Communication in the Cancer Microenvironment as a Target for Therapeutic Interventions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
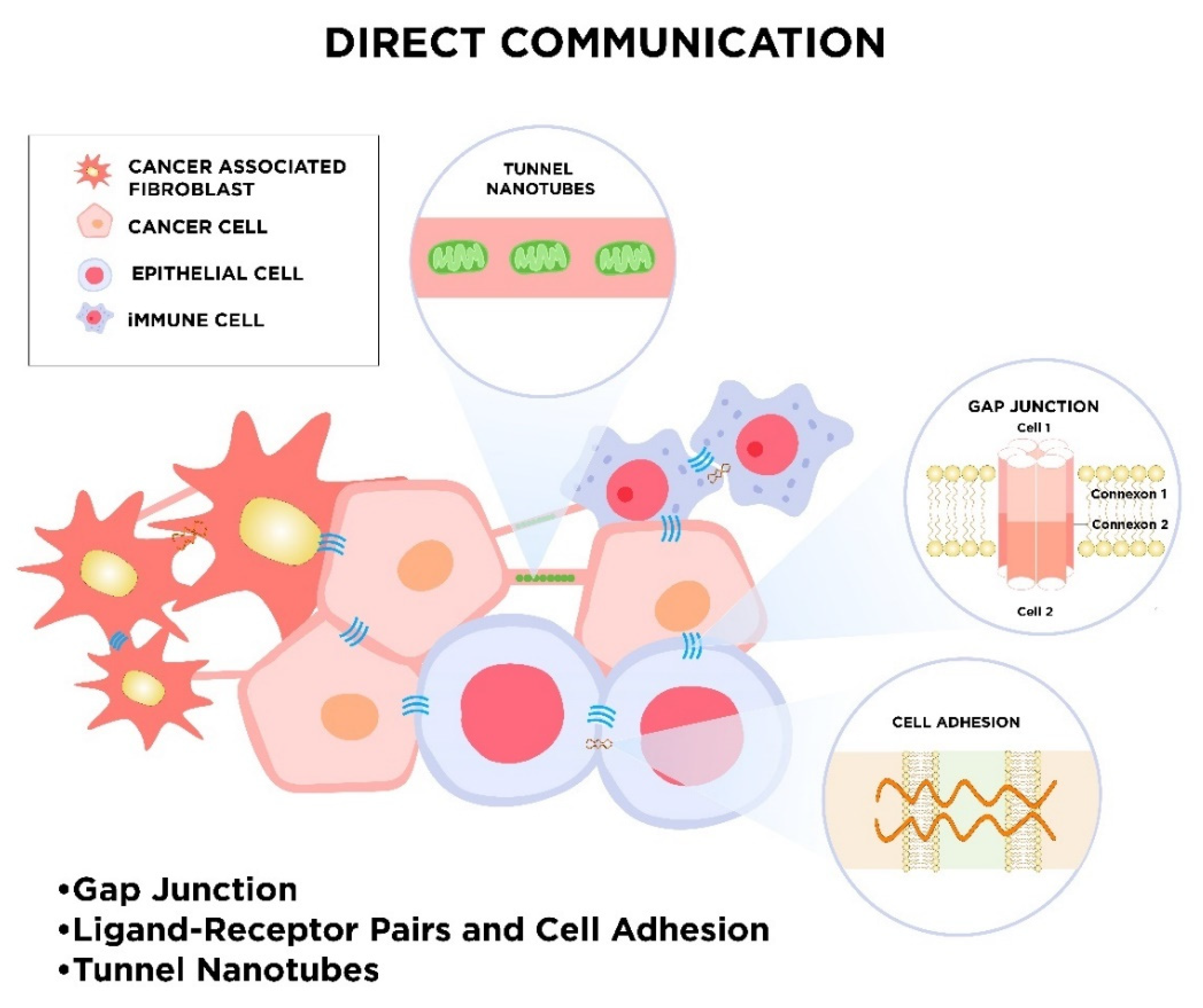
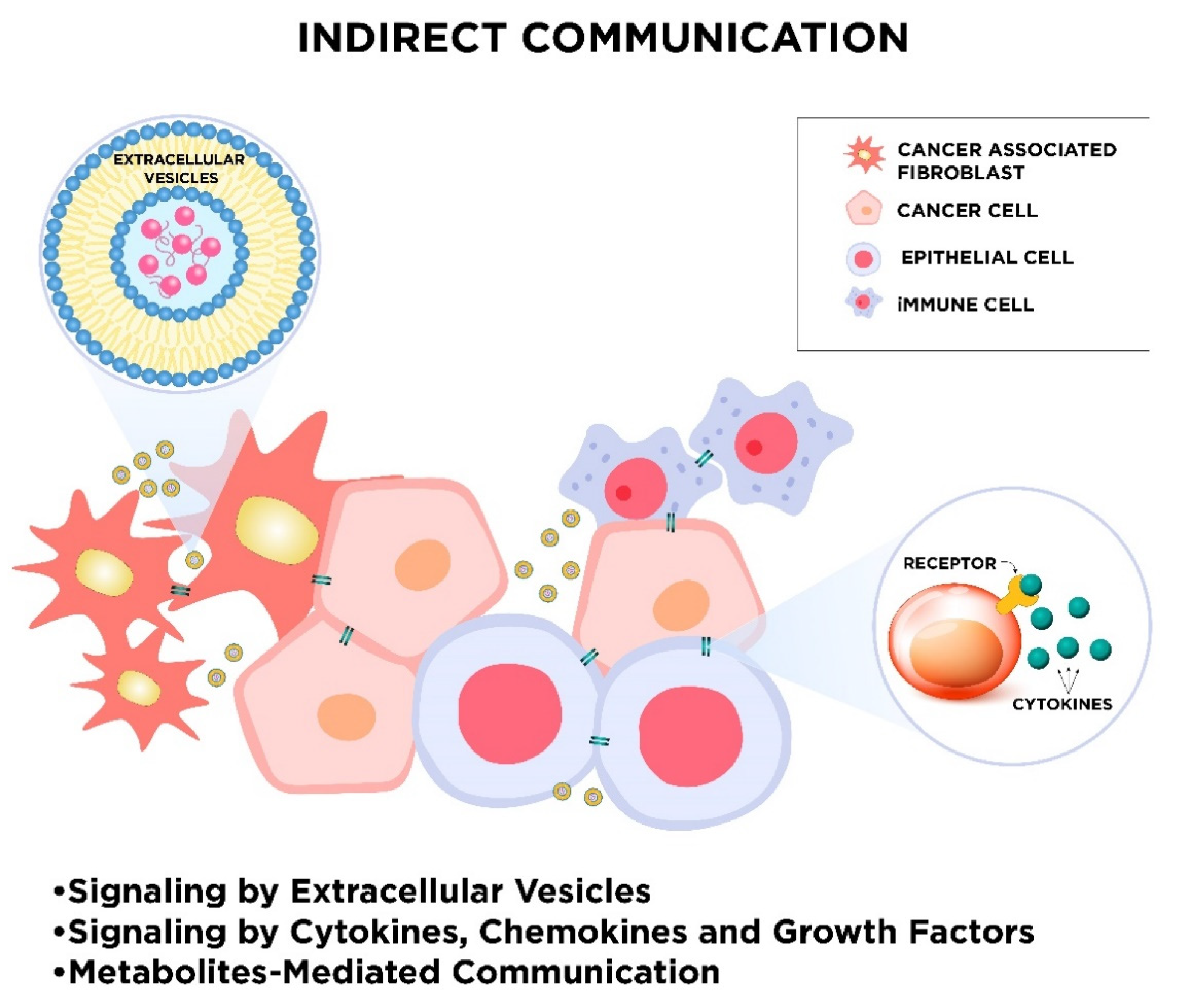
2. Mechanisms of Cellular Communication
2.1. Direct Intercellular Communication
2.1.1. Gap Junctions
2.1.2. Ligand–Receptor Pairs and Cell Adhesion
2.1.3. Tunnel Nanotubes called “Intercellular Bridges”
2.2. Indirect Intercellular Communication
2.2.1. Signaling by Extracellular Vesicles
2.2.2. Signaling by Cytokines, Chemokines, and Growth Factors
2.2.3. Metabolites-Mediated Communication
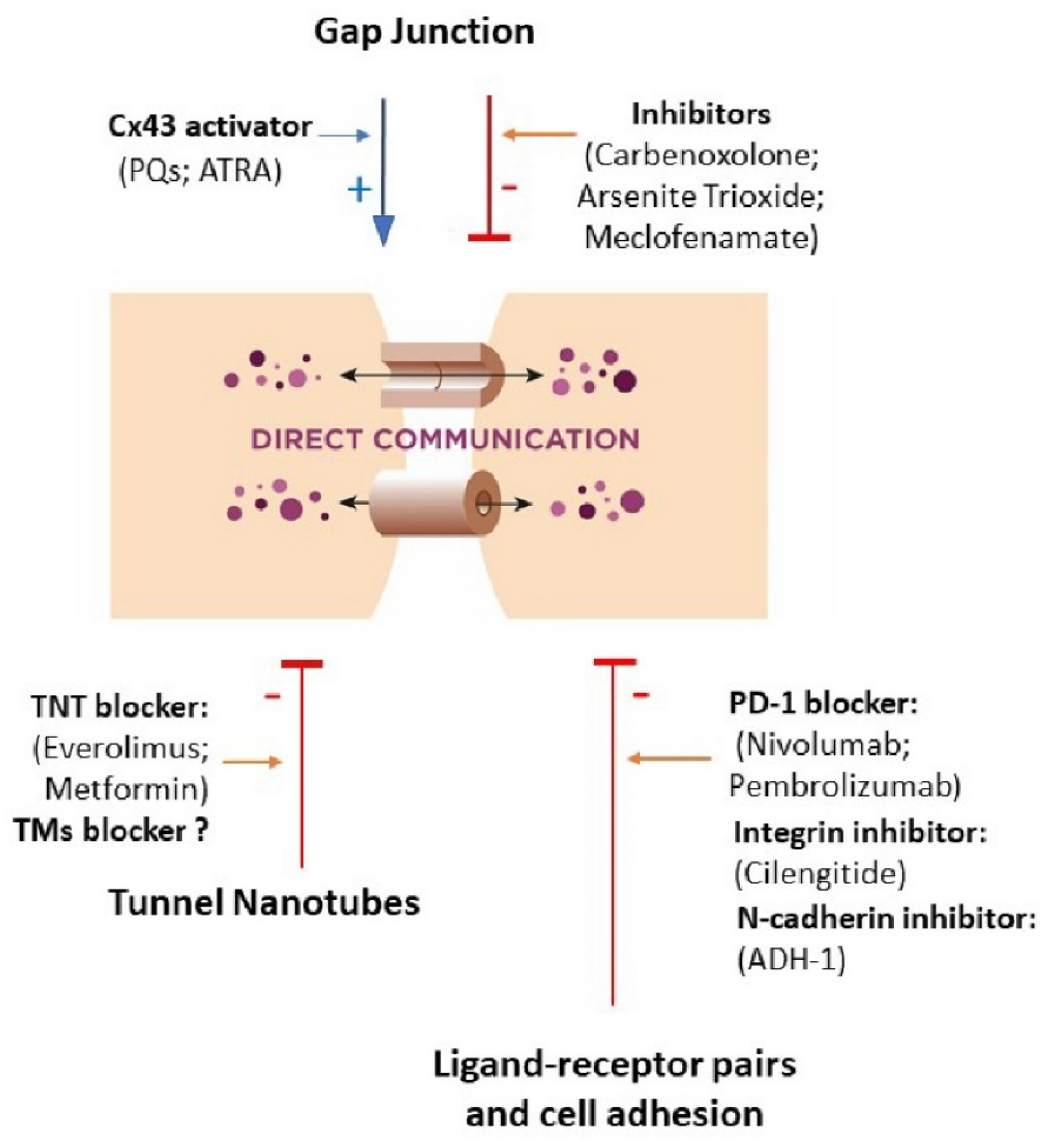
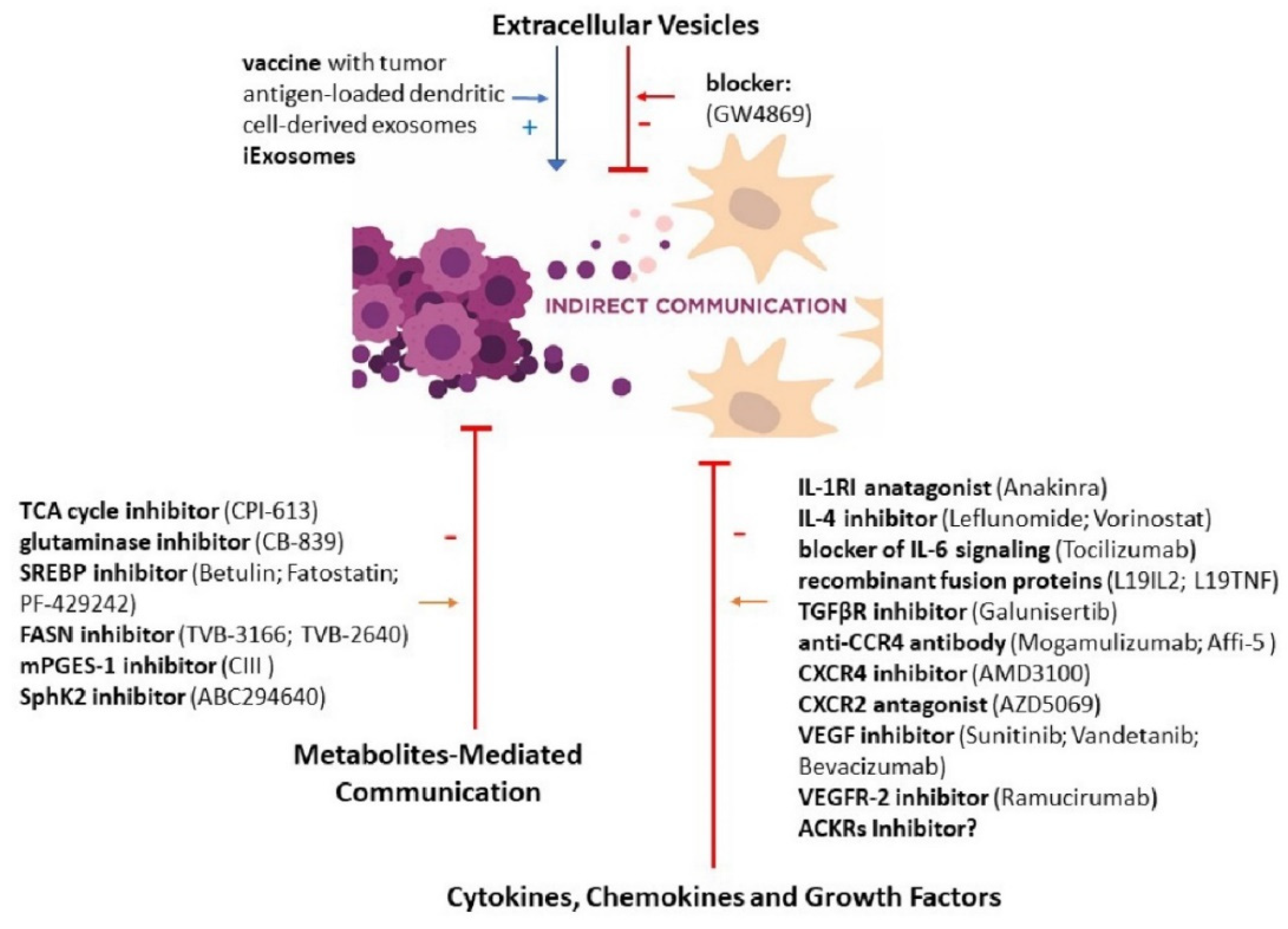
3. New Therapeutic Perspectives Targeted at Cellular Communication
3.1. Gap Junction
3.2. Ligand–Receptor Pairs and Cell Adhesion
3.3. Tunnel Nanotubes
3.4. Extracellular Vesicles
3.5. Cytokines, Chemokines and Growth Factors
3.6. Metabolites-Mediated Communication
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethical Approval
References
- Bode, A.M.; Dong, Z. Cancer prevention research—Then and now. Nat. Rev. Cancer 2009, 9, 508–516. [Google Scholar] [CrossRef]
- Helsper, C.C.W.; van Erp, N.N.F.; Peeters, P.; de Wit, N.N.J. Time to diagnosis and treatment for cancer patients in the Netherlands: Room for improvement? Eur. J. Cancer 2017, 87, 113–121. [Google Scholar] [CrossRef]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Bergfeld, S.A.; DeClerck, Y.A. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev. 2010, 29, 249–261. [Google Scholar] [CrossRef]
- Walker, C.; Mojares, E.; Del Rio Hernandez, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef]
- Taube, J.M.; Galon, J.; Sholl, L.M.; Rodig, S.J.; Cottrell, T.R.; Giraldo, N.A.; Baras, A.S.; Patel, S.S.; Anders, R.A.; Rimm, D.L.; et al. Implications of the tumor immune microenvironment for staging and therapeutics. Modem Pathol. 2018, 31, 214–234. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Polak, R.; de Rooij, B.; Pieters, R.; den Boer, M.L. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef]
- De la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Bourget, I.; Pons, C.; Butet, V.; Hofman, P.; Tartare-Deckert, S.; Feral, C.C.; Meneguzzi, G.; Gaggioli, C. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014, 7, 1664–1678. [Google Scholar] [CrossRef] [PubMed]
- Foster, C.T.; Gualdrini, F.; Treisman, R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017, 31, 2361–2375. [Google Scholar] [CrossRef] [PubMed]
- Scharenberg, M.A.; Pippenger, B.E.; Sack, R.; Zingg, D.; Ferralli, J.; Schenk, S.; Martin, I.; Chiquet-Ehrismann, R. TGF-β-induced differentiation into myofibroblasts involves specific regulation of two MKL1 isoforms. J. Cell Sci. 2014, 127, 1079–1091. [Google Scholar] [CrossRef]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayán, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef]
- Qu, Y.; Dahl, G. Function of the voltage gate of gap junction channels: Selective exclusion of molecules. Proc. Natl. Acad. Sci. USA 2002, 99, 697–702. [Google Scholar] [CrossRef]
- Beckmann, A.; Hainz, N.; Tschernig, T.; Meier, C. Facets of Communication: Gap Junction Ultrastructure and Function in Cancer Stem Cells and Tumor Cells. Cancers (Basel) 2019, 11, 288. [Google Scholar] [CrossRef]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Sorgen, P.L.; Trease, A.J.; Spagnol, G.; Delmar, M.; Nielsen, M.S. Protein(-)Protein Interactions with Connexin 43: Regulation and Function. Int. J. Mol. Sci. 2018, 19, 1428. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W.R.; Kanno, Y. Intercellular communication and the control of tissue growth: Lack of communication between cancer cells. Nature 1966, 209, 1248–1249. [Google Scholar] [CrossRef] [PubMed]
- Babica, P.; Ctverackova, L.; Lencesova, Z.; Trosko, J.E.; Upham, B.L. Chemopreventive Agents Attenuate Rapid Inhibition of Gap Junctional Intercellular Communication Induced by Environmental Toxicants. Nutr. Cancer 2016, 68, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Chen, H.J.; Chen, S.H.; Xue, X.Y.; Chen, H.; Zheng, Q.S.; Wei, Y.; Li, X.D.; Huang, J.B.; Cai, H.; et al. Reduced Connexin 43 expression is associated with tumor malignant behaviors and biochemical recurrence-free survival of prostate cancer. Oncotarget 2016, 7, 67476–67484. [Google Scholar] [CrossRef]
- Yeh, E.S.; Williams, C.J.; Williams, C.B.; Bonilla, I.V.; Klauber-DeMore, N.; Phillips, S.L. Dysregulated connexin 43 in HER2-positive drug resistant breast cancer cells enhances proliferation and migration. Oncotarget 2017, 8, 109358–109369. [Google Scholar] [CrossRef]
