Lipid Regulatory Proteins as Potential Therapeutic Targets for Ovarian Cancer in Obese Women


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
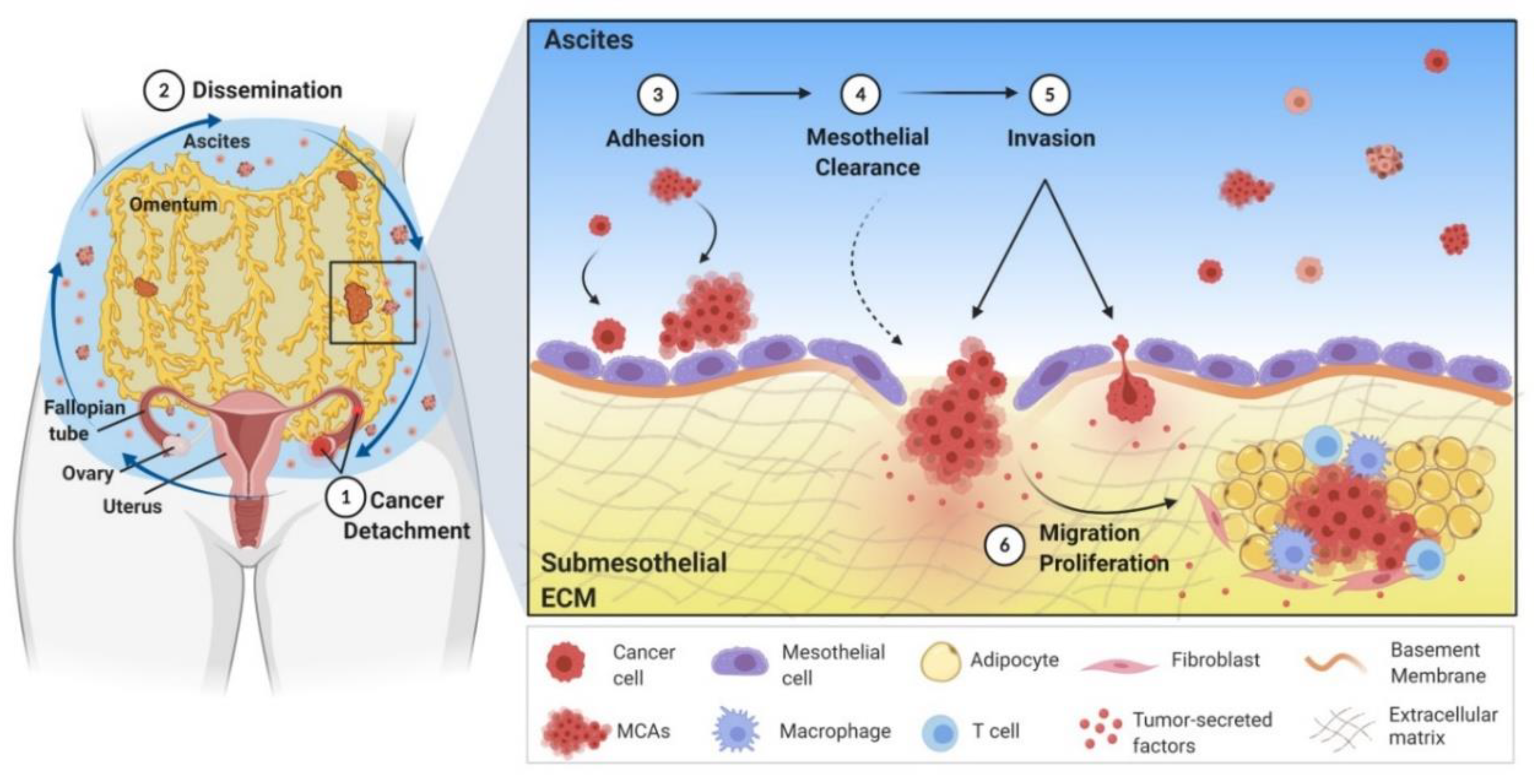
2. Mechanism of Ovarian Cancer Metastasis
3. The Link between Obesity and Ovarian Cancer
3.1. Obesity-Associated Tumor Inflammatory Microenvironment Contributes to Ovarian Cancer Progression and Metastasis
