The Mechanical Microenvironment in Breast Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction to Mechanical Signaling
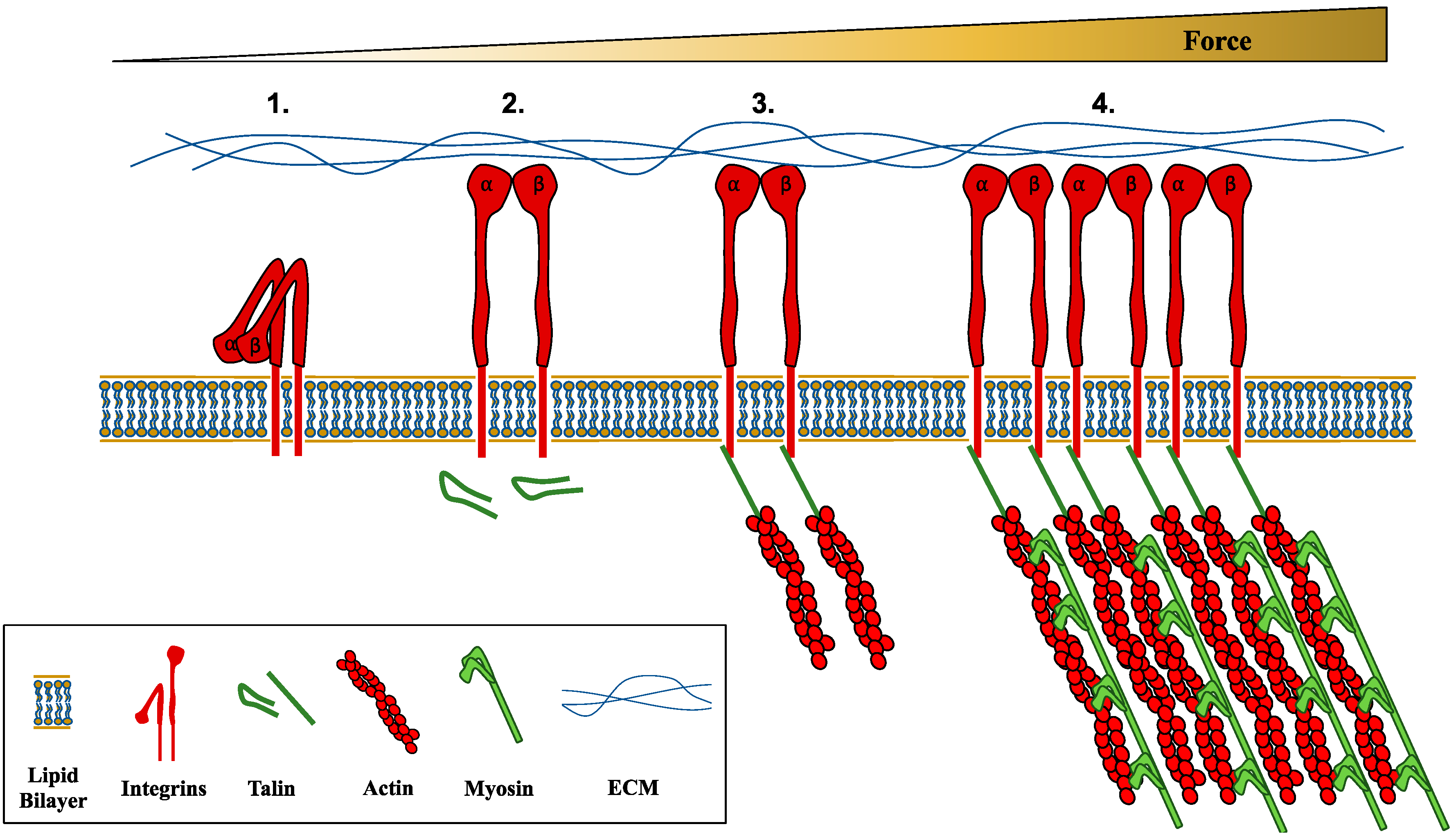
2. Mechanoresponsiveness: Mechanosensation and Mechanotransduction
3. Mechanoreciprocity
4. Mechanical Forces and Signaling in the Normal Breast
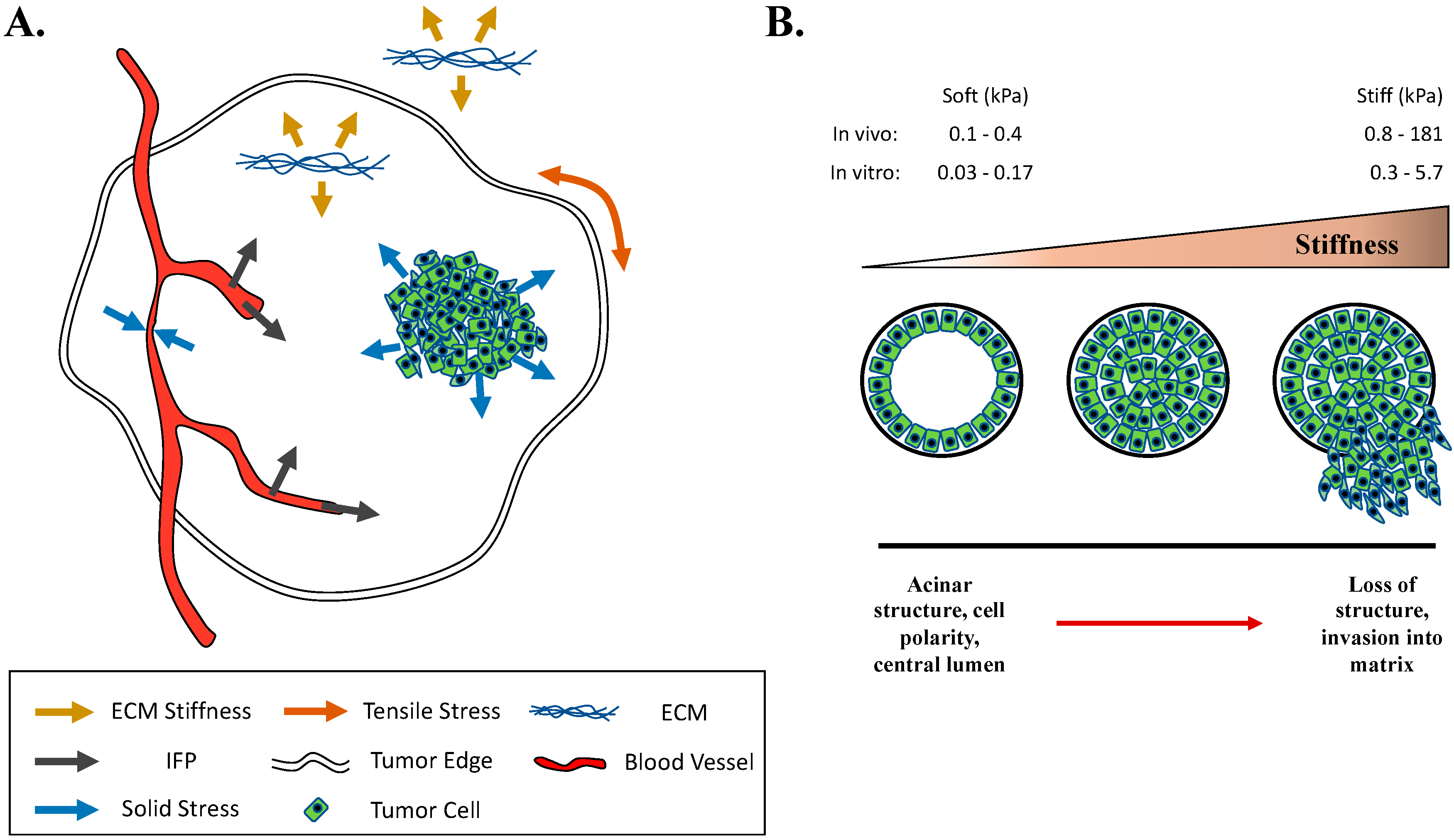
5. Mechanical Forces and Signaling in Breast Cancer
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gillespie, P.G.; Muller, U. Mechanotransduction by hair cells: Models, molecules, and mechanisms. Cell 2009, 139, 33–44. [Google Scholar] [CrossRef]
- Corey, D.P.; Hudspeth, A.J. Kinetics of the receptor current in bullfrog saccular hair cells. J. Neurosci. 1983, 3, 962–976. [Google Scholar] [CrossRef]
- Lyons, J.S.; Joca, H.C.; Law, R.A.; Williams, K.; Kerr, J.P.; Shi, G.; Khairallah, R.J.; Martin, S.S.; Konstantopoulos, K.; Ward, C.W.; et al. Microtubules tune mechanotransduction through NOX2 and TRPV4 to decrease sclerostin abundance in osteocytes. Sci. Signal. 2017, 10, eaan5748. [Google Scholar] [CrossRef] [PubMed]
- Prosser, B.L.; Ward, C.; Lederer, W. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Alenghat, F.J.; Nauli, S.M.; Kolb, R.; Zhou, J.; Ingber, D.E. Global cytoskeletal control of mechanotransduction in kidney epithelial cells. Exp. Cell Res. 2004, 301, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.H.; Lu, W.; Brown, E.M.; Quinn, S.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137. [Google Scholar] [CrossRef]
- Praetorius, H.A.; Spring, K. Bending the MDCK cell primary cilium increases intracellular calcium. J. Membr. Biol. 2001, 184, 71–79. [Google Scholar] [CrossRef]
- Qiu, Y.; Brown, A.C.; Myers, D.R.; Sakurai, Y.; Mannino, R.G.; Tran, R.; Ahn, B.; Hardy, E.T.; Kee, M.F.; Kumar, S.; et al. Platelet mechanosensing of substrate stiffness during clot formation mediates adhesion, spreading, and activation. Proc. Natl. Acad. Sci. USA 2014, 111, 14430–14435. [Google Scholar] [CrossRef]
- Wang, K.-C.; Yeh, Y.-T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.-L.; Li, Y.-S.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef]
- Desai, S.S.; Tung, J.C.; Zhou, V.X.; Grenert, J.P.; Malato, Y.; Rezvani, M.; Espanol-Suner, R.; Willenbring, H.; Weaver, V.M.; Chang, T.T.; et al. Physiological ranges of matrix rigidity modulate primary mouse hepatocyte function in part through hepatocyte nuclear factor 4 alpha. Hepatology 2016, 64, 261–275. [Google Scholar] [CrossRef]
- Kim, S.T.; Takeuchi, K.; Sun, Z.Y.; Touma, M.; Castro, C.E.; Fahmy, A.; Lang, M.J.; Wagner, G.; Reinherz, E.L. The alphabeta T cell receptor is an anisotropic mechanosensor. J. Biol. Chem. 2009, 284, 31028–31037. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H. T cell receptors, mechanosensors, catch bonds and immunotherapy. Prog. Biophys. Mol. Biol. 2020, 153, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Shoham, N.; Gottlieb, R.; Sharabani-Yosef, O.; Zaretsky, U.; Benayahu, D.; Gefen, A. Static mechanical stretching accelerates lipid production in 3T3-L1 adipocytes by activating the MEK signaling pathway. Am. J. Physiol. Physiol. 2012, 302, C429–C441. [Google Scholar] [CrossRef]
- Erickson, G.R.; Alexopoulos, L.G.; Guilak, F. Hyper-osmotic stress induces volume change and calcium transients in chondrocytes by transmembrane, phospholipid, and G-protein pathways. J. Biomech. 2001, 34, 1527–1535. [Google Scholar] [CrossRef]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial–mesenchymal transition and tumour metastasis through a TWIST1–G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef]
- Gudipaty, S.A.; Lindblom, J.; Loftus, P.D.; Redd, M.J.; Edes, K.; Davey, C.F.; Krishnegowda, V.; Rosenblatt, J. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature 2017, 543, 118–121. [Google Scholar] [CrossRef]
- Gasparski, A.; Ozarkar, S.; Beningo, K.A. Transient mechanical strain promotes the maturation of invadopodia and enhances cancer cell invasion in vitro. J. Cell Sci. 2017, 130, 1965–1978. [Google Scholar] [CrossRef]
- Li, Y.; Ge, C.; Long, J.P.; Begun, D.L.; Rodríguez, J.A.; Goldstein, S.A.; Franceschi, R.T. Biomechanical stimulation of osteoblast gene expression requires phosphorylation of the RUNX2 transcription factor. J. Bone Miner. Res. 2012, 27, 1263–1274. [Google Scholar] [CrossRef]
- Marshall, K.L.; Lumpkin, E.A. The molecular basis of mechanosensory transduction. Adv. Exp. Med. Biol. 2012, 739, 142–155. [Google Scholar] [CrossRef]
- Anishkin, A.; Sukharev, S. Water dynamics and dewetting transitions in the small mechanosensitive channel MscS. Biophys. J. 2004, 86, 2883–2895. [Google Scholar] [CrossRef]
- Chiang, C.-S.; Anishkin, A.; Sukharev, S. Gating of the large mechanosensitive channel in situ: Estimation of the spatial scale of the transition from channel population responses. Biophys. J. 2004, 86, 2846–2861. [Google Scholar] [CrossRef]
- Cox, C.D.; Bae, C.; Ziegler, L.; Hartley, S.; Nikolova-Krstevski, V.; Rohde, P.R.; Ng, C.-A.; Sachs, F.; Gottlieb, P.; Martinac, B.; et al. Removal of the mechanoprotective influence of the cytoskeleton reveals PIEZO1 is gated by bilayer tension. Nat. Commun. 2016, 7, 10366. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Spencer, R.H.; Lee, A.T.; Barclay, M.T.; Rees, D.C. Structure of the MscL homolog from mycobacterium tuberculosis: A gated mechanosensitive ion channel. Science 1998, 282, 2220–2226. [Google Scholar] [CrossRef]
- Betanzos, M.; Chiang, C.S.; Guy, H.R.; Sukharev, S. A large iris-like expansion of a mechanosensitive channel protein induced by membrane tension. Nat. Struct. Biol. 2002, 9, 704–710. [Google Scholar] [CrossRef]
- Perozo, E.; Cortes, D.M.; Sompornpisut, P.; Kloda, A.; Martinac, B. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature 2002, 418, 942–948. [Google Scholar] [CrossRef]
- Corry, B.; Rigby, P.; Liu, Z.W.; Martinac, B. Conformational changes involved in MscL channel gating measured using FRET spectroscopy. Biophys. J. 2005, 89, L49–L51. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhou, H.; Chi, S.; Wang, Y.; Wang, J.; Geng, J.; Wu, K.; Liu, W.; Zhang, T.; Dong, M.Q.; et al. Structure and mechanogating mechanism of the Piezo1 channel. Nature 2018, 554, 487–492. [Google Scholar] [CrossRef]
