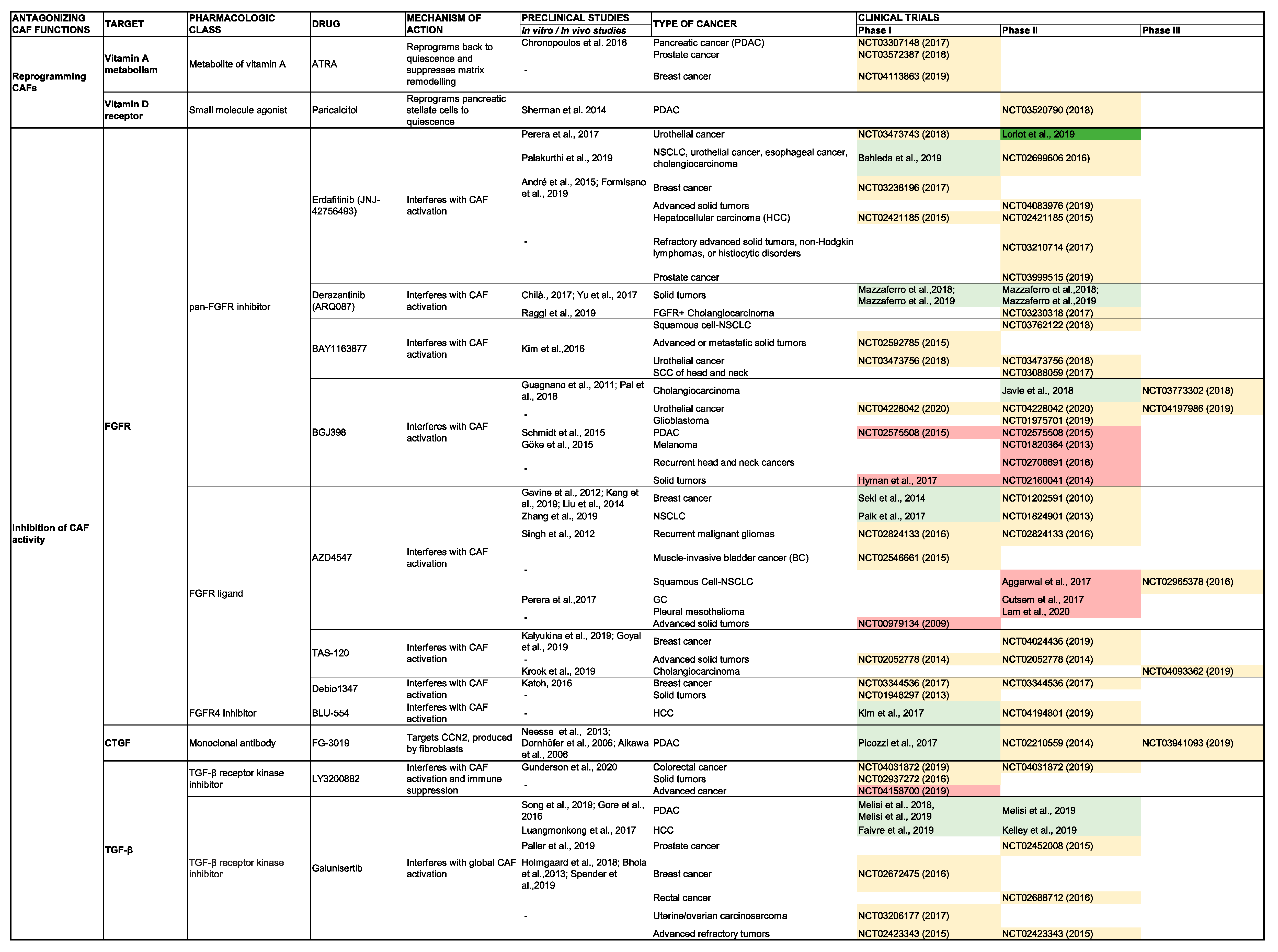
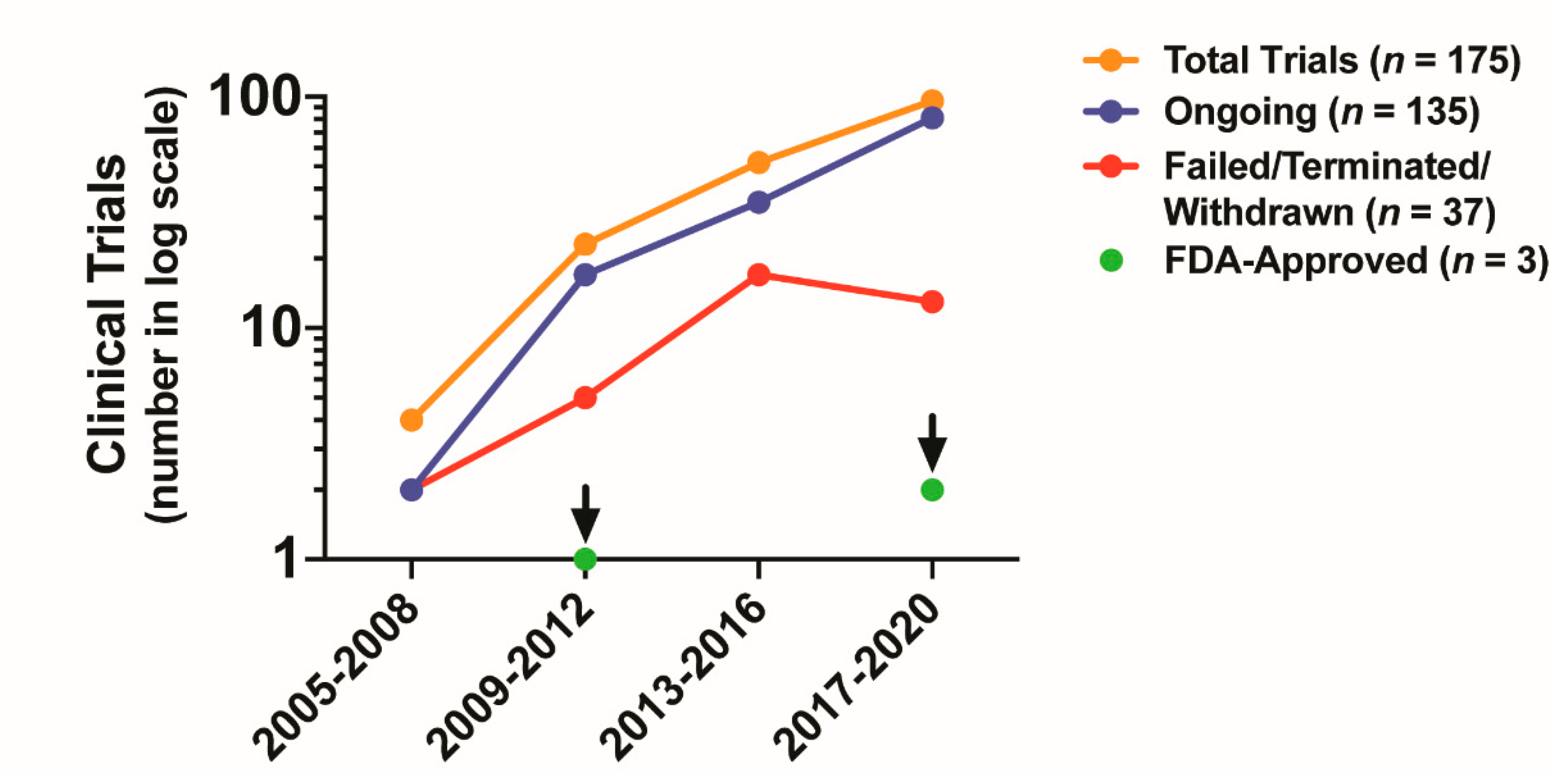
5. Clinical Impact of Targeting CAF Activity
5.1. Antagonizing CAF Activity
5.1.1. Inhibition of the CAF Secretome
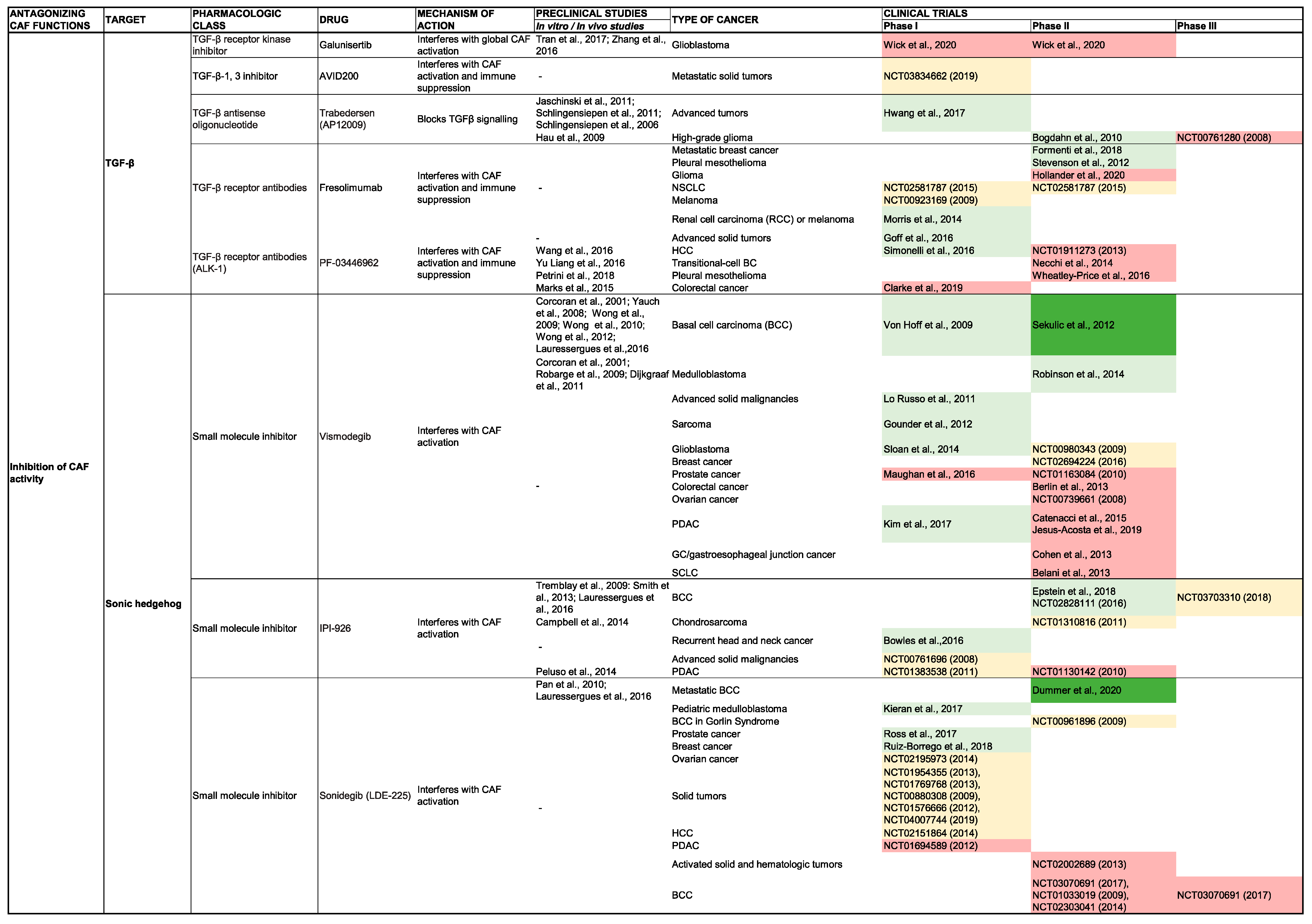
TGF-β
Fibroblast Growth Factor
CCN2
SHH
5.1.2. Reverting CAFs to a Quiescent State
Vitamin D Receptor Agonism
Vitamin A Receptor Agonism
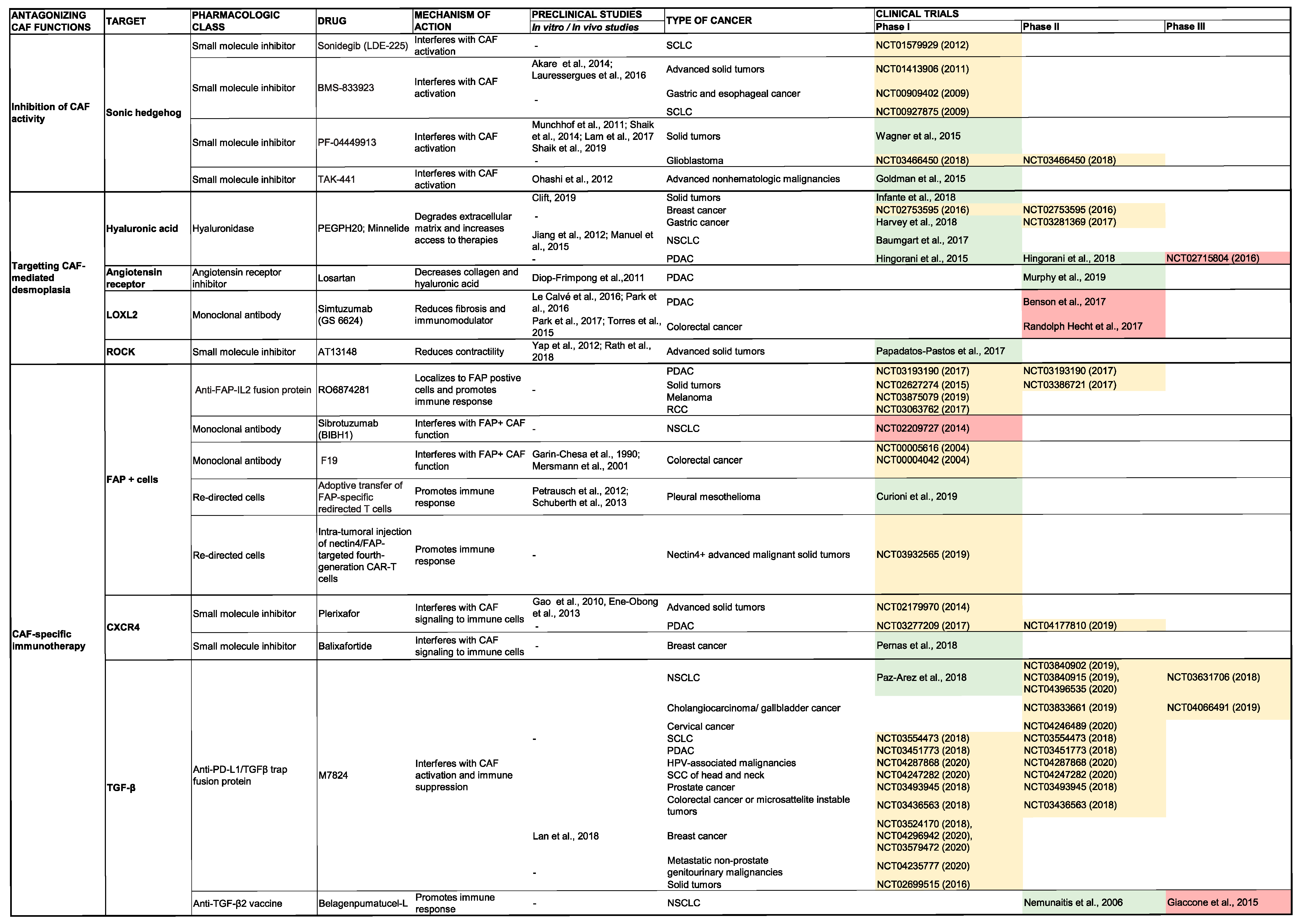
5.2. Inhibition of ECM Production and Stroma Remodeling
5.2.1. Hyaluronic Acid
5.2.2. Collagen
5.2.3. Photothermal Therapy
5.3. CAF-Specific Immunotherapy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Öhlund, D.; Elyada, E.; Tuveson, D.A. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Muller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2019, 10, 232–253. [Google Scholar] [CrossRef]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 5, 539874. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Die cellularpathologie in ihrer begründung auf physiologische und pathologische gewebelehre. Available online: https://www.loc.gov/item/06041231/.1858 (accessed on 1 April 2020).
