Changes in the Tumor Immune Microenvironment during Disease Progression in Patients with Ovarian Cancer

 , ,
, , 
 ,
, 
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Patient Characteristics
2.2. Tumor Recurrence is Associated with Higher Levels of Immune Cell Infiltration
2.3. HLA Expression Is Upregulated in Recurrent Tumors
2.4. Immunosuppressive Molecules Increase in Some Patients While Decreasing in Others
2.5. Increased Stromal Infiltration in the TME
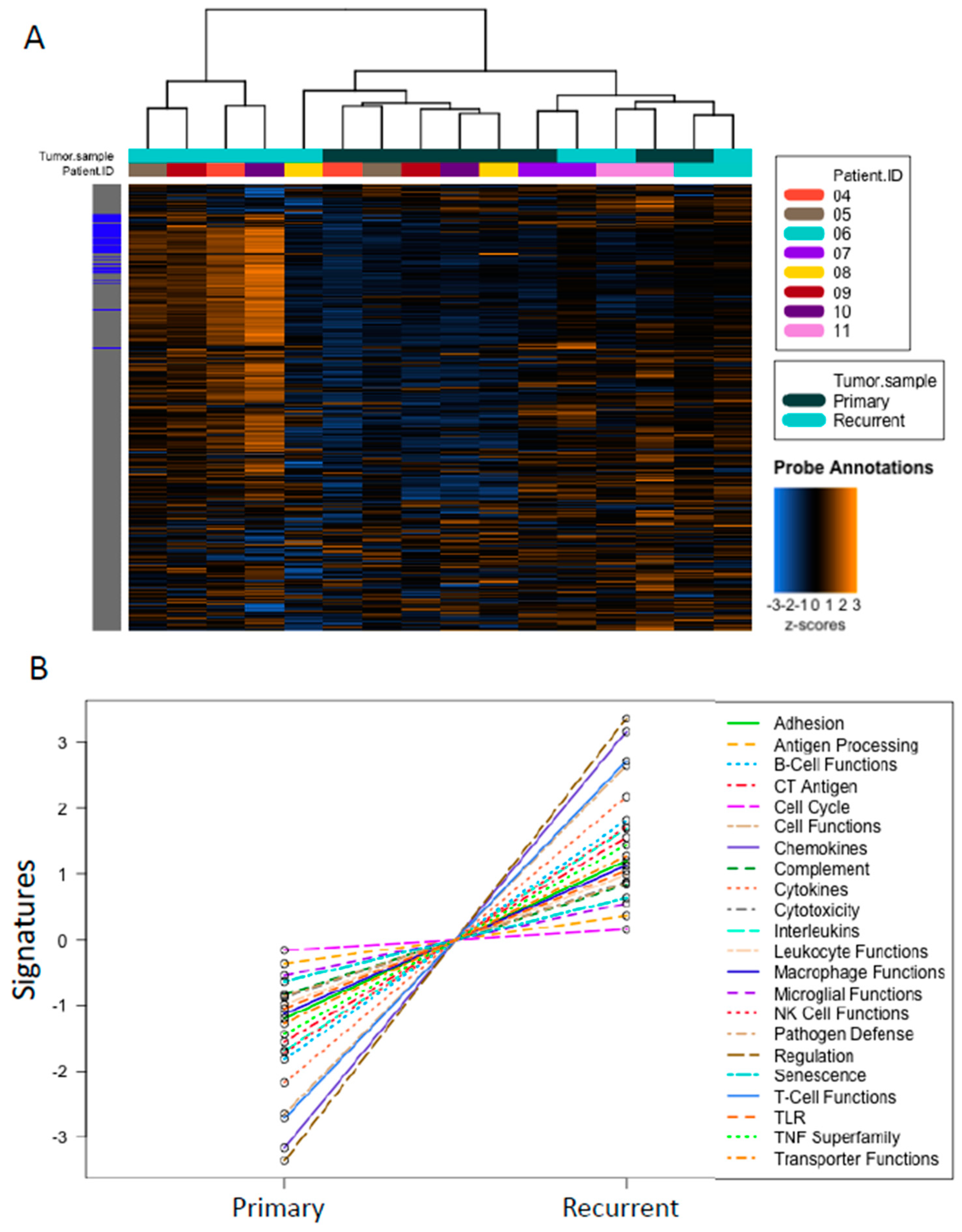
2.6. Higher Cancer- and Immune-Related Gene Expression in Recurrent Tumors
2.7. Indications of Adaptive Immune Resistance
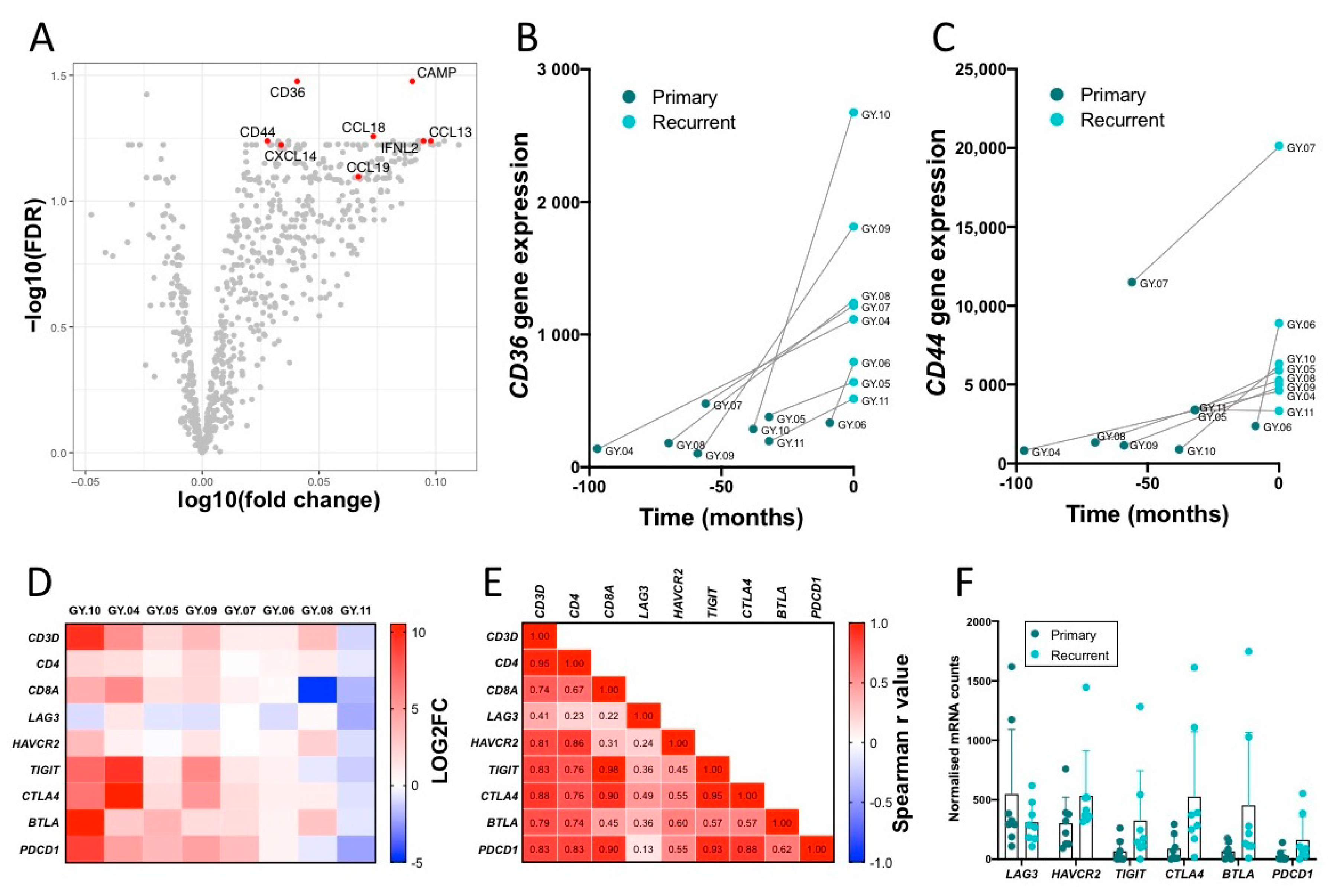
2.8. Top Upregulated Genes, CD36 and CD44
2.9. Infiltrating T Cells and Immune Checkpoint Expression
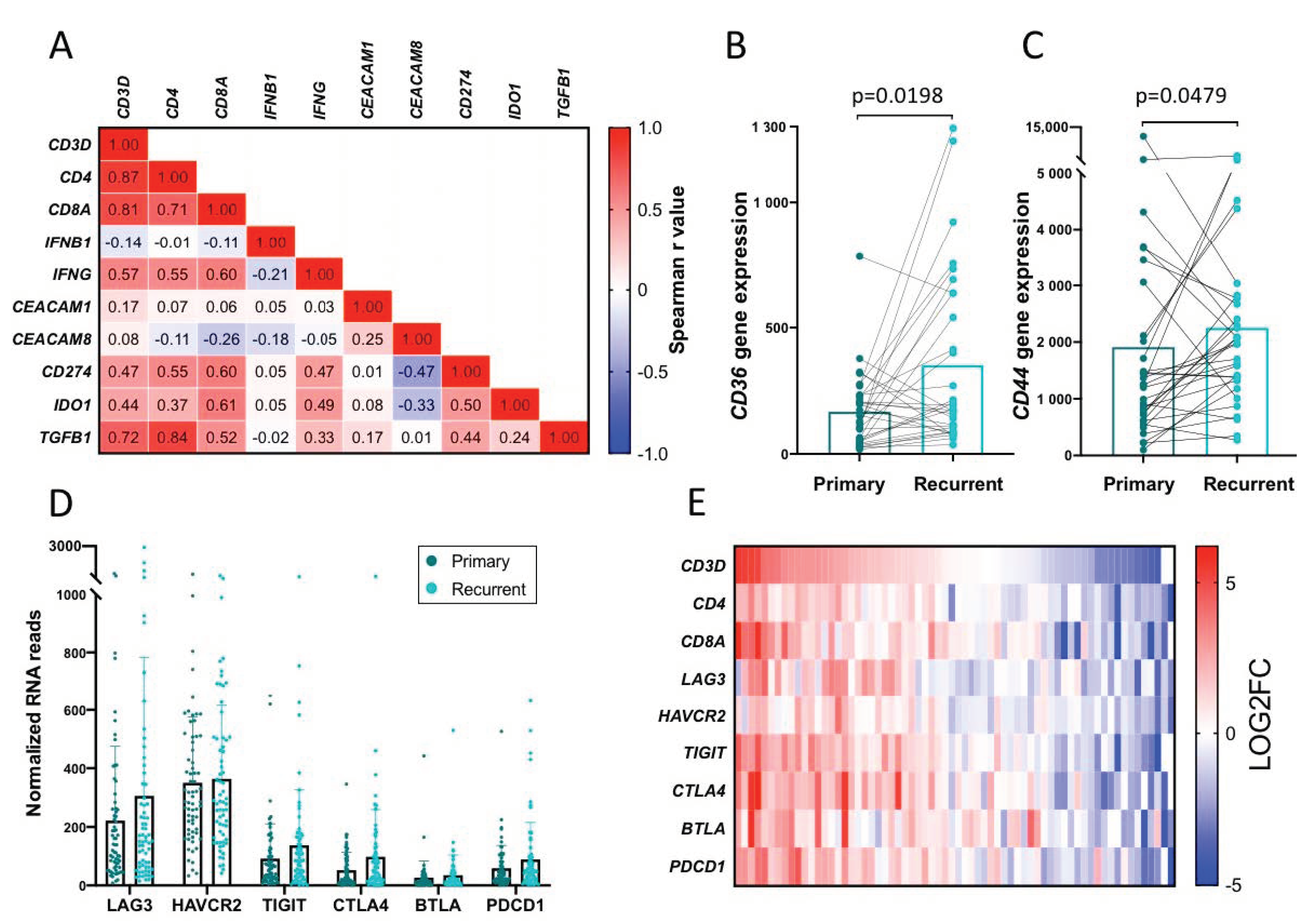
2.10. Validation of Findings in a Separate Cohort
2.11. Expression of Immune Checkpoints on In Vitro Expanded TILs from Recurrent Tumor
3. Discussion
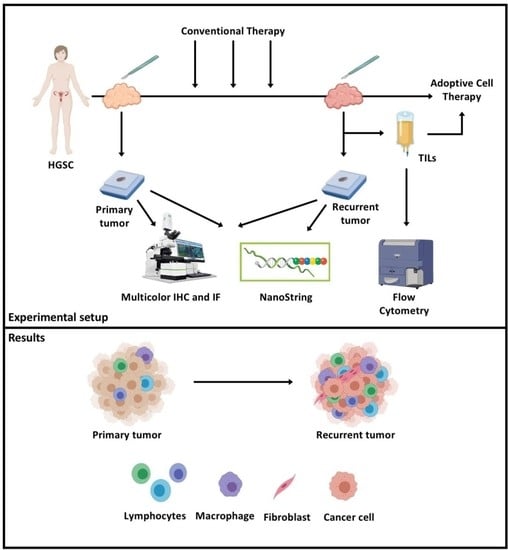
4. Materials and Methods
4.1. Patient Characteristics
4.2. Multicolor Immunohistochemistry/Immunofluorescence
4.3. Imaging and Quantification
4.4. NanoString Specimen Processing
4.5. Flow Cytometry Analysis of TILs from Recurrent Tumors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Hanker, L.C.; Loibl, S.; Burchardi, N.; Pfisterer, J.; Meier, W.; Pujade-Lauraine, E.; Ray-Coquard, I.; Sehouli, J.; Harter, P.; du Bois, A. The impact of second to sixth line therapy on survival of relapsed ovarian cancer after primary taxane/platinum-based therapy. Ann. Oncol. 2012, 23, 2605–2612. [Google Scholar] [CrossRef]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Mullen, M.M.; Kuroki, L.M.; Thaker, P.H. Novel treatment options in platinum-sensitive recurrent ovarian cancer: A review. Gynecol. Oncol. 