- Saotome, K.; Murthy, S.E.; Kefauver, J.M.; Whitwam, T.; Patapoutian, A.; Ward, A.B. Structure of the mechanically activated ion channel Piezo1. Nature 2017, 554, 481–486. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, H.; Zhang, M.; Liu, W.; Deng, T.; Zhao, Q.; Li, Y.; Lei, J.; Li, X.; Xiao, B.; et al. Structure and mechanogating of the mammalian tactile channel PIEZO2. Nature 2019, 573, 225–229. [Google Scholar] [CrossRef]
- Wu, J.; Young, M.; Lewis, A.H.; Martfeld, A.N.; Kalmeta, B.; Grandl, J. Inactivation of mechanically activated piezo1 ion channels is determined by the c-terminal extracellular domain and the inner pore helix. Cell Rep. 2017, 21, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.H.; Grandl, J. Mechanical sensitivity of Piezo1 ion channels can be tuned by cellular membrane tension. eLife 2015, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Gnanasambandam, R.; Bae, C.; Gottlieb, P.; Sachs, F. Ionic selectivity and permeation properties of human PIEZO1 channels. PLoS ONE 2015, 10, e0125503. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Molecular mechanism of TRP channels. Compr. Physiol. 2013, 3, 221–242. [Google Scholar]
- Clapham, D.E.; Runnels, L.W.; Strubing, C. The TRP ion channel family. Nat. Rev. Neurosci. 2001, 2, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, Y.A.; Cox, C.D.; Ridone, P.; Rohde, P.R.; Cordero-Morales, J.F.; Vásquez, V.; Laver, D.R.; Martinac, B. Mammalian TRP ion channels are insensitive to membrane stretch. J. Cell Sci. 2019, 132, jcs238360. [Google Scholar] [CrossRef]
- Christensen, A.P.; Corey, D.P. TRP channels in mechanosensation: Direct or indirect activation? Nat. Rev. Neurosci. 2007, 8, 510–521. [Google Scholar] [CrossRef]
- Liedtke, W.; Choe, Y.; Marti-Renom, M.A.; Bell, A.M.; Denis, C.S.; Sali, A.; Hudspeth, A.J.; Friedman, J.M.; Heller, S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 2000, 103, 525–535. [Google Scholar] [CrossRef]
- Wu, L.; Gao, X.; Brown, R.C.; Heller, S.; O’Neil, R.G. Dual role of the TRPV4 channel as a sensor of flow and osmolality in renal epithelial cells. Am. J. Physiol. Renal Physiol. 2007, 293, F1699–F1713. [Google Scholar] [CrossRef]
- Loukin, S.H.; Zhou, X.; Su, Z.; Saimi, Y.; Kung, C. Wild-type and Brachyolmia-causing mutant TRPV4 channels respond directly to stretch force. J. Biol. Chem. 2010, 285, 27176–27181. [Google Scholar] [CrossRef]
- Morita, H.; Honda, A.; Inoue, R.; Ito, Y.; Abe, K.; Nelson, M.T.; Brayden, J.E. Membrane stretch-induced activation of a TRPM4-like nonselective cation channel in cerebral artery myocytes. J. Pharmacol. Sci. 2007, 103, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Numata, T.; Shimizu, T.; Okada, Y. TRPM7 is a stretch- and swelling-activated cation channel involved in volume regulation in human epithelial cells. Am. J. Physiol. Physiol. 2007, 292, C460–C467. [Google Scholar] [CrossRef] [PubMed]
- Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Martinac, B.; Hamill, O.P. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat. Cell Biol. 2005, 7, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Spassova, M.A.; Hewavitharana, T.; Xu, W.; Soboloff, J.; Gill, D.L. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl. Acad. Sci. USA 2006, 103, 16586–16591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cheng, L.E.; Kittelmann, M.; Li, J.; Petkovic, M.; Cheng, T.; Jin, P.; Guo, Z.; Göpfert, M.C.; Jan, L.Y.; et al. Ankyrin repeats convey force to gate the NOMPC mechanotransduction channel. Cell 2015, 162, 1391–1403. [Google Scholar] [CrossRef]
- Matthews, B.D.; Thodeti, C.K.; Tytell, J.D.; Mammoto, A.; Overby, D.R.; Ingber, D.E. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr. Biol. (Camb.) 2010, 2, 435–442. [Google Scholar] [CrossRef]
- Brohawn, S.G.; Su, Z.; MacKinnon, R. Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc. Natl. Acad. Sci. USA 2014, 111, 3614–3619. [Google Scholar] [CrossRef]
- Brohawn, S.G.; Campbell, E.B.; MacKinnon, R. Physical mechanism for gating and mechanosensitivity of the human TRAAK K+ channel. Nature 2014, 516, 126–130. [Google Scholar] [CrossRef]
- Sun, Z.; Costell, M.; Fässler, R. Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol. 2019, 21, 25–31. [Google Scholar] [CrossRef]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef]
- Lecuit, T.; Yap, A.S. E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat. Cell Biol. 2015, 17, 533–539. [Google Scholar] [CrossRef]
- Pannekoek, W.-J.; De Rooij, J.; Gloerich, M. Force transduction by cadherin adhesions in morphogenesis. F1000Research 2019, 8, 1044. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.; Jang, J.; Ye, F.; Hong, S.J.; Petrich, B.G.; Ulmer, T.S.; Kim, C. Topological adaptation of transmembrane domains to the force-modulated lipid bilayer is a basis of sensing mechanical force. Curr. Biol. 2020, 30, 1614–1625. [Google Scholar] [CrossRef]
- Zhu, J.; Luo, B.-H.; Xiao, T.S.; Zhang, C.; Nishida, N.; Springer, T.A. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol. Cell 2008, 32, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lee, H.; Tong, H.; Schwartz, M.; Zhu, C. Force regulated conformational change of integrin alphaVbeta3. Matrix Biol. 2017, 60, 70–85. [Google Scholar] [CrossRef]
- Li, J.; Springer, T.A. Integrin extension enables ultrasensitive regulation by cytoskeletal force. Proc. Natl. Acad. Sci. USA 2017, 114, 4685–4690. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Garcia, A.J.; Mould, A.P.; Humphries, M.J.; Zhu, C. Demonstration of catch bonds between an integrin and its ligand. J. Cell Biol. 2009, 185, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Goult, B.T.; Klapholz, B.; Hu, X.; Toseland, C.P.; Guo, Y.; Cong, P.; Sheetz, M.P.; Yan, J. The mechanical response of talin. Nat. Commun. 2016, 7, 11966. [Google Scholar] [CrossRef]