- Duval, M.M. Atlas D’embryologie; G. Masson: Paris, France, 1889. [Google Scholar]
- Gabbiani, G.; Ryan, G.B.; Majne, G.; Majno, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Cell. Mol. Life Sci. 1971, 27, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G.; Schmitt-Gräff, A.; Desmoulière, A. Heterogeneity of myofibroblast phenotypic features: An example of fibroblastic cell plasticity. Virchows Arch. 1994, 425, 3–24. [Google Scholar] [CrossRef]
- Seemayer, T.A.; Schurch, W.; Lagacé, R.; Tremblay, G. Myofibroblasts in the stroma of invasive and metastatic carcinoma A possible host response to neoplasia. Am. J. Surg. Pathol. 1979, 3, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Schürch, W.; Seemayer, A.T.; Lagacé, R. Stromal myofibroblasts in primary invasive and metastatic carcinomas. A combined immunological, light and electron microscopic study. Virchows Arch. A Pathol. Anat. Histol. 1981, 391, 125–139. [Google Scholar]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Micallef, L.; Vedrenne, N.; Billet, F.; Coulomb, B.; Darby, A.I.; Desmoulière, A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair 2012, 5, S5. [Google Scholar] [CrossRef]
- Madar, S.; Goldstein, I.; Rotter, V. ’Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Boil. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef]
- Hawinkels, L.J.; Paauwe, M.; Verspaget, H.W.; Wiercinska, E.; van der Zon, J.M.; van der Ploeg, K.; Koelink, P.J.; Lindeman, J.H.N.; Mesker, W.; ten Dijke, P.; et al. Interaction with colon cancer cells hyperactivates TGF-beta signaling in cancer-associated fibroblasts. Oncogene 2014, 33, 97–107. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Pelon, F.; Bourachot, B.; Kieffer, Y.; Magagna, I.; Mermet-Meillon, F.; Bonnet, I.; Costa, A.; Givel, A.-M.; Atieh, Y.; Barbazán, J.; et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.-L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551–7556. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Barrera, L.N.; Evans, A.; Lane, B.; Brumskill, S.; Oldfield, E.F.; Campbell, F.; Andrews, T.; Lu, Z.; Perez-Mancera, P.A.; Liloglou, T.; et al. Fibroblasts from distinct pancreatic pathologies exhibit disease-specific properties. Cancer Res. 2020, 80, 2861–2873. [Google Scholar] [CrossRef]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, E.J.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Boil. 1995, 130, 393–405. [Google Scholar] [CrossRef]
- Tarin, D.; Croft, C.B. Ultrastructural features of wound healing in mouse skin. J. Anat. 1969, 105, 189–190. [Google Scholar]
- Sharpe, P. Neural crest and tooth morphogenesis. Adv. Dent. Res. 2001, 15, 4–7. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2018, 9, 282–301. [Google Scholar] [CrossRef]
- Löhr, M.; Schmidt, C.; Ringel, J.; Kluth, M.; Müller, P.; Nizze, H.; Jesnowski, R. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001, 61, 550–555. [Google Scholar]
- Elenbaas, B.; Weinberg, R.A. Heterotypic Signaling between Epithelial Tumor Cells and Fibroblasts in Carcinoma Formation. Exp. Cell Res. 2001, 264, 169–184. [Google Scholar] [CrossRef]
- Kuzet, S.-E.; Gaggioli, C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res. 2016, 365, 607–619. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-κB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Virós, A.; Hooper, S.; Mitter, R.; Féral, C.C.; Cook, M.; et al. ROCK and JAK1 Signaling Cooperate to Control Actomyosin Contractility in Tumor Cells and Stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Straub, J.M.; New, J.; Hamilton, C.D.; Lominska, C.; Shnayder, Y.; Thomas, S.M. Radiation-induced fibrosis: Mechanisms and implications for therapy. J. Cancer Res. Clin. Oncol. 2015, 141, 1985–1994. [Google Scholar] [CrossRef]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef]
- Haber, P.S.; Keogh, G.W.; Apte, M.V.; Moran, C.S.; Stewart, N.L.; Crawford, D.H.; Pirola, R.C.; McCaughan, G.W.; Ramm, G.A.; Wilson, J.S. Activation of Pancreatic Stellate Cells in Human and Experimental Pancreatic Fibrosis. Am. J. Pathol. 1999, 155, 1087–1095. [Google Scholar] [CrossRef]
- Omary, M.B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J. Clin. Investig. 2007, 117, 50–59. [Google Scholar] [CrossRef]
- Cuiffo, B.G.; Karnoub, A.E. Mesenchymal stem cells in tumor development: Emerging roles and concepts. Cell Adhes. Migr. 2012, 6, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow–derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Du, L.; Lin, L.; Wang, Y. Tumour-associated mesenchymal stem/stromal cells: Emerging therapeutic targets. Nat. Rev. Drug Discov. 2016, 16, 35–52. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, X.; Zhang, L.; Li, W.; Wu, H.; Yuan, X.; Mao, F.; Wang, M.; Zhu, W.; Qian, H.; et al. The IL-6-STAT3 axis mediates a reciprocal crosstalk between cancer-derived mesenchymal stem cells and neutrophils to synergistically prompt gastric cancer progression. Cell Death Dis. 2014, 5, e1295. [Google Scholar] [CrossRef]
- Krstic, J.; Trivanovic, D.; Obradović, H.; Kukolj, T.; Bugarski, D.; Santibanez, J.F. Regulation of Mesenchymal Stem Cell Differentiation by Transforming Growth Factor Beta Superfamily. Curr. Protein Pept. Sci. 2018, 19, 1138–1154. [Google Scholar] [CrossRef]
- Sandoval, P.; Jiménez-Heffernan, J.A.; Rynne-Vidal, A.; Perez-Lozano, M.L.; Gilsanz, A.; Ruiz-Carpio, V.; Reyes, R.; García-Bordas, J.; Stamatakis, K.; Dotor, J.; et al. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J. Pathol. 2013, 231, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Rynne-Vidal, A.; Jiménez-Heffernan, J.A.; Fernández-Chacón, C.; López-Cabrera, M.; Sandoval, P. The Mesothelial Origin of Carcinoma Associated-Fibroblasts in Peritoneal Metastasis. Cancers 2015, 7, 1994–2011. [Google Scholar] [CrossRef]
- Bochet, L.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; Couderc, B.; et al. Adipocyte-Derived Fibroblasts Promote Tumor Progression and Contribute to the Desmoplastic Reaction in Breast Cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- Reilkoff, R.A.; Bucala, R.; Herzog, E.L. Fibrocytes: Emerging effector cells in chronic inflammation. Nat. Rev. Immunol. 2011, 11, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Uchinami, H.; Feirt, N.; Quintana-Bustamante, O.; Segovia, J.C.; Schwabe, R.F.; Brenner, A.D. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J. Hepatol. 2006, 45, 429–438. [Google Scholar] [CrossRef]