- Arshad, M.; Conzelmann, C.; Riaz, M.A.; Noll, T.; Gunduz, D. Inhibition of Cx43 attenuates ERK1/2 activation, enhances the expression of Cav1 and suppresses cell proliferation. Int. J. Mol. Med. 2018, 42, 2811–2818. [Google Scholar] [CrossRef]
- Sinyuk, M.; Mulkearns-Hubert, E.E.; Reizes, O.; Lathia, J. Cancer Connectors: Connexins, Gap Junctions, and Communication. Front. Oncol. 2018, 8, 646. [Google Scholar] [CrossRef]
- Kar, R.; Batra, N.; Riquelme, M.A.; Jiang, J.X. Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 2012, 524, 2–15. [Google Scholar] [CrossRef]
- Sin, W.C.; Crespin, S.; Mesnil, M. Opposing roles of connexin43 in glioma progression. Biochim. Biophys. Acta 2012, 1818, 2058–2067. [Google Scholar] [CrossRef]
- Arora, S.; Heyza, J.R.; Chalfin, E.C.; Ruch, R.J.; Patrick, S.M. Gap Junction Intercellular Communication Positively Regulates Cisplatin Toxicity by Inducing DNA Damage through Bystander Signaling. Cancers (Basel) 2018, 10, 368. [Google Scholar] [CrossRef] [PubMed]
- Mattes, B.; Scholpp, S. Emerging role of contact-mediated cell communication in tissue development and diseases. Histochem. Cell Biol. 2018, 150, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Bosenberg, M.W.; Massague, J. Juxtacrine cell signaling molecules. Curr. Opin. Cell Biol. 1993, 5, 832–838. [Google Scholar] [CrossRef]
- Cuatrecasas, P. Membrane receptors. Annu. Rev. Biochem. 1974, 43, 169–214. [Google Scholar] [CrossRef]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Gonzalez-Mariscal, L.; Tapia, R.; Chamorro, D. Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta 2008, 1778, 729–756. [Google Scholar] [CrossRef]
- Ikenouchi, J.; Matsuda, M.; Furuse, M.; Tsukita, S. Regulation of tight junctions during the epithelium-mesenchyme transition: Direct repression of the gene expression of claudins/occludin by Snail. J. Cell Sci. 2003, 116, 1959–1967. [Google Scholar] [CrossRef]
- Martin, T.A. The role of tight junctions in cancer metastasis. Semin. Cell Dev. Biol. 2014, 36, 224–231. [Google Scholar] [CrossRef]
- Ren, J.; Hamada, J.; Takeichi, N.; Fujikawa, S.; Kobayashi, H. Ultrastructural differences in junctional intercellular communication between highly and weakly metastatic clones derived from rat mammary carcinoma. Cancer Res. 1990, 50, 358–362. [Google Scholar]
- Takeichi, M. Dynamic contacts: Rearranging adherens junctions to drive epithelial remodelling. Nat. Rev. Mol. Cell Biol. 2014, 15, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.Q.; Li, P.; Ge, M.H.; Zhao, X.; Hu, F.J.; Fang, X.H.; Dong, Z.M.; Mao, W.M. Hypermethylation-modulated down-regulation of CDH1 expression contributes to the progression of esophageal cancer. Int. J. Mol. Med. 2011, 27, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.; Johnson, J.P.; Leitl, F.; Jauch, K.W.; Heiss, M.M.; Schildberg, F.W.; Birchmeier, W.; Funke, I. E-cadherin expression in primary and metastatic gastric cancer: Down-regulation correlates with cellular dedifferentiation and glandular disintegration. Cancer Res. 1993, 53, 1690–1695. [Google Scholar]
- Zhai, B.; Yan, H.X.; Liu, S.Q.; Chen, L.; Wu, M.C.; Wang, H.Y. Reduced expression of E-cadherin/catenin complex in hepatocellular carcinomas. World J. Gastroenterol. 2008, 14, 5665–5673. [Google Scholar] [CrossRef] [PubMed]
- Shiina, H.; Breault, J.E.; Basset, W.W.; Enokida, H.; Urakami, S.; Li, L.C.; Okino, S.T.; Deguchi, M.; Kaneuchi, M.; Terashima, M.; et al. Functional Loss of the gamma-catenin gene through epigenetic and genetic pathways in human prostate cancer. Cancer Res. 2005, 65, 2130–2138. [Google Scholar] [CrossRef] [PubMed]
- Dusek, R.L.; Attardi, L.D. Desmosomes: New perpetrators in tumour suppression. Nat. Rev. Cancer 2011, 11, 317–323. [Google Scholar] [CrossRef]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling Nanotubes and Tumor Microtubes in Cancer. Cancers (Basel) 2020, 12, 857. [Google Scholar] [CrossRef]
- Drab, M.; Stopar, D.; Kralj-Iglic, V.; Iglic, A. Inception Mechanisms of Tunneling Nanotubes. Cells 2019, 8, 626. [Google Scholar] [CrossRef]
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T.; Morel, S.; Kwak, B.R.; Girao, H. Role of connexin 43 in different forms of intercellular communication—Gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630. [Google Scholar] [CrossRef]
- Desir, S.; Wong, P.; Turbyville, T.; Chen; Shetty, M.; Clark, C.; Zhai, E.; Romin, Y.; Manova-Todorova, K.; Starr, T.K.; et al. Intercellular Transfer of Oncogenic KRAS via Tunneling Nanotubes Introduces Intracellular Mutational Heterogeneity in Colon Cancer Cells. Cancers (Basel) 2019, 11, 892. [Google Scholar] [CrossRef]