- Del Rio, A.; Perez-Jimenez, R.; Liu, R.; Roca-Cusachs, P.; Fernandez, J.M.; Sheetz, M.P. Stretching single talin rod molecules activates vinculin binding. Science 2009, 323, 638–641. [Google Scholar] [CrossRef]
- Yao, M.; Goult, B.T.; Chen, H.; Cong, P.; Sheetz, M.P.; Yan, J. Mechanical activation of vinculin binding to talin locks talin in an unfolded conformation. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Elosegui-Artola, A.; Oria, R.; Chen, Y.; Kosmalska, A.; Pérez-González, C.; Castro, N.; Zhu, C.; Trepat, X.; Roca-Cusachs, P. Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nature 2016, 18, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Dufort, C.C.; Rossier, O.; Bainer, R.; Mouw, J.K.; Godula, K.; Hudak, J.E.; Lakins, J.N.; Wijekoon, A.C.; Cassereau, L.; et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 2014, 511, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Boettiger, D.; Weaver, V.M.; Hammer, D.A. Integrin clustering is driven by mechanical resistance from the glycocalyx and the substrate. PLoS Comput. Biol. 2009, 5, e1000604. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti-Adam, E.A.; Volberg, T.; Micoulet, A.; Kessler, H.; Geiger, B.; Spatz, J.P. Cell spreading and focal adhesion dynamics are regulated by spacing of integrin ligands. Biophys. J. 2007, 92, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Murrell, M.; Oakes, P.W.; Lenz, M.; Gardel, M.L. Forcing cells into shape: The mechanics of actomyosin contractility. Nat. Rev. Mol. Cell Biol. 2015, 16, 486–498. [Google Scholar] [CrossRef]
- Pandya, P.; Orgaz, J.L.; Sanz-Moreno, V. Actomyosin contractility and collective migration: May the force be with you. Curr. Opin. Cell Biol. 2017, 48, 87–96. [Google Scholar] [CrossRef]
- Tharp, K.M.; Kang, M.S.; Timblin, G.; Dempersmier, J.; Dempsey, G.E.; Zushin, P.-J.H.; Benavides, J.; Choi, C.; Li, C.X.; Jha, A.K.; et al. Actomyosin-mediated tension orchestrates uncoupled respiration in adipose tissues. Cell Metab. 2018, 27, 602–615. [Google Scholar] [CrossRef]
- Takaki, T.; Montagner, M.; Serres, M.P.; Le Berre, M.; Russell, M.R.; Collinson, L.M.; Szuhai, K.; Howell, M.; Boulton, S.J.; Sahai, E.; et al. Actomyosin drives cancer cell nuclear dysmorphia and threatens genome stability. Nat. Commun. 2017, 8, 16013. [Google Scholar] [CrossRef]
- Samuel, M.S.; Lopez, J.I.; McGhee, E.J.; Croft, D.R.; Strachan, D.; Timpson, P.; Munro, J.; Schroder, E.; Zhou, J.; Brunton, V.G.; et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 2011, 19, 776–791. [Google Scholar] [CrossRef]
- Poincloux, R.; Collin, O.; Lizárraga, F.; Romao, M.; Debray, M.; Piel, M.; Chavrier, P. Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proc. Natl. Acad. Sci. USA 2011, 108, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Meckel, K.F.; Laird, L.S.; Chia, C.Y.; Park, J.J.; Olino, K.L.; Tsunedomi, R.; Harada, T.; Iizuka, N.; Hazama, S.; et al. Integrin alpha2 mediates selective metastasis to the liver. Cancer Res. 2009, 69, 7320–7328. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Gibson-Corley, K.N.; Herndon, M.E.; Sun, Y.; Gustafson-Wagner, E.; Teoh-Fitzgerald, M.; Domann, F.E.; Henry, M.D.; Stipp, C.S. Integrin alpha3beta1 can function to promote spontaneous metastasis and lung colonization of invasive breast carcinoma. Mol. Cancer Res. 2014, 12, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Song, L.; Li, J.; Wang, Y.; Yang, C.; Kou, X.; Xiao, B.; Zhang, W.; Li, L.; Liu, S.; et al. Bone sialoprotein-alphavbeta3 integrin axis promotes breast cancer metastasis to the bone. Cancer Sci. 2019, 110, 3157–3172. [Google Scholar] [CrossRef] [PubMed]
- Rakshit, S.; Zhang, Y.; Manibog, K.; Shafraz, O.; Sivasankar, S. Ideal, catch, and slip bonds in cadherin adhesion. Proc. Natl. Acad. Sci. USA 2012, 109, 18815–18820. [Google Scholar] [CrossRef]
- Manibog, K.; Li, H.; Rakshit, S.; Sivasankar, S. Resolving the molecular mechanism of cadherin catch bond formation. Nat. Commun. 2014, 5, 3941. [Google Scholar] [CrossRef]
- Buckley, C.D.; Tan, J.; Anderson, K.L.; Hanein, R.; Volkmann, N.; Weis, W.I.; Nelson, W.J.; Dunn, A.R. The minimal cadherin-catenin complex binds to actin filaments under force. Science 2014, 346, 1254211. [Google Scholar] [CrossRef]
- Ishiyama, N.; Sarpal, R.; Wood, M.N.; Barrick, S.K.; Nishikawa, T.; Hayashi, H.; Kobb, A.B.; Flozak, A.S.; Yemelyanov, A.; Fernandez-Gonzalez, R.; et al. Force-dependent allostery of the α-catenin actin-binding domain controls adherens junction dynamics and functions. Nat. Commun. 2018, 9, 5121. [Google Scholar] [CrossRef]
- Yonemura, S.; Wada, Y.; Watanabe, T.; Nagafuchi, A.; Shibata, M. α-Catenin as a tension transducer that induces adherens junction development. Nature 2010, 12, 533–542. [Google Scholar] [CrossRef]
- Yao, M.; Qiu, W.; Liu, R.; Efremov, A.K.; Cong, P.; Seddiki, R.; Payre, M.; Lim, C.T.; Ladoux, B.; Mege, R.M.; et al. Force-dependent conformational switch of alpha-catenin controls vinculin binding. Nat. Commun. 2014, 5, 4525. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.L.; Bax, N.A.; Buckley, C.D.; I Weis, W.; Dunn, A.R. Vinculin forms a directionally asymmetric catch bond with F-actin. Science 2017, 357, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.A.; Boscher, C.; Chu, Y.S.; Cuvelier, D.; Martinez-Rico, C.; Seddiki, R.; Heysch, J.; Ladoux, B.; Thiery, J.P.; Mege, R.M.; et al. Alpha-Catenin and vinculin cooperate to promote high E-cadherin-based adhesion strength. J. Biol. Chem. 2013, 288, 4957–4969. [Google Scholar] [CrossRef] [PubMed]
- Maruthamuthu, V.; Sabass, B.; Schwarz, U.S.; Gardel, M.L. Cell-ECM traction force modulates endogenous tension at cell–cell contacts. Proc. Natl. Acad. Sci. USA 2011, 108, 4708–4713. [Google Scholar] [CrossRef]
- Price, A.; Cost, A.-L.; Ungewiß, H.; Waschke, J.; Dunn, A.R.; Grashoff, C. Mechanical loading of desmosomes depends on the magnitude and orientation of external stress. Nat. Commun. 2018, 9, 5284. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Li, J.; Hou, B.; Tumova, S.; Muraki, K.; Bruns, A.; Ludlow, M.J.; Sedo, A.; Hyman, A.; McKeown, L.; Young, R.S.; et al. Piezo1 integration of vascular architecture with physiological force. Nature 2014, 515, 279–282. [Google Scholar] [CrossRef]