2019, 152, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goode, E.L.; Block, M.S.; Kalli, K.R.; Vierkant, R.A.; Chen, W.; Fogarty, Z.C.; Gentry-Maharaj, A.; Tołoczko, A.; Hein, A.; Bouligny, A.L.; et al. Dose-Response Association of CD8+ Tumor-Infiltrating Lymphocytes and Survival Time in High-Grade Serous Ovarian Cancer. JAMA Oncol. 2017, 3, e173290. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, D.A.; Webb, J.R.; Nielsen, J.S.; Martin, S.D.; Kroeger, D.R.; Milne, K.; Castellarin, M.; Twumasi-Boateng, K.; Watson, P.H.; Holt, R.A.; et al. Surveillance of the tumor mutanome by T cells during progression from primary to recurrent ovarian cancer. Clin. Cancer Res. 2014, 20, 1125–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomsová, M.; Melichar, B.; Sedláková, I.; Steiner, I. Prognostic significance of CD3+ tumor-infiltrating lymphocytes in ovarian carcinoma. Gynecol. Oncol. 2008, 108, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [Green Version]
- Milne, K.; Köbel, M.; Kalloger, S.E.; Barnes, R.O.; Gao, D.; Gilks, C.B.; Watson, P.H.; Nelson, B.H. Systematic analysis of immune infiltrates in high-grade serous ovarian cancer reveals CD20, FoxP3 and TIA-1 as positive prognostic factors. PLoS ONE 2009, 4, e6412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, J.S.; Sahota, R.A.; Milne, K.; Kost, S.E.; Nesslinger, N.J.; Watson, P.H.; Nelson, B.H. CD20+ tumor-infiltrating lymphocytes have an atypical CD27—Memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin. Cancer Res. 2012, 18, 3281–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.R.; Milne, K.; Kroeger, D.R.; Nelson, B.H. PD-L1 expression is associated with tumor-infiltrating T cells and favorable prognosis in high-grade serous ovarian cancer. Gynecol. Oncol. 