- D’Aloia, A.; Berruti, G.; Costa, B.; Schiller, C.; Ambrosini, R.; Pastori, V.; Martegani, E.; Ceriani, M. RalGPS2 is involved in tunneling nanotubes formation in 5637 bladder cancer cells. Exp. Cell Res. 2018, 362, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Asencio-Barria, C.; Defamie, N.; Saez, J.C.; Mesnil, M.; Godoy, A.S. Direct Intercellular Communications and Cancer: A Snapshot of the Biological Roles of Connexins in Prostate Cancer. Cancers (Basel) 2019, 11, 1370. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Galas, L.; Boulangé-Lecomte, C.; Rioult, D.; Bultelle, F.; Magal, P.; Webb, G.; Le Foll, F. Different modalities of intercellular membrane exchanges mediate cell-to-cell p-glycoprotein transfers in MCF-7 breast cancer cells. J. Biol. Chem. 2012, 287, 7374–7387. [Google Scholar] [CrossRef] [PubMed]
- Thayanithy, V.; Dickson, E.L.; Steer, C.; Subramanian, S.; Lou, E. Tumor-stromal cross talk: Direct cell-to-cell transfer of oncogenic microRNAs via tunneling nanotubes. Transl. Res. 2014, 164, 359–365. [Google Scholar] [CrossRef]
- Nawaz, M.; Fatima, F. Extracellular Vesicles, Tunneling Nanotubes, and Cellular Interplay: Synergies and Missing Links. Front. Mol. Biosci. 2017, 4, 50. [Google Scholar] [CrossRef]
- Murray, L.M.A.; Krasnodembskaya, A.D. Concise Review: Intercellular Communication Via Organelle Transfer in the Biology and Therapeutic Applications of Stem Cells. Stem Cells 2019, 37, 14–25. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, J.; Sun, X.; Zhang, Y. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 2011, 18, 732–742. [Google Scholar] [CrossRef]
- Babenko, V.A.; Silachev, D.N.; Zorova, L.D.; Pevzner, I.B.; Khutornenko, A.A.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Improving the Post-Stroke Therapeutic Potency of Mesenchymal Multipotent Stromal Cells by Cocultivation with Cortical Neurons: The Role of Crosstalk Between Cells. Stem Cells Transl. Med. 2015, 4, 1011–1020. [Google Scholar] [CrossRef]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Osswald, M.; Solecki, G.; Wick, W.; Winkler, F. A malignant cellular network in gliomas: Potential clinical implications. Neuro Oncol. 2016, 18, 479–485. [Google Scholar] [CrossRef]
- Abounit, S.; Delage, E.; Zurzolo, C. Identification and Characterization of Tunneling Nanotubes for Intercellular Trafficking. Curr. Protoc. Cell Biol. 2015, 67, 12.10.11–12.10.21. [Google Scholar] [CrossRef] [PubMed]
- Okafo, G.; Prevedel, L.; Eugenin, E. Tunneling nanotubes (TNT) mediate long-range gap junctional communication: Implications for HIV cell to cell spread. Sci. Rep. 2017, 7, 16660. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-L.; Sun, P.; Li, Y.; Liu, S.-S.; Lu, Y. Exosomes as critical mediators of cell-to-cell communication in cancer pathogenesis and their potential clinical application. Transl. Cancer Res. 2019, 8, 298–311. [Google Scholar] [CrossRef]
- Kanada, M.; Bachmann, M.H.; Contag, C.H. Signaling by Extracellular Vesicles Advances Cancer Hallmarks. Trends Cancer 2016, 2, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Lotvall, J.; Hill, A.F.; Hochberg, F.; Buzas, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]
- Sadallah, S.; Eken, C.; Martin, P.J.; Schifferli, J.A. Microparticles (ectosomes) shed by stored human platelets downregulate macrophages and modify the development of dendritic cells. J. Immunol. 2011, 186, 6543–6552. [Google Scholar] [CrossRef]
- Wu, M.; Wang, G.; Hu, W.; Yao, Y.; Yu, X.F. Emerging roles and therapeutic value of exosomes in cancer metastasis. Mol. Cancer 2019, 18, 53. [Google Scholar] [CrossRef]
- Savina, A.; Fader, C.M.; Damiani, M.T.; Colombo, M.I. Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic 2005, 6, 131–143. [Google Scholar] [CrossRef]
- Oves, M.; Qari, H.; Felemban, N.; Aslam, A.; Khan, A.A.; Rehan, M.; Tabrez, S.; Ahmed, F.; Haque, A.; Khan, I.; et al. Exosomes: A Paradigm in Drug Development against Cancer and Infectious Diseases. J. Nanomater. 2018, 2018. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Bujanda, L. Glypican-1 in exosomes as biomarker for early detection of pancreatic cancer. Ann. Transl. Med. 2016, 4, 64. [Google Scholar] [CrossRef]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.H.; Wan, Y.L.; Lin, Y.; Zhang, W.; Yang, M.; Li, G.L.; Lin, H.M.; Shang, C.Z.; Chen, Y.J.; Min, J. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J. Biol. Chem. 2012, 287, 15874–15885. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wu, S.; Zhang, K.; Qing, Y.; Xu, T. A comprehensive overview of exosomes in ovarian cancer: Emerging biomarkers and therapeutic strategies. J. Ovarian Res. 2017, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Lopez, L.; Blancas, I.; Garrido, J.M.; Mut-Salud, N.; Moya-Jodar, M.; Osuna, A.; Rodriguez-Serrano, F. The role of exosomes on colorectal cancer: A review. J. Gastroenterol. Hepatol. 2018, 33, 792–799. [Google Scholar] [CrossRef]