- Mousawi, F.; Peng, H.; Li, J.; Ponnambalam, S.; Roger, S.; Zhao, H.; Yang, X.; Jiang, L.-H.; Sreenivasan, P. Chemical activation of the Piezo1 channel drives mesenchymal stem cell migration via inducing ATP release and activation of P2 receptor purinergic signaling. Stem Cells 2020, 38, 410–421. [Google Scholar] [CrossRef]
- Pardo-Pastor, C.; Rubio-Moscardó, F.; Vogel-González, M.; Serra, S.A.; Afthinos, A.; Mrkonjic, S.; Destaing, O.; Abenza, J.F.; Fernández-Fernández, J.M.; Trepat, X.; et al. Piezo2 channel regulates RhoA and actin cytoskeleton to promote cell mechanobiological responses. Proc. Natl. Acad. Sci. USA 2018, 115, 1925–1930. [Google Scholar] [CrossRef]
- Han, Y.; Liu, C.; Zhang, N.; Men, H.; Huo, L.; Geng, Q.; Wang, S.; Gao, Y.; Zhang, W.; Zhang, Y.; et al. Mechanosensitive ion channel Piezo1 promotes prostate cancer development through the activation of the Akt/mTOR pathway and acceleration of cell cycle. Int. J. Oncol. 2019, 55, 629–644. [Google Scholar] [CrossRef]
- Plotnikov, S.V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell 2012, 151, 1513–1527. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Swaminathan, V.; Garcia-Mata, R.; O’Brien, E.T.; Superfine, R.; Burridge, K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat. Cell Biol. 2011, 13, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, R.; Rajakyla, E.K.; Vartiainen, M.K.; Treisman, R. An actin-regulated importin alpha/beta-dependent extended bipartite NLS directs nuclear import of MRTF-A. EMBO J. 2010, 29, 3448–3458. [Google Scholar] [CrossRef] [PubMed]
- Plessner, M.; Melak, M.; Chinchilla, P.; Baarlink, C.; Grosse, R. Nuclear F-actin formation and reorganization upon cell spreading. J. Biol. Chem. 2015, 290, 11209–11216. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tytell, J.D.; Ingber, D.E. Mechanotransduction at a distance: Mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 2009, 10, 75–82. [Google Scholar] [CrossRef]
- Isermann, P.; Lammerding, J. Nuclear mechanics and mechanotransduction in health and disease. Curr. Biol. 2013, 23, R1113-21. [Google Scholar] [CrossRef]
- Ito, S.; Okuda, S.; Abe, M.; Fujimoto, M.; Onuki, T.; Nishimura, T.; Takeichi, M. Induced cortical tension restores functional junctions in adhesion-defective carcinoma cells. Nat. Commun. 2017, 8, 1834. [Google Scholar] [CrossRef]
- De Pascalis, C.; Etienne-Manneville, S. Single and collective cell migration: The mechanics of adhesions. Mol. Biol. Cell 2017, 28, 1833–1846. [Google Scholar] [CrossRef]
- Ladoux, B.; Mege, R.M. Mechanobiology of collective cell behaviours. Nat. Rev. Mol. Cell Biol. 2017, 18, 743–757. [Google Scholar] [CrossRef]
- Thorne, J.; Segal, T.R.; Chang, S.; Jorge, S.; Segars, J.H.; Leppert, P. Dynamic reciprocity between cells and their microenvironment in reproduction. Biol. Reprod. 2015, 92, 25. [Google Scholar] [CrossRef]
- Bissell, M.J.; Hall, H.; Parry, G. How does the extracellular matrix direct gene expression? J. Theor. Biol. 1982, 99, 31–68. [Google Scholar] [CrossRef]
- Jorge, S.; Chang, S.; Barzilai, J.J.; Leppert, P.; Segars, J. Mechanical signaling in reproductive tissues: Mechanisms and importance. Reprod. Sci. 2014, 21, 1093–1107. [Google Scholar] [CrossRef]
- Xu, R.; Boudreau, A.; Bissell, M.J. Tissue architecture and function: Dynamic reciprocity via extra- and intra-cellular matrices. Cancer Metastasis Rev. 2009, 28, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Weaver, V.M. The tension mounts: Mechanics meets morphogenesis and malignancy. J. Mammary Gland Biol. Neoplasia 2004, 9, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Van Helvert, S.; Storm, C.; Friedl, P. Mechanoreciprocity in cell migration. Nat. Cell Biol. 2017, 20, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.S.; Alisafaei, F.; Ban, E.; Feng, X.; Hui, C.-Y.; Shenoy, V.B.; Wu, M. Fibrous nonlinear elasticity enables positive mechanical feedback between cells and ECMs. Proc. Natl. Acad. Sci. USA 2016, 113, 14043–14048. [Google Scholar] [CrossRef] [PubMed]
- van Helvert, S.; Friedl, P. Strain stiffening of fibrillar collagen during individual and collective cell migration identified by AFM nanoindentation. ACS Appl. Mater. Interfaces 2016, 8, 21946–21955. [Google Scholar] [CrossRef] [PubMed]
- Gjorevski, N.; Piotrowski, A.S.; Varner, V.D.; Nelson, C.M. Dynamic tensile forces drive collective cell migration through three-dimensional extracellular matrices. Sci. Rep. 2015, 5, 11458. [Google Scholar] [CrossRef]
- Kim, J.; Feng, J.; Jones, C.A.R.; Mao, X.; Sander, L.M.; Levine, H.; Sun, B. Stress-induced plasticity of dynamic collagen networks. Nat. Commun. 2017, 8, 842. [Google Scholar] [CrossRef]
- Solon, J.; Levental, I.; Sengupta, K.; Georges, P.C.; Janmey, P.A. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys. J. 2007, 93, 4453–4461. [Google Scholar] [CrossRef]
- Lo, C.-M.; Wang, H.-B.; Dembo, M.; Wang, Y.-L. Cell movement is guided by the rigidity of the substrate. Biophys. J. 2000, 79, 144–152. [Google Scholar] [CrossRef]
- Vincent, L.G.; Choi, Y.S.; Alonso-Latorre, B.; Del Alamo, J.C.; Engler, A.J. Mesenchymal stem cell durotaxis depends on substrate stiffness gradient strength. Biotechnol. J. 2013, 8, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Sunyer, R.; Conte, V.; Escribano, J.; Elosegui-Artola, A.; Labernadie, A.; Valon, L.; Navajas, D.; Garcia-Aznar, J.M.; Munoz, J.J.; Roca-Cusachs, P.; et al. Collective cell durotaxis emerges from long-range intercellular force transmission. Science 2016, 353, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Tamariz, E.; Grinnell, F. Modulation of fibroblast morphology and adhesion during collagen matrix remodeling. Mol. Biol. Cell 2002, 13, 3915–3929. [Google Scholar] [CrossRef] [PubMed]