- Radisky, D.C.; Kenny, P.A.; Bissell, M.J. Fibrosis and cancer: Do myofibroblasts come also from epithelial cells via EMT? J. Cell. Biochem. 2007, 101, 830–839. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Hosaka, K.; Yang, L.; Seki, T.; Fischer, C.; Dubey, O.; Fredlund, E.; Hartman, J.; Religa, P.; Morikawa, H.; Ishii, Y.; et al. Pericyte–fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E5618–E5627. [Google Scholar] [CrossRef]
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front. Immunol. 2018, 9, 414. [Google Scholar] [CrossRef]
- Miles, F.L.; Sikes, R.A. Insidious changes in stromal matrix fuel cancer progression. Mol. Cancer Res. 2014, 12, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Miyake, K.; Nandi, T.; Yashiro, M.; Onishi, N.; Huang, K.K.; Lin, S.J.; Kalpana, R.; Tay, S.T.; Suzuki, Y.; et al. Activation of Transforming Growth Factor Beta 1 Signaling in Gastric Cancer-associated Fibroblasts Increases Their Motility, via Expression of Rhomboid 5 Homolog 2, and Ability to Induce Invasiveness of Gastric Cancer Cells. Gastroenterology 2017, 153, 191–204.e16. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Bershadsky, A.; Pankov, R.; Yamada, K.M. Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2001, 2, 793–805. [Google Scholar] [CrossRef]
- Labernadie, A.; Kato, T.; Brugués, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.N.; Weston, A.; González-Tarragó, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nature 2017, 19, 224–237. [Google Scholar] [CrossRef]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef]
- Djurec, M.; Graña, O.; Lee, A.; Troulé, K.; Espinet, E.; Cabras, L.; Navas, C.; Blasco, M.T.; Martin-Diaz, L.; Burdiel, M.; et al. Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc. Natl. Acad. Sci. USA 2018, 115, E1147–E1156. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- Sebastian, A.; Hum, N.R.; Martin, K.; Gilmore, S.F.; Peran, I.; Byers, S.W.; Wheeler, E.K.; Coleman, M.; Loots, G.G. Single-Cell Transcriptomic Analysis of Tumor-Derived Fibroblasts and Normal Tissue-Resident Fibroblasts Reveals Fibroblast Heterogeneity in Breast Cancer. Cancers 2020, 12, 1307. [Google Scholar] [CrossRef] [PubMed]
- Vickman, R.E.; Broman, M.M.; Lanman, N.A.; Franco, O.E.; Sudyanti, P.A.G.; Ni, Y.; Ji, Y.; Helfand, B.T.; Ma, J.P.; Paterakos, M.C.; et al. Heterogeneity of human prostate carcinoma-associated fibroblasts implicates a role for subpopulations in myeloid cell recruitment. Prostate 2019, 80, 173–185. [Google Scholar] [CrossRef]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwe, H.; Pircher, A.; Eynde, K.V.D.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, H.; Wan, L.; Wang, Z.; Wang, H.; Ge, C.; Liu, Y.; Hao, Y.; Zhang, D.; Shi, G.; et al. Single cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed]
- Ollila, S.; Domènech-Moreno, E.; Laajanen, K.; Wong, I.P.; Tripathi, S.; Pentinmikko, N.; Gao, Y.; Yan, Y.; Niemelä, E.H.; Wang, T.C.; et al. Stromal Lkb1 deficiency leads to gastrointestinal tumorigenesis involving the IL-11-JAK/STAT3 pathway. J. Clin. Investig. 2017, 128, 402–414. [Google Scholar] [CrossRef]
- Maffini, M.V.; Soto, A.M.; Calabro, J.M.; Ucci, A.A.; Sonnenschein, C. The stroma as a crucial target in rat mammary gland carcinogenesis. J. Cell Sci. 2004, 117, 1495–1502. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Huang, M.; Li, Y.; Zhang, H.; Nan, F. Breast cancer stromal fibroblasts promote the generation of CD44+CD24- cells through SDF-1/CXCR4 interaction. J. Exp. Clin. Cancer Res. 2010, 29, 80. [Google Scholar]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef]
- Mohammadi, H.; Sahai, E. Publisher Correction: Mechanisms and impact of altered tumour mechanics. Nature 2018, 20, 1099. [Google Scholar] [CrossRef]
- Dufort, C.C.; DelGiorno, K.E.; Carlson, M.A.; Osgood, R.J.; Zhao, C.; Huang, Z.; Thompson, C.B.; Connor, R.J.; Thanos, C.D.; Brockenbrough, J.S.; et al. Interstitial Pressure in Pancreatic Ductal Adenocarcinoma Is Dominated by a Gel-Fluid Phase. Biophys. J. 2016, 110, 2106–2119. [Google Scholar] [CrossRef]
- Yeo, C.D.; Kang, N.; Choi, S.Y.; Na Kim, B.; Park, C.K.; Kim, J.W.; Kim, Y.K.; Kim, S.J. The role of hypoxia on the acquisition of epithelial-mesenchymal transition and cancer stemness: A possible link to epigenetic regulation. Korean J. Intern. Med. 2017, 32, 589–599. [Google Scholar] [CrossRef]
- Zhang, X.-D.; Dong, X.-Q.; Xu, J.-L.; Chen, S.-C.; Sun, Z. Hypoxia promotes epithelial-mesenchymal transition of hepatocellular carcinoma cells via inducing Twist1 expression. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3061–3068. [Google Scholar] [PubMed]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Moore-Smith, L.D.; Isayeva, T.; Lee, J.H.; Frost, A.; Ponnazhagan, S. Silencing of TGF-beta1 in tumor cells impacts MMP-9 in tumor microenvironment. Sci. Rep. 2017, 7, 8678. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Kawana, K.; Tomio, K.; Yamashita, A.; Isobe, Y.; Nagasaka, K.; Koga, K.; Inoue, T.; Nishida, H.; Kojima, S.; et al. Matrix Metalloproteinase (MMP)-9 in Cancer-Associated Fibroblasts (CAFs) Is Suppressed by Omega-3 Polyunsaturated Fatty Acids In Vitro and In Vivo. PLoS ONE 2014, 9, e89605. [Google Scholar] [CrossRef] [PubMed]
- Dayer, C.; Stamenkovic, I. Recruitment of Matrix Metalloproteinase-9 (MMP-9) to the Fibroblast Cell Surface by Lysyl Hydroxylase 3 (LH3) Triggers Transforming Growth Factor-beta (TGF-beta) Activation and Fibroblast Differentiation. J. Biol. Chem. 2015, 290, 13763–13778. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Boil. 2015, 44–46, 94–112. [Google Scholar] [CrossRef]
- Miyazaki, K.; Oyanagi, J.; Hoshino, D.; Togo, S.; Kumagai, H.; Miyagi, Y. Cancer cell migration on elongate protrusions of fibroblasts in collagen matrix. Sci. Rep. 2019, 9, 292. [Google Scholar] [CrossRef]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; Macdonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef]
- Ferrara, N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010, 21, 21–26. [Google Scholar] [CrossRef]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFbeta1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 11924. [Google Scholar] [CrossRef]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.e27. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br. J. Cancer. 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Li, M.; Cao, L.M.; Gu, Q.H.; Deng, P.B.; Tan, Y.; Hu, C.P. Snail1-dependent cancer-associated fibroblasts induce epithelial-mesenchymal transition in lung cancer cells via exosomes. QJM Int. J. Med. 2019, 112, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef]