2016, 141, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aust, S.; Felix, S.; Auer, K.; Bachmayr-Heyda, A.; Kenner, L.; Dekan, S.; Meier, S.M.; Gerner, C.; Grimm, C.; Pils, D. Absence of PD-L1 on tumor cells is associated with reduced MHC i expression and PD-L1 expression increases in recurrent serous ovarian cancer. Sci. Rep. 2017, 7, 42929. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.R.; Milne, K.; Nelson, B.H. PD-1 and CD103 Are Widely Coexpressed on Prognostically Favorable Intraepithelial CD8 T Cells in Human Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinartz, S.; Schumann, T.; Finkernagel, F.; Wortmann, A.; Jansen, J.M.; Meissner, W.; Krause, M.; Schwörer, A.M.; Wagner, U.; Müller-Brüsselbach, S.; et al. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: Correlation of CD163 expression, cytokine levels and early relapse. Int. J. Cancer 2014, 134, 32–42. [Google Scholar] [CrossRef]
- Drakes, M.L.; Stiff, P.J. Regulation of Ovarian Cancer Prognosis by Immune Cells in the Tumor Microenvironment. Cancers 2018, 10, 302. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.J.; Kasper, S.; Vokes, E.E.; Even, C.; et al. CheckMate 141: 1-Year Update and Subgroup Analysis of Nivolumab as First-Line Therapy in Patients with Recurrent/Metastatic Head and Neck Cancer. Oncologist 2018, 23, 1079–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and antitumor activity of Anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.R.; Moore, K.M.; Gulley, J.L.; Park, H.; Hamilton, E.P.; Kelly, K.; Mita, A.C.; Taylor, M.H.; Chand, V.; Grewal, J.; et al. Efficacy and Safety of Avelumab for Patients With Recurrent or Refractory Ovarian Cancer. JAMA Oncol. 2019, 5, 393. [Google Scholar] [CrossRef] [Green Version]