- Lowry, M.C.; Gallagher, W.M.; O’Driscoll, L. The Role of Exosomes in Breast Cancer. Clin. Chem. 2015, 61, 1457–1465. [Google Scholar] [CrossRef]
- Tucci, M.; Mannavola, F.; Passarelli, A.; Stucci, L.S.; Cives, M.; Silvestris, F. Exosomes in melanoma: A role in tumor progression, metastasis and impaired immune system activity. Oncotarget 2018, 9, 20826–20837. [Google Scholar] [CrossRef]
- Pan, J.; Ding, M.; Xu, K.; Yang, C.; Mao, L.J. Exosomes in diagnosis and therapy of prostate cancer. Oncotarget 2017, 8, 97693–97700. [Google Scholar] [CrossRef]
- Corcoran, C.; Rani, S.; O’Brien, K.; O’Neill, A.; Prencipe, M.; Sheikh, R.; Webb, G.; McDermott, R.; Watson, W.; Crown, J.; et al. Docetaxel-resistance in prostate cancer: Evaluating associated phenotypic changes and potential for resistance transfer via exosomes. PLoS ONE 2012, 7, e50999. [Google Scholar] [CrossRef]
- Kong, J.N.; He, Q.; Wang, G.; Dasgupta, S.; Dinkins, M.B.; Zhu, G.; Kim, A.; Spassieva, S.; Bieberich, E. Guggulsterone and bexarotene induce secretion of exosome-associated breast cancer resistance protein and reduce doxorubicin resistance in MDA-MB-231 cells. Int. J. Cancer 2015, 137, 1610–1620. [Google Scholar] [CrossRef]
- Xiao, X.; Yu, S.; Li, S.; Wu, J.; Ma, R.; Cao, H.; Zhu, Y.; Feng, J. Exosomes: Decreased sensitivity of lung cancer A549 cells to cisplatin. PLoS ONE 2014, 9, e89534. [Google Scholar] [CrossRef]
- Takahashi, K.; Yan, I.K.; Kogure, T.; Haga, H.; Patel, T. Extracellular vesicle-mediated transfer of long non-coding RNA ROR modulates chemosensitivity in human hepatocellular cancer. FEBS Open Bio 2014, 4, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Z.; Beiter, T.; Schluesener, H.J. Nanovesicular vaccines: Exosomes. Arch. Immunol. Ther. Exp. (Warsz) 2005, 53, 329–335. [Google Scholar] [PubMed]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Exosome and mesenchymal stem cell cross-talk in the tumor microenvironment. Semin. Immunol. 2018, 35, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Y.; Cao, X. The exosomes in tumor immunity. Oncoimmunology 2015, 4, e1027472. [Google Scholar] [CrossRef]
- Sethi, G.; Shanmugam, M.K.; Ramachandran, L.; Kumar, A.P.; Tergaonkar, V. Multifaceted link between cancer and inflammation. Biosci. Rep. 2012, 32, 1–15. [Google Scholar] [CrossRef]
- Amedei, A.; Prisco, D.; MM, D.E. The use of cytokines and chemokines in the cancer immunotherapy. Recent Pat. Anticancer Drug Discov. 2013, 8, 126–142. [Google Scholar] [CrossRef]
- Mantovani, A.; Barajon, I.; Garlanda, C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol. Rev. 2018, 281, 57–61. [Google Scholar] [CrossRef]
- Li, Z.; Chen, L.; Qin, Z. Paradoxical roles of IL-4 in tumor immunity. Cell. Mol. Immunol. 2009, 6, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Middleton, K.; Jones, J.; Lwin, Z.; Coward, J.I. Interleukin-6: An angiogenic target in solid tumours. Crit. Rev. Oncol. Hematol. 2014, 89, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Ben-Baruch, A. The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev. 2006, 25, 357–371. [Google Scholar] [CrossRef]
- Mannino, M.H.; Zhu, Z.; Xiao, H.; Bai, Q.; Wakefield, M.R.; Fang, Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015, 367, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Junius, S.; Benoy, I.; Van Dam, P.; Vermeulen, P.; Van Marck, E.; Huget, P.; Dirix, L.Y. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int. J. Cancer 2003, 103, 642–646. [Google Scholar] [CrossRef]
- Lu, X. Impact of IL-12 in Cancer. Curr. Cancer Drug Targets 2017, 17, 682–697. [Google Scholar] [CrossRef]
- Setrerrahmane, S.; Xu, H. Tumor-related interleukins: Old validated targets for new anti-cancer drug development. Mol. Cancer 2017, 16, 153. [Google Scholar] [CrossRef]
- Choudhry, H.; Helmi, N.; Abdulaal, W.H.; Zeyadi, M.; Zamzami, M.A.; Wu, W.; Mahmoud, M.M.; Warsi, M.K.; Rasool, M.; Jamal, M.S. Prospects of IL-2 in Cancer Immunotherapy. Biomed Res. Int. 2018, 2018, 9056173. [Google Scholar] [CrossRef]