- Bhome, R.; Goh, R.W.; Bullock, M.D.; Pillar, N.; Thirdborough, S.M.; Mellone, M.; Mirnezami, R.; Galea, D.; Veselkov, K.; Gu, Q.; et al. Exosomal microRNAs derived from colorectal cancer-associated fibroblasts: Role in driving cancer progression. Aging 2017, 9, 2666–2694. [Google Scholar] [CrossRef]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signaling in Breast Cancer Cell Migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef]
- Martin, Y.D.P.; Park, D.; Ramachandran, A.; Ombrato, L.; Calvo, F.; Chakravarty, P.; Spencer-Dene, B.; Derzsi, S.; Hill, C.S.; Sahai, E.; et al. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep. 2015, 13, 2456–2469. [Google Scholar] [CrossRef]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.-F.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massagué, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef]
- Yu, B.; Wu, K.; Wang, X.; Zhang, J.; Wang, L.; Jiang, Y.; Zhu, X.; Chen, W.-T.; Yan, M. Periostin secreted by cancer-associated fibroblasts promotes cancer stemness in head and neck cancer by activating protein tyrosine kinase 7. Cell Death Dis. 2018, 9, 1082. [Google Scholar] [CrossRef]
- Mateo, F.; Meca-Cortés, Ó.; Celia-Terrassa, T.; Fernández, Y.; Abasolo, I.; Sánchez-Cid, L.; Bermudo, R.; Sagasta, A.; Rodriguez-Carunchio, L.; Pons, M.; et al. SPARC mediates metastatic cooperation between CSC and non-CSC prostate cancer cell subpopulations. Mol. Cancer 2014, 13, 237. [Google Scholar] [CrossRef]
- Aguado, B.; Bushnell, G.G.; Rao, S.S.; Jeruss, J.S.; Shea, L.D. Engineering the pre-metastatic niche. Nat. Biomed. Eng. 2017, 1, 77. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.G.; Duyverman, A.M.M.J.; Kohno, M.; Snuderl, M.; Steller, E.J.A.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar] [CrossRef] [PubMed]
- Malanchi, I.; Santamaria-Martínez, A.; Susanto, E.; Peng, H.; Lehr, H.-A.; Delaloye, J.-F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2011, 481, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Harris, A.L.; Sivridis, E. Comparison of Metabolic Pathways between Cancer Cells and Stromal Cells in Colorectal Carcinomas: A Metabolic Survival Role for Tumor-Associated Stroma. Cancer Res. 2006, 66, 632–637. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Curry, J.M.; Ko, Y.-H.; Lin, Z.; Tuluc, M.; Cognetti, D.; Birbe, R.C.; Pribitkin, E.; Bombonati, A.; Pestell, R.G.; et al. Oncogenes and inflammation rewire host energy metabolism in the tumor microenvironment: RAS and NFκB target stromal MCT4. Cell Cycle 2013, 12, 2580–2597. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Whitaker-Menezes, D.; Dasgupta, A.; Philp, N.J.; Lin, Z.; Gandara, R.; Sneddon, S.F.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: Stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle 2012, 11, 1108–1117. [Google Scholar] [CrossRef]
- Hutcheson, J.; Balaji, U.; Porembka, M.R.; Wachsmann, M.B.; McCue, P.A.; Knudsen, E.S.; Witkiewicz, A.K. Immunologic and Metabolic Features of Pancreatic Ductal Adenocarcinoma Define Prognostic Subtypes of Disease. Clin. Cancer Res. 2016, 22, 3606–3617. [Google Scholar] [CrossRef]
- Mishra, R.; Haldar, S.; Placencio, V.; Madhav, A.; Rohena-Rivera, K.; Agarwal, P.; Duong, F.; Angara, B.; Tripathi, M.; Liu, Z.; et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J. Clin. Investig. 2018, 128, 4472–4484. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S.; Majumdar, S.; Azabdaftari, G.; Attwood, K.; Scioneaux, R.; Subramani, D.; Manhardt, C.; Lorusso, G.D.; Willard, S.S.; Thompson, H.; et al. Serum Glutamate Levels Correlate with Gleason Score and Glutamate Blockade Decreases Proliferation, Migration, and Invasion and Induces Apoptosis in Prostate Cancer Cells. Clin. Cancer Res. 2012, 18, 5888–5901. [Google Scholar] [CrossRef] [PubMed]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10, 4375. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-T.; Zhu, M.; Lance, P. Stromal COX-2 signaling activated by deoxycholic acid mediates proliferation and invasiveness of colorectal epithelial cancer cells. Biochem. Biophys. Res. Commun. 2012, 425, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741. [Google Scholar] [CrossRef]
- Clemente, C.G.; Mihm Jr, M.C.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Koch, M.; Beckhove, P.; Winkel, J.O.D.; Autenrieth, D.; Wagner, P.; Nummer, D.; Specht, S.; Antolovic, D.; Galindo, L.; Schmitz-Winnenthal, F.H.; et al. Tumor Infiltrating T Lymphocytes in Colorectal Cancer. Ann. Surg. 2006, 244, 986–993. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T Cells to protect tumour cells. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Fu, Z.; Zuo, Y.; Li, N.; Xu, W.; Li, D.; Chen, H.; Zheng, S. The crosstalk: Tumor-infiltrating lymphocytes rich in regulatory T cells suppressed cancer-associated fibroblasts. Acta Oncol. 2013, 52, 1760–1770. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Ovcinnikovs, V.; Ross, E.M.; Petersone, L.; Edner, N.M.; Heuts, F.; Ntavli, E.; Kogimtzis, A.; Kennedy, A.; Wang, C.J.; Bennett, C.L.; et al. CTLA-4 mediated transendocytosis of co-stimulatory molecules primarily targets migratory dendritic cells. Sci. Immunol. 2019, 4, eaaw0902. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Martínez, E.; Planès, R.; Anselmi, G.; Reynolds, M.; Menezes, S.; Adiko, A.C.; Saveanu, L.; Guermonprez, P. Cross-presentation of cell-associated antigens by MHC class I in dendritic cell subsets. Front. Immunol. 2015, 6, 363. [Google Scholar] [CrossRef] [PubMed]
- Broeke, T.T.; Wubbolts, R.; Stoorvogel, W. MHC Class II Antigen Presentation by Dendritic Cells Regulated through Endosomal Sorting. Cold Spring Harb. Perspect. Boil. 2013, 5, a016873. [Google Scholar] [CrossRef] [PubMed]
- Kobie, J.J.; Wu, R.S.; Kurt, R.A.; Lou, S.; Adelman, M.K.; Whitesell, L.J.; Ramanathapuram, L.V.; Arteaga, C.L.; Akporiaye, E.T. Transforming growth factor β inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res. 2003, 63, 1860–1864. [Google Scholar]