- Carey, S.P.; Goldblatt, Z.E.; Martin, K.E.; Romero, B.; Williams, R.M.; Reinhart-King, C.A. Local extracellular matrix alignment directs cellular protrusion dynamics and migration through Rac1 and FAK. Integr. Biol. (Camb.) 2016, 8, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.; Te Lindert, M.; Krause, M.; Alexander, S.; Te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P.; et al. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084. [Google Scholar] [CrossRef]
- Wolf, K.; Wu, Y.I.; Liu, Y.; Geiger, J.; Tam, E.; Overall, C.; Stack, M.S.; Friedl, P. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 2007, 9, 893–904. [Google Scholar] [CrossRef]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Toh, R.; Shinohara, M.; Takaya, T.; Yamashita, T.; Masuda, S.; Kawashima, S.; Yokoyama, M.; Yagi, N. An X-Ray diffraction study on mouse cardiac cross-bridge function in vivo: Effects of adrenergic {beta}-stimulation. Biophys. J. 2006, 90, 1723–1728. [Google Scholar] [CrossRef]
- Prosser, B.L.; Khairallah, R.J.; Ziman, A.P.; Ward, C.; Lederer, W. X-ROS signaling in the heart and skeletal muscle: Stretch-dependent local ROS regulates [Ca2+]i. J. Mol. Cell. Cardiol. 2012, 58, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.P.; Robison, P.; Shi, G.; Bogush, A.I.; Kempema, A.M.; Hexum, J.K.; Becerra, N.; Harki, D.A.; Martin, S.S.; Raiteri, R.; et al. Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat. Commun. 2015, 6, 8526. [Google Scholar] [CrossRef] [PubMed]
- Gardinier, J.D.; Townend, C.W.; Jen, K.-P.; Wu, Q.; Duncan, R.L.; Wang, L. In situ permeability measurement of the mammalian lacunar–canalicular system. Bone 2010, 46, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Price, C.; Zhou, X.; Li, W.; Wang, L. Real-time measurement of solute transport within the lacunar-canalicular system of mechanically loaded bone: Direct evidence for load-induced fluid flow. J. Bone Miner. Res. 2010, 26, 277–285. [Google Scholar] [CrossRef]
- Lyons, J.S.; Iyer, S.R.; Lovering, R.M.; Ward, C.W.; Stains, J.P. Novel multi-functional fluid flow device for studying cellular mechanotransduction. J. Biomech. 2016, 49, 4173–4179. [Google Scholar] [CrossRef]
- Mammoto, T.; Ingber, D.E. Mechanical control of tissue and organ development. Development 2010, 137, 1407–1420. [Google Scholar] [CrossRef]
- Mammoto, A.; Mammoto, T.; Ingber, D.E. Mechanosensitive mechanisms in transcriptional regulation. J. Cell Sci. 2012, 125 Pt 13, 3061–3073. [Google Scholar] [CrossRef]
- Alcaraz, J.; Nelson, C.M.; Bissell, M.J. Biomechanical approaches for studying integration of tissue structure and function in mammary epithelia. J. Mammary Gland. Biol. Neoplasia 2004, 9, 361–374. [Google Scholar] [CrossRef][Green Version]
- Schedin, P.; Keely, P.J. Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb. Perspect. Biol. 2010, 3, a003228. [Google Scholar] [CrossRef]
- Marti, A.; Feng, Z.; Altermatt, H.J.; Jaggi, R. Milk accumulation triggers apoptosis of mammary epithelial cells. Eur. J. Cell Biol. 1997, 73, 158–165. [Google Scholar]
- Quaglino, A.; Salierno, M.; Pellegrotti, J.V.; Rubinstein, N.; Kordon, E.C. Mechanical strain induces involution-associated events in mammary epithelial cells. BMC Cell Biol. 2009, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Macias, H.; Hinck, L. Mammary gland development. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 533–557. [Google Scholar] [CrossRef] [PubMed]
- Nia, H.T.; Liu, H.; Seano, G.; Datta, M.; Jones, D.; Rahbari, N.; Incio, J.; Chauhan, V.; Jung, K.; Martin, J.D.; et al. Solid stress and elastic energy as measures of tumour mechanopathology. Nat. Biomed. Eng. 2016, 1, 0004. [Google Scholar] [CrossRef] [PubMed]
- Krouskop, T.A.; Wheeler, T.M.; Kallel, F.; Garra, B.S.; Hall, T. Elastic moduli of breast and prostate tissues under compression. Ultrason Imaging 1998, 20, 260–274. [Google Scholar] [CrossRef]
- Gefen, A.; Dilmoney, B. Mechanics of the normal woman’s breast. Technol. Health Care 2007, 15, 259–271. [Google Scholar] [CrossRef]
- Enomoto, K.-I.; Furuya, K.; Maeno, T.; Edwards, C.; Oka, T. Mechanically induced electrical responses in murine mammary epithelial cells in primary culture. FEBS Lett. 1987, 223, 82–86. [Google Scholar] [CrossRef]
- Furuya, K.; Enomoto, K.-I. Real-time imaging of intracellular calcium change with simultaneous single channel recording in mammary epithelial cells. Brain Res. Bull. 1990, 25, 779–781. [Google Scholar] [CrossRef]
- Enomoto, K.; Furuya, K.; Yamagishi, S.; Maeno, T. Mechanically induced electrical and intracellular calcium responses in normal and cancerous mammary cells. Cell Calcium 1992, 13, 501–511. [Google Scholar] [CrossRef]
- Furuya, K.; Enomoto, K.-I.; Yamagishi, S. Spontaneous calcium oscillations and mechanically and chemically induced calcium responses in mammary epithelial cells. Pflug. Arch. 1993, 422, 295–304. [Google Scholar] [CrossRef]
- Enomoto, K.-I.; Furuya, K.; Yamagishi, S.; Oka, T.; Maeno, T. The increase in the intracellular Ca2+ concentration induced by mechanical stimulation is propagated via release of pyrophosphorylated nucleotides in mammary epithelial cells. Pflug. Arch. 1994, 427, 533–542. [Google Scholar] [CrossRef]
- Guo, Y.; Steele, H.E.; Li, B.-Y.; Na, S. Fluid flow-induced activation of subcellular AMPK and its interaction with FAK and Src. Arch. Biochem. Biophys. 2020, 679, 108208. [Google Scholar] [CrossRef] [PubMed]
- Polacheck, W.; German, A.E.; Mammoto, A.; Ingber, N.E.; Kamm, R.D. Mechanotransduction of fluid stresses governs 3D cell migration. Proc. Natl. Acad. Sci. USA 2014, 111, 2447–2452. [Google Scholar] [CrossRef] [PubMed]
- Rajakyla, E.K.; Lehtimaki, J.I.; Acheva, A.; Schaible, N.; Lappalainen, P.; Krishnan, R.; Tojkander, S. Assembly of peripheral actomyosin bundles in epithelial cells is dependent on the CaMKK2/AMPK pathway. Cell Rep. 2020, 30, 4266–4280. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Brand-Saberi, B. Epithelial-mesenchymal transitions in cancer progression. Acta Anat. 1996, 156, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Desai, R.; Solski, P.A.; Der, C.J.; Keely, P.J. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J. Cell Biol. 2003, 163, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Emerman, J.T.; Pitelka, D.R. Maintenance and induction of morphological differentiation in dissociated mammary epithelium on floating collagen membranes. Vitro 1977, 13, 316–328. [Google Scholar] [CrossRef]