- Martin, M.; Wei, H.; Lu, T. Targeting microenvironment in cancer therapeutics. Oncotarget 2016, 7, 52575–52583. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-beta-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Liu, S.; Chen, S.; Zeng, J. TGF-β signaling: A complex role in tumorigenesis (Review). Mol. Med. Rep. 2018, 17, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Ségui, B. The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef] [PubMed]
- Ion, G.N.D.; Nitulescu, G.M.; Popescu, C.I. Targeting TRAIL. Bioorg Med. Chem. Lett. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Old, L.J.; Schreiber, R.D. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002, 13, 95–109. [Google Scholar] [CrossRef]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef]
- Salcedo, R.; Ponce, M.L.; Young, H.A.; Wasserman, K.; Ward, J.M.; Kleinman, H.K.; Oppenheim, J.J.; Murphy, W.J. Human endothelial cells express CCR2 and respond to MCP-1: Direct role of MCP-1 in angiogenesis and tumor progression. Blood 2000, 96, 34–40. [Google Scholar] [CrossRef]
- Payne, A.S.; Cornelius, L.A. The role of chemokines in melanoma tumor growth and metastasis. J. Investig. Dermatol. 2002, 118, 915–922. [Google Scholar] [CrossRef]
- Zheng, N.; Zhou, Q.; Wang, Z.; Wei, W. Recent advances in SCF ubiquitin ligase complex: Clinical implications. Biochim. Biophys. Acta 2016, 1866, 12–22. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69 (Suppl. 3), 4–10. [Google Scholar] [CrossRef]
- Zeng, F.; Harris, R.C. Epidermal growth factor, from gene organization to bedside. Semin. Cell Dev. Biol. 2014, 28, 2–11. [Google Scholar] [CrossRef]
- Jutten, B.; Rouschop, K.M. EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle 2014, 13, 42–51. [Google Scholar] [CrossRef]
- Newell, L.F.; Holtan, S.G. Placental growth factor: What hematologists need to know. Blood Rev. 2017, 31, 57–62. [Google Scholar] [CrossRef]
- Heldin, C.H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef]
- Vella, V.; Malaguarnera, R.; Nicolosi, M.L.; Morrione, A.; Belfiore, A. Insulin/IGF signaling and discoidin domain receptors: An emerging functional connection. Biochim. Biophys. Acta Mol. Cell Res. 2019, 118522. [Google Scholar] [CrossRef]
- Ramilowski, J.A.; Goldberg, T.; Harshbarger, J.; Kloppmann, E.; Lizio, M.; Satagopam, V.P.; Itoh, M.; Kawaji, H.; Carninci, P.; Rost, B.; et al. A draft network of ligand–receptor-mediated multicellular signalling in human. Nat. Commun. 2015, 6, 7866. [Google Scholar] [CrossRef]
- Zhou, J.X.; Taramelli, R.; Pedrini, E.; Knijnenburg, T.; Huang, S. Extracting Intercellular Signaling Network of Cancer Tissues using Ligand-Receptor Expression Patterns from Whole-tumor and Single-cell Transcriptomes. Sci. Rep. 2017, 7, 8815. [Google Scholar] [CrossRef]
- Sarkar, S.; Sabhachandani, P.; Stroopinsky, D.; Palmer, K.; Cohen, N.; Rosenblatt, J.; Avigan, D.; Konry, T. Dynamic analysis of immune and cancer cell interactions at single cell level in microfluidic droplets. Biomicrofluidics 2016, 10, 054115. [Google Scholar] [CrossRef]
- Yuan, D.; Tao, Y.; Chen, G.; Shi, T. Systematic expression analysis of ligand-receptor pairs reveals important cell-to-cell interactions inside glioma. Cell Commun. Signal. 2019, 17, 48. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Lucchetti, D.; Ricciardi Tenore, C.; Colella, F.; Sgambato, A. Extracellular Vesicles and Cancer: A Focus on Metabolism, Cytokines, and Immunity. Cancers (Basel) 2020, 12, 171. [Google Scholar] [CrossRef]
- Santi, A.; Caselli, A.; Ranaldi, F.; Paoli, P.; Mugnaioni, C.; Michelucci, E.; Cirri, P. Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim. Biophys. Acta 2015, 1853, 3211–3223. [Google Scholar] [CrossRef]
- Curtis, M.; Kenny, H.A.; Ashcroft, B.; Mukherjee, A.; Johnson, A.; Zhang, Y.; Helou, Y.; Batlle, R.; Liu, X.; Gutierrez, N.; et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab. 2019, 29, 141–155. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Shender, V.O.; Pavlyukov, M.S.; Ziganshin, R.H.; Arapidi, G.P.; Kovalchuk, S.I.; Anikanov, N.A.; Altukhov, I.A.; Alexeev, D.G.; Butenko, I.O.; Shavarda, A.L.; et al. Proteome-metabolome profiling of ovarian cancer ascites reveals novel components involved in intercellular communication. Mol. Cell. Proteom. 2014, 13, 3558–3571. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Tang, Z.; Shen, Q.; Xie, H.; Zhou, X.; Li, J.; Feng, J.; Liu, H.; Wang, W.; Zhang, S.; Ni, S. Elevated expression of FABP3 and FABP4 cooperatively correlates with poor prognosis in non-small cell lung cancer (NSCLC). Oncotarget 2016, 7, 46253–46262. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Chang, S.H.; Liu, C.H.; Conway, R.; Han, D.K.; Nithipatikom, K.; Trifan, O.C.; Lane, T.F.; Hla, T. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc. Natl. Acad. Sci. USA 2004, 101, 591–596. [Google Scholar] [CrossRef]