- Varga, A.; Piha-Paul, S.; Ott, P.A.; Mehnert, J.M.; Berton-Rigaud, D.; Morosky, A.; Yang, P.; Ruman, J.; Matei, D. Pembrolizumab in patients with programmed death ligand 1–positive advanced ovarian cancer: Analysis of KEYNOTE-028. Gynecol. Oncol. 2019, 152, 243–250. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.M.; et al. Antitumor Activity and Safety of Pembrolizumab in Patients with Advanced Recurrent Ovarian Cancer: Results from the Phase 2 KEYNOTE-100 Study. Ann. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef]
- Guo, Z.; Cheng, D.; Xia, Z.; Luan, M.; Wu, L.; Wang, G.; Zhang, S. Combined TIM-3 blockade and CD137 activation affords the long-term protection in a murine model of ovarian cancer. J. Transl. Med. 2013, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.-Y.; Francois, A.; McGray, A.R.; Miliotto, A.; Odunsi, K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology 2017, 6, e1249561. [Google Scholar] [CrossRef] [Green Version]
- Inaba, T.; Ino, K.; Kajiyama, H.; Yamamoto, E.; Shibata, K.; Nawa, A.; Nagasaka, T.; Akimoto, H.; Takikawa, O.; Kikkawa, F. Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol. Oncol. 2009, 115, 185–192. [Google Scholar] [CrossRef]
- Takao, M.; Okamoto, A.; Nikaido, T.; Urashima, M.; Takakura, S.; Saito, M.; Saito, M.; Okamoto, S.; Takikawa, O.; Sasaki, H.; et al. Increased synthesis of indoleamine-2,3-dioxygenaseprotein is positively associated with impaired survival in patients with serous-type, but not with other types of, ovarian cancer. Oncol. Rep. 2007, 17, 1333–1339. [Google Scholar] [CrossRef]
- van Baren, N.; Van den Eynde, B.J. Tryptophan-degrading enzymes in tumoral immune resistance. Front. Immunol. 2015, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chodon, T.; Lugade, A.A.; Battaglia, S.; Odunsi, K. Emerging Role and Future Directions of Immunotherapy in Advanced Ovarian Cancer. Hematol. Oncol. Clin. N. Am. 2018, 32, 1025–1039. [Google Scholar] [CrossRef]