- Kenkel, J.A.; Tseng, W.W.; Davidson, M.G.; Tolentino, L.L.; Choi, O.; Bhattacharya, N.; Seeley, E.S.; Winer, D.A.; Reticker-Flynn, E.N.; Engleman, E.G. An Immunosuppressive Dendritic Cell Subset Accumulates at Secondary Sites and Promotes Metastasis in Pancreatic Cancer. Cancer Res. 2017, 77, 4158–4170. [Google Scholar] [CrossRef]
- Cao, W.; Ramakrishnan, R.; Tuyrin, V.A.; Veglia, F.; Condamine, T.; Amoscato, A.; Mohammadyani, D.; Johnson, J.J.; Zhang, L.M.; Klein-Seetharaman, J.; et al. Oxidized Lipids Block Antigen Cross-Presentation by Dendritic Cells in Cancer. J. Immunol. 2014, 192, 2920–2931. [Google Scholar] [CrossRef]
- Cheng, J.-T.; Deng, Y.-N.; Yi, H.-M.; Wang, G.-Y.; Fu, B.-S.; Chen, W.-J.; Liu, W.; Tai, Y.; Peng, Y.-W.; Zhang, Q. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis 2016, 5, e198. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, C.; Sun, A.; Zheng, Y.; Wang, L.; Cao, X. Tumor-Educated CD11bhighIalowRegulatory Dendritic Cells Suppress T Cell Response through Arginase I. J. Immunol. 2009, 182, 6207–6216. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2019, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Zin, A.A.M.; Ang, K.C.; Ch’Ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2020, 9. [Google Scholar] [CrossRef]
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019, 10, 273. [Google Scholar] [CrossRef]
- Leek, R.D.; Hunt, N.C.; Landers, R.J.; Lewis, C.E.; Royds, J.A.; Harris, A.L. Macrophage infiltration is associated with VEGF and EGFR expression in breast cancer. J. Pathol. J. Pathol. Soc. Great Br. Irel. 2000, 190, 430–436. [Google Scholar] [CrossRef]
- Higashino, N.; Koma, Y.-I.; Hosono, M.; Takase, N.; Okamoto, M.; Kodaira, H.; Nishio, M.; Shigeoka, M.; Kakeji, Y.; Yokozaki, H. Fibroblast activation protein-positive fibroblasts promote tumor progression through secretion of CCL2 and interleukin-6 in esophageal squamous cell carcinoma. Lab. Investig. 2019, 99, 777–792. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Wu, F.; Zhou, Y.; Bao, Z.; Li, H.; Zheng, P.; Zhao, S. Gastric cancer-derived mesenchymal stromal cells trigger M2 macrophage polarization that promotes metastasis and EMT in gastric cancer. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Ziani, L.; Ben Safta-Saadoun, T.; Gourbeix, J.; Cavalcanti, A.; Robert, C.; Favre, G.; Chouaib, S.; Thiery, J. Melanoma-associated fibroblasts decrease tumor cell susceptibility to NK cell-mediated killing through matrix-metalloproteinases secretion. Oncotarget 2017, 8, 19780–19794. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, Y.; Gallegos, V.; Sorrelle, N.; Zaid, M.M.; Toombs, J.; Du, W.; Wright, S.; Hagopian, M.; Wang, Z.; et al. Targeting TGFbetaR2-mutant tumors exposes vulnerabilities to stromal TGFbeta blockade in pancreatic cancer. EMBO Mol. Med. 2019, 11, e10515. [Google Scholar] [CrossRef]
- Li, T.; Yi, S.; Liu, W.; Jia, C.; Wang, G.; Hua, X.; Tai, Y.; Zhang, Q.; Chen, G. Colorectal carcinoma-derived fibroblasts modulate natural killer cell phenotype and antitumor cytotoxicity. Med. Oncol. 2013, 30, 663. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-Associated Neutrophils in Cancer: Going Pro. Cancers 2019, 11, 564. [Google Scholar] [CrossRef] [PubMed]
- Soto-Pérez-De-Celis, E.; Chavarri-Guerra, Y.; Leon-Rodriguez, E.; Gamboa-Dominguez, A. Tumor-Associated Neutrophils in Breast Cancer Subtypes. Asian Pac. J. Cancer Prev. 2017, 18, 2689–2694. [Google Scholar]
- Munir, H.; Jones, J.O.; Janowitz, T.; Martins, C.P.; Welsh, S.J.; Shields, J.D. Stromal Amyloid β drives Neutrophil extracellular trap formation to augment tumour growth. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Tesi, R. MDSC; the Most Important Cell You Have Never Heard Of. Trends Pharmacol. Sci. 2019, 40, 4–7. [Google Scholar] [CrossRef]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+CD115+Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef]
- Xiang, H.; Ramil, C.P.; Hai, J.; Zhang, C.; Wang, H.; Watkins, A.A.; Afshar, R.; Georgiev, P.; Sze, M.A.; Song, X.S.; et al. Cancer-Associated Fibroblasts Promote Immunosuppression by Inducing ROS-Generating Monocytic MDSCs in Lung Squamous Cell Carcinoma. Cancer Immunol. Res. 2020, 8, 436–450. [Google Scholar] [CrossRef]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.M.; Gillen, A.E.; Cittelly, D.M.; Tan, A.-C.; Sams, S.B.; Pillai, M.M.; Elias, A.D.; Robinson, W.A.; et al. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin. Cancer Res. 2016, 23, 1710–1721. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2016, 36, 1770–1778. [Google Scholar] [CrossRef]
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics 2018, 8, 3932–3948. [Google Scholar] [CrossRef]
- Leung, C.S.; Yeung, T.-L.; Yip, K.-P.; Wong, K.-K.; Ho, S.Y.; Mangala, L.S.; Sood, A.K.; Lopez-Berestein, G.; Sheng, J.; Wong, S.T.; et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J. Clin. Investig. 2017, 128, 589–606. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Kemi, N.; Eskuri, M.; Herva, A.; Leppänen, J.; Huhta, H.; Helminen, O.; Saarnio, J.; Karttunen, T.; Kauppila, J. Tumour-stroma ratio and prognosis in gastric adenocarcinoma. Br. J. Cancer 2018, 119, 435–439. [Google Scholar] [CrossRef]
- Graizel, D.; Zlotogorski-Hurvitz, A.; Tsesis, I.; Rosen, E.; Kedem, R.; Vered, M. Oral cancer-associated fibroblasts predict poor survival: Systematic review and meta-analysis. Oral Dis. 2019, 26, 733–744. [Google Scholar] [CrossRef]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, W.; Zhang, X.; Huang, B.; Chen, A.; He, Y.; Wang, J.; Li, X. Galunisertib inhibits glioma vasculogenic mimicry formation induced by astrocytes. Sci. Rep. 2016, 6, 23056. [Google Scholar] [CrossRef]
- Liang, Y.; Zhu, F.; Zhang, H.; Chen, D.; Zhang, X.; Gao, Q.; Lia, Y. Conditional ablation of TGF-beta signaling inhibits tumor progression and invasion in an induced mouse bladder cancer model. Sci. Rep. 2016, 6, 29479. [Google Scholar] [CrossRef]
- Luangmonkong, T.; Suriguga, S.; Bigaeva, E.; Boersema, M.; Oosterhuis, R.; De Jong, K.P.; Schuppan, D.; Mutsaers, H.A.M.; Olinga, P. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br. J. Pharmacol. 2017, 174, 3107–3117. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.C.; Wan, Z.; Sheard, M.A.; Sun, J.; Jackson, J.R.; Malvar, J.; Xu, Y.; Wang, L.; Sposto, R.; Kim, E.S.; et al. TGFbetaR1 Blockade with Galunisertib (LY2157299) Enhances Anti-Neuroblastoma Activity of the Anti-GD2 Antibody Dinutuximab (ch14.18) with Natural Killer Cells. Clin. Cancer Res. 2017, 23, 804–813. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef]