- Buchanan, F.G.; Wang, D.; Bargiacchi, F.; DuBois, R.N. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J. Biol. Chem. 2003, 278, 35451–35457. [Google Scholar] [CrossRef]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.B.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef]
- Heusinkveld, M.; de Vos van Steenwijk, P.J.; Goedemans, R.; Ramwadhdoebe, T.H.; Gorter, A.; Welters, M.J.; van Hall, T.; van der Burg, S.H. M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J. Immunol. 2011, 187, 1157–1165. [Google Scholar] [CrossRef]
- Chen, J.Y.; Li, C.F.; Kuo, C.C.; Tsai, K.K.; Hou, M.F.; Hung, W.C. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014, 16, 410. [Google Scholar] [CrossRef]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef]
- Kuwata, S.; Ohkubo, K.; Kumamoto, S.; Yamaguchi, N.; Izuka, N.; Murota, K.; Tsujiuchi, T.; Iwamori, M.; Fukushima, N. Extracellular lipid metabolism influences the survival of ovarian cancer cells. Biochem. Biophys. Res. Commun. 2013, 439, 280–284. [Google Scholar] [CrossRef]
- Visentin, B.; Vekich, J.A.; Sibbald, B.J.; Cavalli, A.L.; Moreno, K.M.; Matteo, R.G.; Garland, W.A.; Lu, Y.; Yu, S.; Hall, H.S.; et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 2006, 9, 225–238. [Google Scholar] [CrossRef]
- Anelli, V.; Gault, C.R.; Snider, A.J.; Obeid, L.M. Role of sphingosine kinase-1 in paracrine/transcellular angiogenesis and lymphangiogenesis in vitro. FASEB J. 2010, 24, 2727–2738. [Google Scholar] [CrossRef]
- Kanaporis, G.; Brink, P.R.; Valiunas, V. Gap junction permeability: Selectivity for anionic and cationic probes. Am. J. Physiol. Cell Physiol. 2011, 300, C600–C609. [Google Scholar] [CrossRef]
- Wu, J.I.; Wang, L.H. Emerging roles of gap junction proteins connexins in cancer metastasis, chemoresistance and clinical application. J. Biomed. Sci. 2019, 26, 8. [Google Scholar] [CrossRef]
- Tang, B.; Peng, Z.H.; Yu, P.W.; Yu, G.; Qian, F. Expression and significance of Cx43 and E-cadherin in gastric cancer and metastatic lymph nodes. Med. Oncol. 2011, 28, 502–508. [Google Scholar] [CrossRef]
- Alaga, K.C.; Crawford, M.; Dagnino, L.; Laird, D.W. Aberrant Cx43 Expression and Mislocalization in Metastatic Human Melanomas. J. Cancer 2017, 8, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Patel, A.; Klubo-Gwiezdzinska, J.; Bauer, A.; Vasko, V. Inhibition of gap junction transfer sensitizes thyroid cancer cells to anoikis. Endocr. Relat. Cancer 2011, 18, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Liu, X.; Yang, L.; Qi, K.; Zhang, H.; Zhang, J.; Huang, Z.; Wang, H. All-trans retinoic acid enhances bystander effect of suicide gene therapy in the treatment of breast cancer. Oncol. Rep. 2016, 35, 1868–1874. [Google Scholar] [CrossRef] [PubMed]
- Heiniger, B.; Gakhar, G.; Prasain, K.; Hua, D.H.; Nguyen, T.A. Second-generation substituted quinolines as anticancer drugs for breast cancer. Anticancer Res. 2010, 30, 3927–3932. [Google Scholar]
- Wang, H.; Tian, L.; Liu, J.; Goldstein, A.; Bado, I.; Zhang, W.; Arenkiel, B.R.; Li, Z.; Yang, M.; Du, S.; et al. The Osteogenic Niche Is a Calcium Reservoir of Bone Micrometastases and Confers Unexpected Therapeutic Vulnerability. Cancer Cell 2018, 34, 823–839. [Google Scholar] [CrossRef]
- Yang, W.H.; Cha, J.H.; Xia, W.; Lee, H.H.; Chan, L.C.; Wang, Y.N.; Hsu, J.L.; Ren, G.; Hung, M.C. Juxtacrine Signaling Inhibits Antitumor Immunity by Upregulating PD-L1 Expression. Cancer Res. 2018, 78, 3761–3768. [Google Scholar] [CrossRef]
- Dang, T.O.; Ogunniyi, A.; Barbee, M.S.; Drilon, A. Pembrolizumab for the treatment of PD-L1 positive advanced or metastatic non-small cell lung cancer. Expert Rev. Anticancer Ther. 2016, 16, 13–20. [Google Scholar] [CrossRef]
- Sundar, R.; Cho, B.-C.; Brahmer, J.R.; Soo, R.A. Nivolumab in NSCLC: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2015, 7, 85–96. [Google Scholar] [CrossRef]
- Philips, G.K.; Atkins, M. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int. Immunol. 2015, 27, 39–46. [Google Scholar] [CrossRef]
- Wang, C.; Yu, X.; Wang, W. A meta-analysis of efficacy and safety of antibodies targeting PD-1/PD-L1 in treatment of advanced nonsmall cell lung cancer. Medicine (Baltimore) 2016, 95, e5539. [Google Scholar] [CrossRef]