- Lee, E.Y.; Parry, G.; Bissell, M.J. Modulation of secreted proteins of mouse mammary epithelial cells by the collagenous substrata. J. Cell Biol. 1984, 98, 146–155. [Google Scholar] [CrossRef]
- Parry, G.; Lee, E.; Farson, D.; Koval, M.; Bissell, M. Collagenous substrata regulate the nature and distribution of glycosaminoglycans produced by differentiated cultures of mouse mammary epithelial cells. Exp. Cell Res. 1985, 156, 487–499. [Google Scholar] [CrossRef]
- Gehler, S.; Baldassarre, M.; Lad, Y.; Leight, J.L.; Wozniak, M.A.; Riching, K.M.; Eliceiri, K.W.; Weaver, V.M.; Calderwood, D.A.; Keely, P.J.; et al. Filamin A-beta1 integrin complex tunes epithelial cell response to matrix tension. Mol. Biol. Cell 2009, 20, 3224–3238. [Google Scholar] [CrossRef]
- Mammoto, A.; Huang, S.; Ingber, D.E. Filamin links cell shape and cytoskeletal structure to Rho regulation by controlling accumulation of p190RhoGAP in lipid rafts. J. Cell Sci. 2007, 120 Pt 3, 456–467. [Google Scholar] [CrossRef]
- Boyd, N.; Guo, H.; Martin, L.J.; Sun, L.; Stone, J.; Fishell, E.; Jong, R.A.; Hislop, G.; Chiarelli, A.; Minkin, S.; et al. Mammographic density and the risk and detection of breast cancer. N. Engl. J. Med. 2007, 356, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Pollan, M.; Ascunce, N.; Ederra, M.; Murillo, A.; Erdozáin, N.; Alés-Martínez, J.E.; Pastor-Barriuso, R. Mammographic density and risk of breast cancer according to tumor characteristics and mode of detection: A Spanish population-based case-control study. Breast Cancer Res. 2013, 15, R9. [Google Scholar] [CrossRef] [PubMed]
- Nazari, S.S.; Mukherjee, P. An overview of mammographic density and its association with breast cancer. Breast Cancer 2018, 25, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.P.; Martin, L.J.; Hanna, W.; Banerjee, D.; Miller, N.; Fishell, E.; Khokha, R.; Boyd, N.F. Growth factors and stromal matrix proteins associated with mammographic densities. Cancer Epidemiol. Biomark. Prev. 2001, 10, 243–248. [Google Scholar]
- Alowami, S.; Troup, S.; Al-Haddad, S.; Kirkpatrick, I.; Watson, P.H. Mammographic density is related to stroma and stromal proteoglycan expression. Breast Cancer Res. 2003, 5, R129–R135. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK–ERK linkage. Oncogene 2009, 28, 4326–4343. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.; White, J.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef]
- Neuhouser, M.L.; Aragaki, A.K.; Prentice, R.L.; Manson, J.E.; Chlebowski, R.; Carty, C.L.; Ochs-Balcom, H.M.; Thomson, C.A.; Caan, B.J.; Tinker, L.F.; et al. Overweight, obesity, and postmenopausal invasive breast cancer risk: A secondary analysis of the women’s health initiative randomized clinical trials. JAMA Oncol. 2015, 1, 611–621. [Google Scholar] [CrossRef]
- Strong, A.L.; A Strong, T.; Rhodes, L.; A Semon, J.; Zhang, X.; Shi, Z.; Zhang, S.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A.; et al. Obesity associated alterations in the biology of adipose stem cells mediate enhanced tumorigenesis by estrogen dependent pathways. Breast Cancer Res. 2013, 15, R102. [Google Scholar] [CrossRef]
- Soguel, L.; Durocher, F.; Tchernof, A.; Diorio, C. Adiposity, breast density, and breast cancer risk: Epidemiological and biological considerations. Eur. J. Cancer Prev. 2017, 26, 511–520. [Google Scholar] [CrossRef]
- Shoham, N.; Gefen, A. Mechanotransduction in adipocytes. J. Biomech. 2012, 45, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.; Li, Q.; Melnichouk, O.; Huszti, E.; Martin, L.J.; Gunasekara, A.; Mawdsley, G.; Yaffe, M.J.; Minkin, S. Evidence that breast tissue stiffness is associated with risk of breast cancer. PLoS ONE 2014, 9, e100937. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Chan, S.; Zhang, Y.; Li, S.; Chang, R.-F.; Su, M.-Y. Evaluation of breast stiffness measured by ultrasound and breast density measured by MRI using a prone-supine deformation model. Biomark. Res. 2019, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Ma, M.; Du, Z.; Liu, Z.; Gong, X. Quantitative evaluation of tissue stiffness around lesion by sound touch elastography in the diagnosis of benign and malignant breast lesions. PLoS ONE 2019, 14, e0219943. [Google Scholar] [CrossRef]
- Choi, W.J.; Kim, H.H.; Cha, J.H.; Shin, H.J.; Kim, H.; Chae, E.Y.; Hong, M.J. Predicting prognostic factors of breast cancer using shear wave elastography. Ultrasound Med. Biol. 2014, 40, 269–274. [Google Scholar] [CrossRef]
- Evans, A.; Whelehan, P.; Thomson, K.; McLean, D.; Brauer, K.; Purdie, C.; Baker, L.; Jordan, L.; Rauchhaus, P.; Thompson, A.; et al. Invasive breast cancer: Relationship between shear-wave elastographic findings and histologic prognostic factors. Radiology 2012, 263, 673–677. [Google Scholar] [CrossRef]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M.; et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. (Camb.) 2015, 7, 1120–1134. [Google Scholar] [CrossRef]
- López, J.I.; Kang, I.; You, W.-K.; McDonald, N.M.; Weaver, V.M. In situforce mapping of mammary gland transformation. Integr. Biol. 2011, 3, 910–921. [Google Scholar] [CrossRef]
- Tanter, M.; Bercoff, J.; Athanasiou, A.; Deffieux, T.; Gennisson, J.-L.; Montaldo, G.; Muller, M.; Tardivon, A.; Fink, M. Quantitative assessment of breast lesion viscoelasticity: Initial clinical results using supersonic shear imaging. Ultrasound Med. Biol. 2008, 34, 1373–1386. [Google Scholar] [CrossRef]
- Zhou, J.; Zhan, W.; Chang, C.; Zhang, X.; Jia, Y.; Dong, Y.; Zhou, C.; Sun, J.; Grant, E.G. Breast lesions: Evaluation with shear wave elastography, with special emphasis on the “Stiff Rim” sign. Radiology 2014, 272, 63–72. [Google Scholar] [CrossRef]