- Spender, L.C.; Ferguson, G.J.; Hughes, G.D.; Davies, B.R.; Goldberg, F.W.; Herrera, B.; Taylor, R.G.; Strathearn, L.S.; Sansom, O.J.; Barry, S.T.; et al. Preclinical Evaluation of AZ12601011 and AZ12799734, Inhibitors of Transforming Growth Factor β Superfamily Type 1 Receptors. Mol. Pharmacol. 2018, 95, 222–234. [Google Scholar] [CrossRef]
- Song, B.; Park, S.-H.; Zhao, J.C.; Fong, K.-W.; Li, S.; Lee, Y.; Yang, Y.A.; Sridhar, S.; Lu, X.; Abdulkadir, S.A.; et al. Targeting FOXA1-mediated repression of TGF-β signaling suppresses castration-resistant prostate cancer progression. J. Clin. Investig. 2018, 129, 569–582. [Google Scholar] [CrossRef]
- Paller, C.; Pu, H.; Begemann, D.E.; Wade, C.A.; Hensley, P.J.; Kyprianou, N. TGF-beta receptor I inhibitor enhances response to enzalutamide in a pre-clinical model of advanced prostate cancer. Prostate 2019, 79, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Lahn, M.M.; Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, E.K.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I Study of GC1008 (Fresolimumab): A Human Anti-Transforming Growth Factor-Beta (TGFβ) Monoclonal Antibody in Patients with Advanced Malignant Melanoma or Renal Cell Carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Formenti, S.C.; Lee, P.; Adams, S.; Goldberg, J.D.; Li, X.; Xie, M.W.; Ratikan, J.A.; Felix, C.; Hwang, L.; Faull, K.F.; et al. Focal Irradiation and Systemic TGFbeta Blockade in Metastatic Breast Cancer. Clin. Cancer Res. 2018, 24, 2493–2504. [Google Scholar] [CrossRef]
- Schlingensiepen, K.-H.; Jaschinski, F.; Lang, S.A.; Moser, C.; Geissler, E.K.; Schlitt, H.J.; Kielmanowicz, M.; Schneider, A. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011, 102, 1193–1200. [Google Scholar] [CrossRef]
- Hwang, L.; Ng, K.; Wang, W.; Trieu, V.N. OT-101: An anti-TGF-beta-2 antisense- primed tumors to subsequent chemotherapies. J. Clin. Oncol. 2016, 34, e15727. [Google Scholar] [CrossRef]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci. Transl. Med. 2018, 10, eaan5488. [Google Scholar] [CrossRef]
- Strauss, J.; Heery, C.R.; Schlom, J.; Madan, R.A.; Cao, L.; Kang, Z.; Lamping, E.; Marté, J.L.; Donahue, R.N.; Grenga, I.; et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFβ, in Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 1287–1295. [Google Scholar] [CrossRef]
- Haugsten, E.M.; Wiedlocha, A.; Olsnes, S.; Wesche, J. Roles of Fibroblast Growth Factor Receptors in Carcinogenesis. Mol. Cancer Res. 2010, 8, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.F.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Bahleda, R.; Italiano, A.; Hierro, C.; Mita, A.C.; Cervantes, A.; Chan, N.; Awad, M.M.; Calvo, E.; Moreno, V.; Govindan, R.; et al. Multicenter Phase I Study of Erdafitinib (JNJ-42756493), Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced or Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 4888–4897. [Google Scholar] [CrossRef]
- Seckl, M.; Badman, P.D.; Liu, X.; MacPherson, I.; Zubairi, I.H.; Baird, R.; Garcia-Corbacho, J.; Cresti, N.; Plummer, E.R.; Armstrong, A.C.; et al. RADICAL trial: A phase Ib/IIa study to assess the safety and efficacy of AZD4547 in combination with either anastrozole or letrozole in ER positive breast cancer patients progressing on these aromatase inhibitors (AIs). J. Clin. Oncol. 2017, 35, 1059. [Google Scholar] [CrossRef]
- Lam, W.-S.; Creaney, J.; Chen, F.K.; Chin, W.L.; Muruganandan, S.; Arunachalam, S.; Attia, M.S.; Read, C.; Murray, K.; Millward, M.; et al. A phase II trial of single oral FGF inhibitor, AZD4547, as second or third line therapy in malignant pleural mesothelioma. Lung Cancer 2020, 140, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Redman, M.W.; Lara, P.N.; Borghaei, H.; Hoffman, P.; Bradley, J.D.; Newman, A.J.; Feldman, M.J.; Minichiello, K.; Miao, J.; et al. SWOG S1400D (NCT02965378), a Phase II Study of the Fibroblast Growth Factor Receptor Inhibitor AZD4547 in Previously Treated Patients With Fibroblast Growth Factor Pathway–Activated Stage IV Squamous Cell Lung Cancer (Lung-MAP Substudy). J. Thorac. Oncol. 2019, 14, 1847–1852. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Bang, Y.-J.; Mansoor, W.; Petty, R.D.; Chao, Y.; Cunningham, D.; Ferry, D.R.; Smith, N.R.; Frewer, P.; Ratnayake, J.; et al. A randomized, open-label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann. Oncol. 2017, 28, 1316–1324. [Google Scholar] [CrossRef]
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.-W.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.-Y.; et al. First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1696–1707. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. All in the CCN family: Essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 2006, 119, 4803–4810. [Google Scholar] [CrossRef] [PubMed]
- Hutchenreuther, J.; Vincent, K.; Norley, C.; Racanelli, M.; Gruber, S.B.; Johnson, T.M.; Fullen, D.R.; Raskin, L.; Perbal, B.; Holdsworth, D.W.; et al. Activation of cancer-associated fibroblasts is required for tumor neovascularization in a murine model of melanoma. Matrix Boil. 2018, 74, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Picozzi, V.J.; Pipas, J.M.; Koong, A.; Giaccia, A.; Bahary, N.; Krishnamurthi, S.S.; Lopez, C.D.; O’Dwyer, P.J.; Modelska, K.; Poolman, V.; et al. FG-3019, a human monoclonal antibody to CTGF, with gemcitabine/erlotinib in patients with locally advanced or metastatic pancreatic ductal adenocarcinoma. J. Clin. Oncol. 2013, 31, 213. [Google Scholar] [CrossRef]
- Rudin, C.M. Vismodegib. Clin. Cancer Res. 2012, 18, 3218–3222. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.; Hamid, O.; et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: Final update of the pivotal ERIVANCE BCC study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog–Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br. J. Cancer 2019, 122, 498–505. [Google Scholar] [CrossRef]
- Berlin, J.D.; Bendell, J.C.; Hart, L.L.; Firdaus, I.; Gore, I.; Hermann, R.C.; Mulcahy, M.F.; Zalupski, M.M.; Mackey, H.M.; Yauch, R.L.; et al. A Randomized Phase II Trial of Vismodegib versus Placebo with FOLFOX or FOLFIRI and Bevacizumab in Patients with Previously Untreated Metastatic Colorectal Cancer. Clin. Cancer Res. 2012, 19, 258–267. [Google Scholar] [CrossRef]