- Kristeleit, R.; Davidenko, I.; Shirinkin, V.; El-Khouly, F.; Bondarenko, I.; Goodheart, M.J.; Gorbunova, V.; Penning, C.A.; Shi, J.G.; Liu, X.; et al. A randomised, open-label, phase 2 study of the IDO1 inhibitor epacadostat (INCB024360) versus tamoxifen as therapy for biochemically recurrent (CA-125 relapse)–only epithelial ovarian cancer, primary peritoneal carcinoma, or fallopian tube cancer. Gynecol. Oncol. 2017, 146, 484–490. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Kreuzinger, C.; Geroldinger, A.; Smeets, D.; Braicu, E.I.; Sehouli, J.; Koller, J.; Wolf, A.; Darb-Esfahani, S.; Joehrens, K.; Vergote, I.; et al. A complex network of tumor microenvironment in human high-grade serous ovarian cancer. Clin. Cancer Res. 2017, 23, 7621–7632. [Google Scholar] [CrossRef] [Green Version]
- Ojalvo, L.S.; Thompson, E.D.; Wang, T.L.; Meeker, A.K.; Shih, I.M.; Fader, A.N.; Cimino-Mathews, A.; Emens, L.A. Tumor-associated macrophages and the tumor immune microenvironment of primary and recurrent epithelial ovarian cancer. Hum. Pathol. 2018, 74, 135–147. [Google Scholar] [CrossRef]
- Nonaka, H.; Saga, Y.; Fujiwara, H.; Akimoto, H.; Yamada, A.; Kagawa, S.; Takei, Y.; Machida, S.; Takikawa, O.; Suzuki, M. Indoleamine 2,3-dioxygenase promotes peritoneal dissemination of ovarian cancer through inhibition of natural killercell function and angiogenesis promotion. Int. J. Oncol. 2011, 38, 113–120. [Google Scholar] [CrossRef]
- De Jong, R.A.; Nijman, H.W.; Boezen, H.M.; Volmer, M.; Ten Hoor, K.A.; Krijnen, J.; Van Der Zee, A.G.J.; Hollema, H.; Kema, I.P. Serum tryptophan and kynurenine concentrations as parameters for indoleamine 2,3-dioxygenase activity in patients with endometrial, ovarian, and vulvar cancer. Int. J. Gynecol. Cancer 2011, 21, 1320–1327. [Google Scholar] [CrossRef]