- Besse, B.; Tsao, L.C.; Chao, D.T.; Fang, Y.; Soria, J.C.; Almokadem, S.; Belani, C.P. Phase Ib safety and pharmacokinetic study of volociximab, an anti-alpha5beta1 integrin antibody, in combination with carboplatin and paclitaxel in advanced non-small-cell lung cancer. Ann. Oncol. 2013, 24, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Sarkari, A.; Munster, M.; Zeevi, E.; Giladi, M.; Lou, E. Abstract 5156: In vitro application of tumor-treating fields to suppress tunneling nanotubes in mesothelioma. Cancer Res. 2019, 79, 5156. [Google Scholar] [CrossRef]
- Sarkari, A.; Zhai, E.; Lou, E. Tumor-Treating Fields (TTFields) Suppress Tunneling Nanotube Formation in Malignant Mesothelioma. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, E662. [Google Scholar] [CrossRef]
- Desir, S.; O’Hare, P.; Vogel, R.; Sperduto, W.; Sarkari, A.; Dickson, E.; Wong, P.; Nelson, A.; Fong, Y.; Steer, C.; et al. Chemotherapy-Induced Tunneling Nanotubes Mediate Intercellular Drug Efflux in Pancreatic Cancer. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Desir, S.; Dickson, E.L.; Vogel, R.I.; Thayanithy, V.; Wong, P.; Teoh, D.; Geller, M.A.; Steer, C.J.; Subramanian, S.; Lou, E. Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells. Oncotarget 2016, 7, 43150–43161. [Google Scholar] [CrossRef] [PubMed]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hanggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. 2017, 19, 1316–1326. [Google Scholar] [CrossRef]
- Haderk, F.; Schulz, R.; Iskar, M.; Cid, L.L.; Worst, T.; Willmund, K.V.; Schulz, A.; Warnken, U.; Seiler, J.; Benner, A.; et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci. Immunol. 2017, 2, eaah5509. [Google Scholar] [CrossRef]
- Tian, W.; Liu, S.; Li, B. Potential Role of Exosomes in Cancer Metastasis. Biomed Res. Int. 2019, 2019, 4649705. [Google Scholar] [CrossRef]
- Dong, W.-W.; Mou, Q.; Chen, J.; Cui, J.-T.; Li, W.-M.; Xiao, W.-H. Differential expression of Rab27A/B correlates with clinical outcome in hepatocellular carcinoma. World J. Gastroenterol. 2012, 18, 1806–1813. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef]
- Marleau, A.M.; Chen, C.-S.; Joyce, J.A.; Tullis, R.H. Exosome removal as a therapeutic adjuvant in cancer. J. Transl. Med. 2012, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.-L.; Chen, K.-C.; Hsieh, J.-T.; Shen, T.-L. Exosomes in cancer development and clinical applications. Cancer Sci. 2018, 109, 2364–2374. [Google Scholar] [CrossRef] [PubMed]
- Iero, M.; Valenti, R.; Huber, V.; Filipazzi, P.; Parmiani, G.; Fais, S.; Rivoltini, L. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ. 2008, 15, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Rahbarghazi, R.; Jabbari, N.; Sani, N.A.; Asghari, R.; Salimi, L.; Kalashani, S.A.; Feghhi, M.; Etemadi, T.; Akbariazar, E.; Mahmoudi, M.; et al. Tumor-derived extracellular vesicles: Reliable tools for Cancer diagnosis and clinical applications. Cell Commun. Signal. 2019, 17, 73. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Shtam, T.A.; Kovalev, R.A.; Varfolomeeva, E.Y.; Makarov, E.M.; Kil, Y.V.; Filatov, M.V. Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun. Signal. 2013, 11, 88. [Google Scholar] [CrossRef]
- Rodon, J.; Carducci, M.A.; Sepulveda-Sánchez, J.M.; Azaro, A.; Calvo, E.; Seoane, J.; Braña, I.; Sicart, E.; Gueorguieva, I.; Cleverly, A.L.; et al. First-in-human dose study of the novel transforming growth factor-β receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin. Cancer Res. 2015, 21, 553–560. [Google Scholar] [CrossRef]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef]
- Fuji, S.; Utsunomiya, A.; Inoue, Y.; Miyagi, T.; Owatari, S.; Sawayama, Y.; Moriuchi, Y.; Choi, I.; Shindo, T.; Yoshida, S.-I.; et al. Outcomes of patients with relapsed aggressive adult T-cell leukemia-lymphoma: Clinical effectiveness of anti-CCR4 antibody and allogeneic hematopoietic stem cell transplantation. Haematologica 2018, 103, e211–e214. [Google Scholar] [CrossRef]
- Allison, S.J. Kidney cancer: CCR4: A new target for RCC. Nat. Rev. Nephrol. 2017, 13, 192. [Google Scholar] [CrossRef]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed]
- Shinriki, S.; Jono, H.; Ueda, M.; Ota, K.; Ota, T.; Sueyoshi, T.; Oike, Y.; Ibusuki, M.; Hiraki, A.; Nakayama, H.; et al. Interleukin-6 signalling regulates vascular endothelial growth factor-C synthesis and lymphangiogenesis in human oral squamous cell carcinoma. J. Pathol. 2011, 225, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, V.; Busser, B.; Vanwonterghem, L.; Michallet, S.; Ferroudj, S.; Cokol, M.; Coll, J.-L.; Ozturk, M.; Hurbin, A. Synergistic activity of vorinostat combined with gefitinib but not with sorafenib in mutant KRAS human non-small cell lung cancers and hepatocarcinoma. Onco Targets Ther. 2016, 9, 6843–6855. [Google Scholar] [CrossRef] [PubMed]