- Cohen, D.J.; Christos, P.J.; Kindler, H.L.; Catenacci, D.V.T.; Bekaii-Saab, T.B.; Tahiri, S.; Janjigian, Y.Y.; Gibson, M.K.; Chan, E.; Rajdev, L.; et al. Vismodegib (V), a hedgehog (HH) pathway inhibitor, combined with FOLFOX for first-line therapy of patients (pts) with advanced gastric and gastroesophageal junction (GEJ) carcinoma: A New York Cancer Consortium led phase II randomized study. J. Clin. Oncol. 2013, 31, 4011. [Google Scholar] [CrossRef]
- Belani, C.P.; Dahlberg, S.E.; Rudin, C.M.; Fleisher, M.; Chen, H.X.; Takebe, N.; Ramalingam, S.S.; Schiller, J.H. Three-arm randomized phase II study of cisplatin and etoposide (CE) versus CE with either vismodegib (V) or cixutumumab (Cx) for patients with extensive stage-small cell lung cancer (ES-SCLC) (ECOG 1508). J. Clin. Oncol. 2013, 31, 7508. [Google Scholar] [CrossRef]
- Kaye, S.B.; Fehrenbacher, L.; Holloway, R.; Amit, A.; Karlan, B.; Slomovitz, B.; Sabbatini, P.; Fu, L.; Yauch, R.L.; Chang, I.; et al. A Phase II, Randomized, Placebo-Controlled Study of Vismodegib as Maintenance Therapy in Patients with Ovarian Cancer in Second or Third Complete Remission. Clin. Cancer Res. 2012, 18, 6509–6518. [Google Scholar] [CrossRef]
- Ruiz-Borrego, M.; Jimenez, B.; Antolín, S.; García-Saenz, J.A.; Corral, J.; Jerez, Y.; Trigo, J.; Urruticoechea, A.; Colom, H.; Gonzalo, N.; et al. A phase Ib study of sonidegib (LDE225), an oral small molecule inhibitor of smoothened or Hedgehog pathway, in combination with docetaxel in triple negative advanced breast cancer patients: GEICAM/2012–12 (EDALINE) study. Investig. New Drugs 2019, 37, 98–108. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; García, R.; Ghassemi, S.; Fabry, B.; et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef]
- Banerjee, S.; Modi, S.; McGinn, O.; Zhao, X.; Dudeja, V.; Ramakrishnan, S.; Saluja, A.K. Impaired Synthesis of Stromal Components in Response to Minnelide Improves Vascular Function, Drug Delivery, and Survival in Pancreatic Cancer. Clin. Cancer Res. 2015, 22, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, U.; Mustafi, R.; Haider, H.I.; Khalil, A.; Souris, J.S.; Joseph, L.; Hart, J.; Konda, V.J.; Zhang, W.; Pekow, J.; et al. Losartan and Vitamin D Inhibit Colonic Tumor Development in a Conditional Apc-Deleted Mouse Model of Sporadic Colon Cancer. Cancer Prev. Res. 2019, 12, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cao, J.; Melamed, A.; Worley, M.; Gockley, A.; Jones, D.; Nia, H.T.; Zhang, Y.; Stylianopoulos, T.; Kumar, A.S.; et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2019, 116, 2210–2219. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Liu, X.; Ran, W.; Meng, J.; Zhai, Y.; Zhang, P.; Yin, Q.; Yu, H.; Zhang, Z.; Li, Y. Regulating cancer associated fibroblasts with losartan-loaded injectable peptide hydrogel to potentiate chemotherapy in inhibiting growth and lung metastasis of triple negative breast cancer. Biomaterials 2017, 144, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1020–1027. [Google Scholar] [CrossRef]
- Benson, A.B.; Wainberg, Z.A.; Hecht, J.R.; Vyushkov, D.; Dong, H.; Bendell, J.; Kudrik, F. A Phase II Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab or Placebo in Combination with Gemcitabine for the First-Line Treatment of Pancreatic Adenocarcinoma. Oncologist 2017, 22, 241. [Google Scholar] [CrossRef]
- Hecht, J.R.; Benson, A.B., 3rd; Vyushkov, D.; Yang, Y.; Bendell, J.; Verma, U. A Phase II, Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab in Combination with FOLFIRI for the Second-Line Treatment of Metastatic KRAS Mutant Colorectal Adenocarcinoma. Oncologist 2017, 22, 243. [Google Scholar] [CrossRef]
- Boluda, A.N.; Vaquero, J.; Laurent, G.; Renault, G.; Bazzi, R.; Donnadieu, E.; Roux, S.; Fouassier, L.; Gazeau, F. Photothermal Depletion of Cancer-Associated Fibroblasts Normalizes Tumor Stiffness in Desmoplastic Cholangiocarcinoma. ACS Nano 2020, 14, 5738–5753. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Pernas, S.; Martín, M.; Kaufman, P.A.; Gil-Martin, M.; Pardo, P.G.; Lopez-Tarruella, S.; Manso, L.; Ciruelos, E.M.; Perez-Fidalgo, J.A.; Hernando, C.; et al. Balixafortide plus eribulin in HER2-negative metastatic breast cancer: A phase 1, single-arm, dose-escalation trial. Lancet Oncol. 2018, 19, 812–824. [Google Scholar] [CrossRef]
- Loeffler, M.; Krüger, J.A.; Niethammer, A.G.; Reisfeld, R.A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Investig. 2006, 116, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J.; Dillman, R.O.; Schwarzenberger, P.O.; Senzer, N.; Cunningham, C.; Cutler, J.; Tong, A.; Kumar, P.; Pappen, B.; Hamilton, C.; et al. Phase II Study of Belagenpumatucel-L, a Transforming Growth Factor Beta-2 Antisense Gene-Modified Allogeneic Tumor Cell Vaccine in Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2006, 24, 4721–4730. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Bazhenova, L.; Nemunaitis, J.; Tan, M.; Juhász, E.; Ramlau, R.; Heuvel, M.V.D.; Lal, R.; Kloecker, G.; Eaton, K.; et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur. J. Cancer 2015, 51, 2321–2329. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganguly, D.; Chandra, R.; Karalis, J.; Teke, M.; Aguilera, T.; Maddipati, R.; Wachsmann, M.B.; Ghersi, D.; Siravegna, G.; Zeh, H.J., III; et al. Cancer-Associated Fibroblasts: Versatile Players in the Tumor Microenvironment. Cancers 2020, 12, 2652. https://doi.org/10.3390/cancers12092652
Ganguly D, Chandra R, Karalis J, Teke M, Aguilera T, Maddipati R, Wachsmann MB, Ghersi D, Siravegna G, Zeh HJ III, et al. Cancer-Associated Fibroblasts: Versatile Players in the Tumor Microenvironment. Cancers. 2020; 12(9):2652. https://doi.org/10.3390/cancers12092652
Chicago/Turabian StyleGanguly, Debolina, Raghav Chandra, John Karalis, Martha Teke, Todd Aguilera, Ravikanth Maddipati, Megan B. Wachsmann, Dario Ghersi, Giulia Siravegna, Herbert J. Zeh, III, and et al. 2020. "Cancer-Associated Fibroblasts: Versatile Players in the Tumor Microenvironment" Cancers 12, no. 9: 2652. https://doi.org/10.3390/cancers12092652
APA StyleGanguly, D., Chandra, R., Karalis, J., Teke, M., Aguilera, T., Maddipati, R., Wachsmann, M. B., Ghersi, D., Siravegna, G., Zeh, H. J., III, Brekken, R., Ting, D. T., & Ligorio, M. (2020). Cancer-Associated Fibroblasts: Versatile Players in the Tumor Microenvironment. Cancers, 12(9), 2652. https://doi.org/10.3390/cancers12092652