- Mills, A.M.; Peres, L.C.; Meiss, A.; Ring, K.L.; Modesitt, S.C.; Abbott, S.E.; Alberg, A.J.; Bandera, E.V.; Barnholtz-Sloan, J.; Bondy, M.L.; et al. Targetable Immune Regulatory Molecule Expression in High-Grade Serous Ovarian Carcinomas in African American Women. Int. J. Gynecol. Pathol. 2019, 38, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H.; Svane, I.M. Indoleamine 2,3-dioxygenase vaccination. Oncoimmunology 2015, 4, 983770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, T.F. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non–T-Cell–Inflamed Tumor Microenvironment. Semin. Oncol. 2015, 42, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, P.S.; Karanikas, V.; Evers, S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, M.; Westergaard, M.C.W.; Milne, K.; Nielsen, M.; Borch, T.H.; Poulsen, L.G.; Hendel, H.W.; Kennedy, M.; Briggs, G.; Ledoux, S.; et al. Adoptive cell therapy with tumor-infiltrating lymphocytes in patients with metastatic ovarian cancer: A pilot study. Oncoimmunology 2018, 7, e1502905. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, N.; Giese, N.A.; Giese, T.; Poschke, I.; Offringa, R.; Werner, J.; Ryschich, E. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin. Cancer Res. 2014, 20, 3422–3433. [Google Scholar] [CrossRef] [Green Version]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.L.; Carretta, M.; Kalvisa, A.; Siersbæk, M.S.; Simões, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen density regulates the activity of tumor-infiltrating T cells. J. Immunother. Cancer 2019, 7, 68. [Google Scholar] [CrossRef] [Green Version]