- Ren, W.-W.; Li, X.-L.; He, Y.-P.; Li, D.-D.; Wang, D.; Zhao, C.-K.; Bo, X.-W.; Liu, B.-J.; Yue, W.-W.; Xu, H.-X.; et al. Two-dimensional shear wave elastography of breast lesions: Comparison of two different systems. Clin. Hemorheol. Microcirc. 2017, 66, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.; Whelehan, P.; Thomson, K.; McLean, D.; Brauer, K.; Purdie, C.; Jordan, L.B.; Baker, L.; Thompson, A. Quantitative shear wave ultrasound elastography: Initial experience in solid breast masses. Breast Cancer Res. 2010, 12, R104. [Google Scholar] [CrossRef] [PubMed]
- Tozaki, M.; Fukuma, E. Pattern classification of ShearWaveTM Elastography images for differential diagnosis between benign and malignant solid breast masses. Acta Radiol. 2011, 52, 1069–1075. [Google Scholar] [CrossRef]
- Youk, J.H.; Gweon, H.M.; Son, E.J. Shear-wave elastography in breast ultrasonography: The state of the art. Ultrasonography 2017, 36, 300–309. [Google Scholar] [CrossRef]
- Moon, J.H.; Hwang, J.Y.; Park, J.S.; Koh, S.H.; Park, S.Y. Impact of region of interest (ROI) size on the diagnostic performance of shear wave elastography in differentiating solid breast lesions. Acta Radiol. 2018, 59, 657–663. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Koshy, S.; Da Cunha, C.B.; Shin, J.-W.; Verbeke, C.S.; Allison, K.H.; Mooney, D.J. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat. Mater. 2014, 13, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Ondeck, M.G.; Kumar, A.; Placone, J.K.; Plunkett, C.M.; Matte, B.F.; Wong, K.C.; Fattet, L.; Yang, J.; Engler, A.J. Dynamically stiffened matrix promotes malignant transformation of mammary epithelial cells via collective mechanical signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 3502–3507. [Google Scholar] [CrossRef] [PubMed]
- Stowers, R.S.; Allen, S.C.; Sanchez, K.; Davis, C.L.; Ebelt, N.; Berg, C.V.D.; Suggs, L.J. Extracellular matrix stiffening induces a malignant phenotypic transition in breast epithelial cells. Cell. Mol. Bioeng. 2016, 10, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Young, J.S.; Llumsden, C.E.; Stalker, A.L. The significance of the “tissue pressure” of normal testicular and of neoplastic (Brown-Pearce carcinoma) tissue in the rabbit. J. Pathol. Bacteriol. 1950, 62, 313–333. [Google Scholar] [CrossRef]
- Boucher, Y.; Jain, R.K. Microvascular pressure is the principal driving force for interstitial hypertension in solid tumors: Implications for vascular collapse. Cancer Res. 1992, 52, 5110–5114. [Google Scholar]
- Less, J.R.; Posner, M.C.; Boucher, Y.; Borochovitz, D.; Wolmark, N.; Jain, R.K. Interstitial hypertension in human breast and colorectal tumors. Cancer Res. 1992, 52, 6371–6374. [Google Scholar]
- Nathanson, S.D.; Nelson, L. Interstitial fluid pressure in breast cancer, benign breast conditions, and breast parenchyma. Ann. Surg. Oncol. 1994, 1, 333–338. [Google Scholar] [CrossRef]
- Boucher, Y.; Baxter, L.T.; Jain, R.K. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: Implications for therapy. Cancer Res. 1990, 50, 4478–4484. [Google Scholar]
- Hobbs, S.K.; Monsky, W.L.; Yuan, F.; Roberts, W.G.; Griffith, L.G.; Torchilin, V.P.; Jain, R.K. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc. Natl. Acad. Sci. USA 1998, 95, 4607–4612. [Google Scholar] [CrossRef] [PubMed]
- Griffon-Etienne, G.; Boucher, Y.; Brekken, C.; Suit, H.D.; Jain, R.K. Taxane-induced apoptosis decompresses blood vessels and lowers interstitial fluid pressure in solid tumors: Clinical implications. Cancer Res. 1999, 59, 3776–3782. [Google Scholar] [PubMed]
- Milosevic, M.; Fyles, A.; Hedley, D.; Pintilie, M.; Levin, W.; Manchul, L.; Hill, R. Interstitial fluid pressure predicts survival in patients with cervix cancer independent of clinical prognostic factors and tumor oxygen measurements. Cancer Res. 2001, 61, 6400–6405. [Google Scholar] [PubMed]
- Baxter, L.T.; Jain, R.K. Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc. Res. 1989, 37, 77–104. [Google Scholar] [CrossRef]
- Butler, T.P.; Grantham, F.H.; Gullino, P.M. Bulk transfer of fluid in the interstitial compartment of mammary tumors. Cancer Res. 1975, 35 Pt 1, 3084–3088. [Google Scholar]
- Chary, S.R.; Jain, R.K. Direct measurement of interstitial convection and diffusion of albumin in normal and neoplastic tissues by fluorescence photobleaching. Proc. Natl. Acad. Sci. USA 1989, 86, 5385–5389. [Google Scholar] [CrossRef]
- Dafni, H.; Israely, T.; Bhujwalla, Z.M.; E Benjamin, L.; Neeman, M. Overexpression of vascular endothelial growth factor 165 drives peritumor interstitial convection and induces lymphatic drain: Magnetic resonance imaging, confocal microscopy, and histological tracking of triple-labeled albumin. Cancer Res. 2002, 62, 6731–6739. [Google Scholar]
- Tang, K.; Li, S.; Li, P.; Xia, Q.; Yang, R.; Li, T.; Li, L.; Jiang, Y.; Qin, X.; Yang, H.; et al. Shear stress stimulates integrin beta1 trafficking and increases directional migration of cancer cells via promoting deacetylation of microtubules. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118676. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski-Daspit, A.S.; Tien, J.; Nelson, C.M. Interstitial fluid pressure regulates collective invasion in engineered human breast tumors via Snail, vimentin, and E-cadherin. Integr. Biol. 2016, 8, 319–331. [Google Scholar] [CrossRef]
- Novak, C.M.; Horst, E.N.; Taylor, C.C.; Liu, C.Z.; Mehta, G. Fluid shear stress stimulates breast cancer cells to display invasive and chemoresistant phenotypes while upregulating PLAU in a 3D bioreactor. Biotechnol. Bioeng. 2019, 116, 3084–3097. [Google Scholar] [CrossRef]
- Shieh, A.; Rozansky, H.A.; Hinz, B.; Swartz, M.A. Tumor cell invasion is promoted by interstitial flow-induced matrix priming by stromal fibroblasts. Cancer Res. 2011, 71, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Polacheck, W.; Charest, J.L.; Kamm, R.D. Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 11115–11120. [Google Scholar] [CrossRef] [PubMed]
- Nia, H.T.; Datta, M.; Seano, G.; Huang, P.; Munn, L.L.; Jain, R.K. Quantifying solid stress and elastic energy from excised or in situ tumors. Nat. Protoc. 2018, 13, 1091–1105. [Google Scholar] [CrossRef]
- Helmlinger, G.; Netti, P.; Lichtenbeld, H.C.; Melder, R.J.; Jain, R.K. Solid stress inhibits the growth of multicellular tumor spheroids. Nat. Biotechnol. 1997, 15, 778–783. [Google Scholar] [CrossRef]