- Rock, E.P.; Goodman, V.; Jiang, J.X.; Mahjoob, K.; Verbois, S.L.; Morse, D.; Dagher, R.; Justice, R.; Pazdur, R. Food and Drug Administration Drug Approval Summary: Sunitinib Malate for the Treatment of Gastrointestinal Stromal Tumor and Advanced Renal Cell Carcinoma. Oncologist 2007, 12, 107–113. [Google Scholar] [CrossRef]
- Chau, N.G.; Haddad, R.I. Vandetanib for the Treatment of Medullary Thyroid Cancer. Clin. Cancer Res. 2013, 19, 524–529. [Google Scholar] [CrossRef]
- Raouf, S.; Bertelli, G.; Ograbek, A.; Field, P.; Tran, I. Real-world use of bevacizumab in metastatic colorectal, metastatic breast, advanced ovarian and cervical cancer: A systematic literature review. Future Oncol. 2019, 15, 543–561. [Google Scholar] [CrossRef]
- Gardner, V.; Madu, C.; Lu, Y. Anti-VEGF Therapy in Cancer: A Double-Edged Sword. In Physiologic and Pathologic Angiogenesis. Signaling Mechanisms and Targeted Therapy; InTechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Pardee, T.S.; Lee, K.; Luddy, J.; Maturo, C.; Rodriguez, R.; Isom, S.; Miller, L.D.; Stadelman, K.M.; Levitan, D.; Hurd, D.; et al. A phase I study of the first-in-class antimitochondrial metabolism agent, CPI-613, in patients with advanced hematologic malignancies. Clin. Cancer Res. 2014, 20, 5255–5264. [Google Scholar] [CrossRef]
- Lycan, T.W.; Pardee, T.S.; Petty, W.J.; Bonomi, M.; Alistar, A.; Lamar, Z.S.; Isom, S.; Chan, M.D.; Miller, A.A.; Ruiz, J. A Phase II Clinical Trial of CPI-613 in Patients with Relapsed or Refractory Small Cell Lung Carcinoma. PLoS ONE 2016, 11, e0164244. [Google Scholar] [CrossRef]
- Kamisuki, S.; Mao, Q.; Abu-Elheiga, L.; Gu, Z.; Kugimiya, A.; Kwon, Y.; Shinohara, T.; Kawazoe, Y.; Sato, S.-i.; Asakura, K.; et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem. Biol. 2009, 16, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Król, S.K.; Kiełbus, M.; Rivero-Müller, A.; Stepulak, A. Comprehensive review on betulin as a potent anticancer agent. Biomed Res. Int. 2015, 2015, 584189. [Google Scholar] [CrossRef] [PubMed]
- Siqingaowa; Sekar, S.; Gopalakrishnan, V.; Taghibiglou, C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem. Biophys. Res. Commun. 2017, 488, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Li, J.; Guo, D. SCAP/SREBPs are Central Players in Lipid Metabolism and Novel Metabolic Targets in Cancer Therapy. Curr. Top. Med. Chem. 2018, 18, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Dean, E.; Falchook, G.; Patel, M.; Brenner, A.; Infante, J.; Arkenau, H.-T.; Borazanci, E.; Lopez, J.; Pant, S.; Schmid, P.; et al. Preliminary activity in the first in human study of the first-in-class fatty acid synthase (FASN) inhibitor, TVB-2640. J. Clin. Oncol. 2016, 34, 2512. [Google Scholar] [CrossRef]
- Kock, A.; Larsson, K.; Bergqvist, F.; Eissler, N.; Elfman, L.H.M.; Raouf, J.; Korotkova, M.; Johnsen, J.I.; Jakobsson, P.-J.; Kogner, P. Inhibition of Microsomal Prostaglandin E Synthase-1 in Cancer-Associated Fibroblasts Suppresses Neuroblastoma Tumor Growth. EBioMedicine 2018, 32, 84–92. [Google Scholar] [CrossRef]
- Britten, C.D.; Garrett-Mayer, E.; Chin, S.H.; Shirai, K.; Ogretmen, B.; Bentz, T.A.; Brisendine, A.; Anderton, K.; Cusack, S.L.; Maines, L.W.; et al. A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 4642–4650. [Google Scholar] [CrossRef]
- Eugenin, E. Role of cell-to-cell communication in cancer: New features, insights, and directions. Cancer Rep. 2019. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dominiak, A.; Chełstowska, B.; Olejarz, W.; Nowicka, G. Communication in the Cancer Microenvironment as a Target for Therapeutic Interventions. Cancers 2020, 12, 1232. https://doi.org/10.3390/cancers12051232
Dominiak A, Chełstowska B, Olejarz W, Nowicka G. Communication in the Cancer Microenvironment as a Target for Therapeutic Interventions. Cancers. 2020; 12(5):1232. https://doi.org/10.3390/cancers12051232
Chicago/Turabian StyleDominiak, Agnieszka, Beata Chełstowska, Wioletta Olejarz, and Grażyna Nowicka. 2020. "Communication in the Cancer Microenvironment as a Target for Therapeutic Interventions" Cancers 12, no. 5: 1232. https://doi.org/10.3390/cancers12051232
APA StyleDominiak, A., Chełstowska, B., Olejarz, W., & Nowicka, G. (2020). Communication in the Cancer Microenvironment as a Target for Therapeutic Interventions. Cancers, 12(5), 1232. https://doi.org/10.3390/cancers12051232