- Reis, P.P.; Waldron, L.; Goswami, R.S.; Xu, W.; Xuan, Y.; Perez-Ordonez, B.; Gullane, P.; Irish, J.; Jurisica, I.; Kamel-Reid, S. mRNA transcript quantification in archival samples using multiplexed, color-coded probes. BMC Biotechnol. 2011, 11, 46. [Google Scholar] [CrossRef] [Green Version]
- Malkov, V.A.; Serikawa, K.A.; Balantac, N.; Watters, J.; Geiss, G.; Mashadi-Hossein, A.; Fare, T. Multiplexed measurements of gene signatures in different analytes using the Nanostring nCounterTM Assay System. BMC Res. Notes 2009, 2, 80. [Google Scholar] [CrossRef] [Green Version]
- Talhouk, A.; Kommoss, S.; Mackenzie, R.; Cheung, M.; Leung, S.; Chiu, D.S.; Kalloger, S.E.; Huntsman, D.G.; Chen, S.; Intermaggio, M.; et al. Single-patient molecular testing with nanostring ncounter data using a reference-based strategy for batch effect correction. PLoS ONE 2016, 11, e0153844. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov ID NCT01611558. Phase II Study of Ipilimumab Monotherapy in Recurrent Platinum-Sensitive Ovarian Cancer—Study Results. 2012. Available online: https://clinicaltrials.gov/ct2/show/NCT01611558 (accessed on 10 August 2020).
- Smazynski, J.; Hamilton, P.T.; Thornton, S.; Milne, K.; Wouters, M.C.A.; Webb, J.R.; Nelson, B.H. The immune suppressive factors CD155 and PD-L1 show contrasting expression patterns and immune correlates in ovarian and other cancers. Gynecol. Oncol. 2020, 158, 167–177. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov ID NCT04570839. COM701 in Combination With BMS-986207 and Nivolumab in Subjects with Advanced Solid Tumors. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04570839 (accessed on 20 November 2020).
- ClinicalTrials.gov ID NCT04354246. COM902 (A TIGIT Inhibitor) in Subjects with Advanced Malignancies. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04354246 (accessed on 20 November 2020).