- Cheng, G.; Tse, J.; Jain, R.K.; Munn, L. Micro-environmental mechanical stress controls tumor spheroid size and morphology by suppressing proliferation and inducing apoptosis in cancer cells. PLoS ONE 2009, 4, e4632. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef]
- Padera, T.P.; Stoll, B.R.; Tooredman, J.B.; Capen, D.; di Tomaso, E.; Jain, R.K. Pathology: Cancer cells compress intratumour vessels. Nature 2004, 427, 695. [Google Scholar] [CrossRef]
- Chauhan, V.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Martin, J.D.; Snuderl, M.; Mpekris, F.; Jain, S.R.; Jain, R.K. Coevolution of solid stress and interstitial fluid pressure in tumors during progression: Implications for vascular collapse. Cancer Res. 2013, 73, 3833–3841. [Google Scholar] [CrossRef]
- Fovargue, D.; Fiorito, M.; Capilnasiu, A.; Nordsletten, D.; Lee, J.; Sinkus, R. Towards noninvasive estimation of tumour pressure by utilising MR elastography and nonlinear biomechanical models: A simulation and phantom study. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Sanchez, M.E.; Barbier, S.; Whitehead, J.; Bealle, G.; Michel, A.; Latorre-Ossa, H.; Rey, C.; Fouassier, L.; Claperon, A.; Brulle, L.; et al. Mechanical induction of the tumorigenic beta-catenin pathway by tumour growth pressure. Nature 2015, 523, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Seano, G.; Nia, H.T.; Emblem, K.E.; Datta, M.; Ren, J.; Krishnan, S.; Kloepper, J.; Pinho, M.C.; Ho, W.W.; Ghosh, M.; et al. Solid stress in brain tumours causes neuronal loss and neurological dysfunction and can be reversed by lithium. Nat. Biomed. Eng. 2019, 3, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Tse, J.M.; Cheng, G.; Tyrrell, J.A.; Wilcox-Adelman, S.A.; Boucher, Y.; Jain, R.K.; Munn, L.L. Mechanical compression drives cancer cells toward invasive phenotype. Proc. Natl. Acad. Sci. USA 2011, 109, 911–916. [Google Scholar] [CrossRef]
- Shi, Q.; Ghosh, R.; Engelke, H.; Rycroft, C.H.; Cassereau, L.; Sethian, J.A.; Weaver, V.M.; Liphardt, J.T. Rapid disorganization of mechanically interacting systems of mammary acini. Proc. Natl. Acad. Sci. USA 2013, 111, 658–663. [Google Scholar] [CrossRef]
- Ricca, B.L.; Venugopalan, G.; Furuta, S.; Tanner, K.; Orellana, W.A.; Reber, C.D.; Brownfield, D.G.; Bissell, M.J.; Fletcher, D. Transient external force induces phenotypic reversion of malignant epithelial structures via nitric oxide signaling. eLife 2018, 7, e26161. [Google Scholar] [CrossRef]
- Indra, I.; Gasparski, A.N.; Beningo, K.A. An in vitro correlation of metastatic capacity and dual mechanostimulation. PLoS ONE 2018, 13, e0207490. [Google Scholar] [CrossRef]
- Xu, J.; Mathur, J.; Vessieres, E.; Hammack, S.; Nonomura, K.; Favre, J.; Grimaud, L.; Petrus, M.; Francisco, A.; Li, J.; et al. GPR68 senses flow and is essential for vascular physiology. Cell 2018, 173, 762–775. [Google Scholar] [CrossRef]
- Wiley, S.Z.; Sriram, K.; Salmerón, C.; Insel, P.A. GPR68: An emerging drug target in cancer. Int. J. Mol. Sci. 2019, 20, 559. [Google Scholar] [CrossRef] [PubMed]
- Scholz, N. Cancer cell mechanics: Adhesion G protein-coupled receptors in action? Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Jin, R.; Qu, G.; Wang, X.; Li, Z.; Yuan, Z.; Zhao, C.; Siwko, S.; Shi, T.; Wang, P.; et al. GPR116, an adhesion G-protein-coupled receptor, promotes breast cancer metastasis via the Galphaq-p63RhoGEF-Rho GTPase pathway. Cancer Res. 2013, 73, 6206–6218. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Chen, W.; Lou, J.; Rittase, W.; Li, K. Mechanosensing through immunoreceptors. Nat. Immunol. 2019, 20, 1269–1278. [Google Scholar] [CrossRef]
- Hu, K.H.; Butte, M.J. T cell activation requires force generation. J. Cell Biol. 2016, 213, 535–542. [Google Scholar] [CrossRef]
- Basu, R.; Whitlock, B.M.; Husson, J.; Le Floc’H, A.; Jin, W.; Oyler-Yaniv, A.; Dotiwala, F.; Giannone, G.; Hivroz, C.; Biais, N.; et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell 2016, 165, 100–110. [Google Scholar] [CrossRef]
- Saitakis, M.; Dogniaux, S.; Goudot, C.; Bufi, N.; Asnacios, S.; Maurin, M.; Randriamampita, C.; Asnacios, A.; Hivroz, C. Different TCR-induced T lymphocyte responses are potentiated by stiffness with variable sensitivity. eLife 2017, 6, 6. [Google Scholar] [CrossRef]
- Wan, Z.; Zhang, S.; Fan, Y.; Liu, K.; Du, F.; Davey, A.M.; Zhang, H.; Han, W.; Xiong, C.; Liu, W.; et al. B cell activation is regulated by the stiffness properties of the substrate presenting the antigens. J. Immunol. 2013, 190, 4661–4675. [Google Scholar] [CrossRef]
- Balzer, E.M.; Whipple, R.A.; Cho, E.; Matrone, M.A.; Martin, S. Antimitotic chemotherapeutics promote adhesive responses in detached and circulating tumor cells. Breast Cancer Res. Treat. 2009, 121, 65–78. [Google Scholar] [CrossRef]
- Chakrabarti, K.R.; Hessler, L.; Bhandary, L.; Martin, S. Molecular pathways: New signaling considerations when targeting cytoskeletal balance to reduce tumor growth. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 5209–5214. [Google Scholar] [CrossRef]
- Khairallah, R.J.; Shi, G.; Sbrana, F.; Prosser, B.L.; Borroto, C.; Mazaitis, M.J.; Hoffman, E.P.; Mahurkar, A.; Sachs, F.; Sun, Y.; et al. Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci. Signal. 2012, 5, ra56. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pratt, S.J.P.; Lee, R.M.; Martin, S.S. The Mechanical Microenvironment in Breast Cancer. Cancers 2020, 12, 1452. https://doi.org/10.3390/cancers12061452
Pratt SJP, Lee RM, Martin SS. The Mechanical Microenvironment in Breast Cancer. Cancers. 2020; 12(6):1452. https://doi.org/10.3390/cancers12061452
Chicago/Turabian StylePratt, Stephen J.P., Rachel M. Lee, and Stuart S. Martin. 2020. "The Mechanical Microenvironment in Breast Cancer" Cancers 12, no. 6: 1452. https://doi.org/10.3390/cancers12061452
APA StylePratt, S. J. P., Lee, R. M., & Martin, S. S. (2020). The Mechanical Microenvironment in Breast Cancer. Cancers, 12(6), 1452. https://doi.org/10.3390/cancers12061452