- Rachidi, S.M.; Qin, T.; Sun, S.; Zheng, W.J.; Li, Z. Molecular Profiling of Multiple Human Cancers Defines an Inflammatory Cancer-Associated Molecular Pattern and Uncovers KPNA2 as a Uniform Poor Prognostic Cancer Marker. PLoS ONE 2013, 8, e57911. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Franco, F.; Tsui, Y.-C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.-H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef]
- Bartakova, A.; Michalova, K.; Presl, J.; Vlasak, P.; Kostun, J.; Bouda, J. CD44 as a cancer stem cell marker and its prognostic value in patients with ovarian carcinoma. J. Obstet. Gynaecol. 2018, 38, 110–114. [Google Scholar] [CrossRef]
- Sillanpää, S.; Anttila, M.A.; Voutilainen, K.; Tammi, R.H.; Tammi, M.I.; Saarikoski, S.V.; Kosma, V.M. CD44 Expression Indicates Favorable Prognosis in Epithelial Ovarian Cancer. Clin. Cancer Res. 2003, 9, 5318–5324. [Google Scholar]
- Puvanenthiran, S.; Essapen, S.; Haagsma, B.; Bagwan, I.; Green, M.; Khelwatty, S.A.; Seddon, A.; Modjtahedi, H. Co-expression and prognostic significance of the HER family members, EGFRvIII, c-MET, CD44 in patients with ovarian cancer. Oncotarget 2018, 9, 19662–19674. [Google Scholar] [CrossRef] [Green Version]
- Baaten, B.J.G.; Tinoco, R.; Chen, A.T.; Bradley, L.M. Regulation of antigen-experienced T cells: Lessons from the quintessential memory marker CD44. Front. Immunol. 2012, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Estess, P.; DeGrendele, H.C.; Pascual, V.; Siegelman, M.H. Functional activation of lymphocyte CD44 in peripheral blood is a marker of autoimmune disease activity. J. Clin. Investig. 1998, 102, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Talhouk, A.; George, J.; Wang, C.; Budden, T.; Tan, T.Z.; Chiu, D.S.; Kommoss, S.; Leong, H.S.; Chen, S.; Intermaggio, M.P.; et al. Development and Validation of the Gene Expression Predictor of High-grade Serous Ovarian Carcinoma Molecular SubTYPE (PrOTYPE). Clin. Cancer Res. 2020, 26, 5411–5423. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Westergaard, M.C.W.; Milne, K.; Pedersen, M.; Hasselager, T.; Olsen, L.R.; Anglesio, M.S.; Borch, T.H.; Kennedy, M.; Briggs, G.; Ledoux, S.; et al. Changes in the Tumor Immune Microenvironment during Disease Progression in Patients with Ovarian Cancer. Cancers 2020, 12, 3828. https://doi.org/10.3390/cancers12123828
Westergaard MCW, Milne K, Pedersen M, Hasselager T, Olsen LR, Anglesio MS, Borch TH, Kennedy M, Briggs G, Ledoux S, et al. Changes in the Tumor Immune Microenvironment during Disease Progression in Patients with Ovarian Cancer. Cancers. 2020; 12(12):3828. https://doi.org/10.3390/cancers12123828
Chicago/Turabian StyleWestergaard, Marie Christine Wulff, Katy Milne, Magnus Pedersen, Thomas Hasselager, Lars Rønn Olsen, Michael S. Anglesio, Troels Holz Borch, Mia Kennedy, Gillian Briggs, Stacey Ledoux, and et al. 2020. "Changes in the Tumor Immune Microenvironment during Disease Progression in Patients with Ovarian Cancer" Cancers 12, no. 12: 3828. https://doi.org/10.3390/cancers12123828
APA StyleWestergaard, M. C. W., Milne, K., Pedersen, M., Hasselager, T., Olsen, L. R., Anglesio, M. S., Borch, T. H., Kennedy, M., Briggs, G., Ledoux, S., Kreuzinger, C., Decken, I. v. d., Donia, M., Castillo-Tong, D. C., Nelson, B. H., & Svane, I. M. (2020). Changes in the Tumor Immune Microenvironment during Disease Progression in Patients with Ovarian Cancer. Cancers, 12(12), 3828. https://doi.org/10.3390/cancers12123828