3.2. Obese Adipose Tissue Promotes Ovarian Cancer Progression by Altered Exogenous Lipid Transport and Uptake
3.3. Obesity Promotes Ovarian Cancer Progression by Altered de novo Lipogenesis
4. Sterol Regulatory Element Binding Protein (SREBP)-Regulated de novo Lipogenesis in Cancer
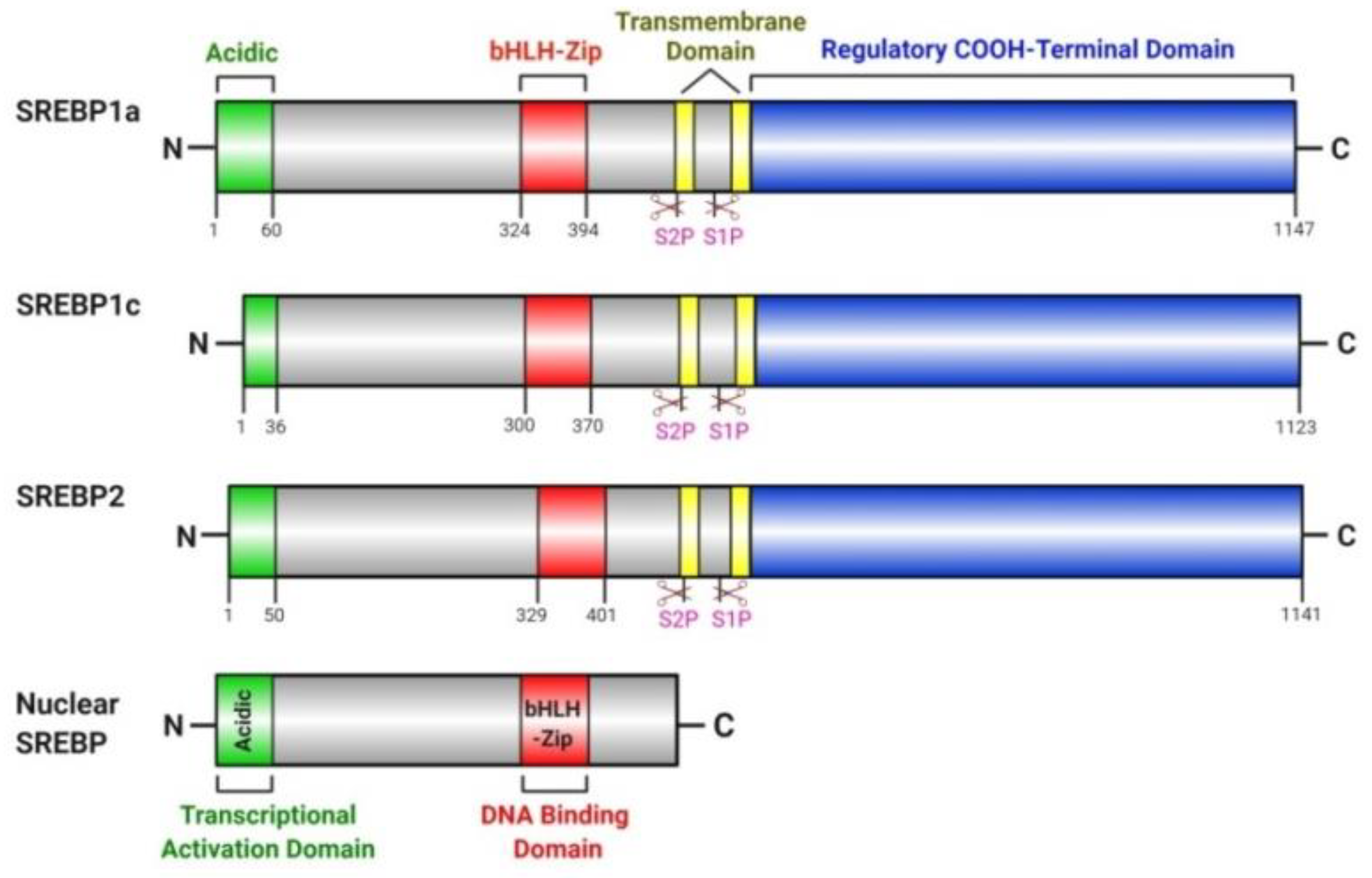
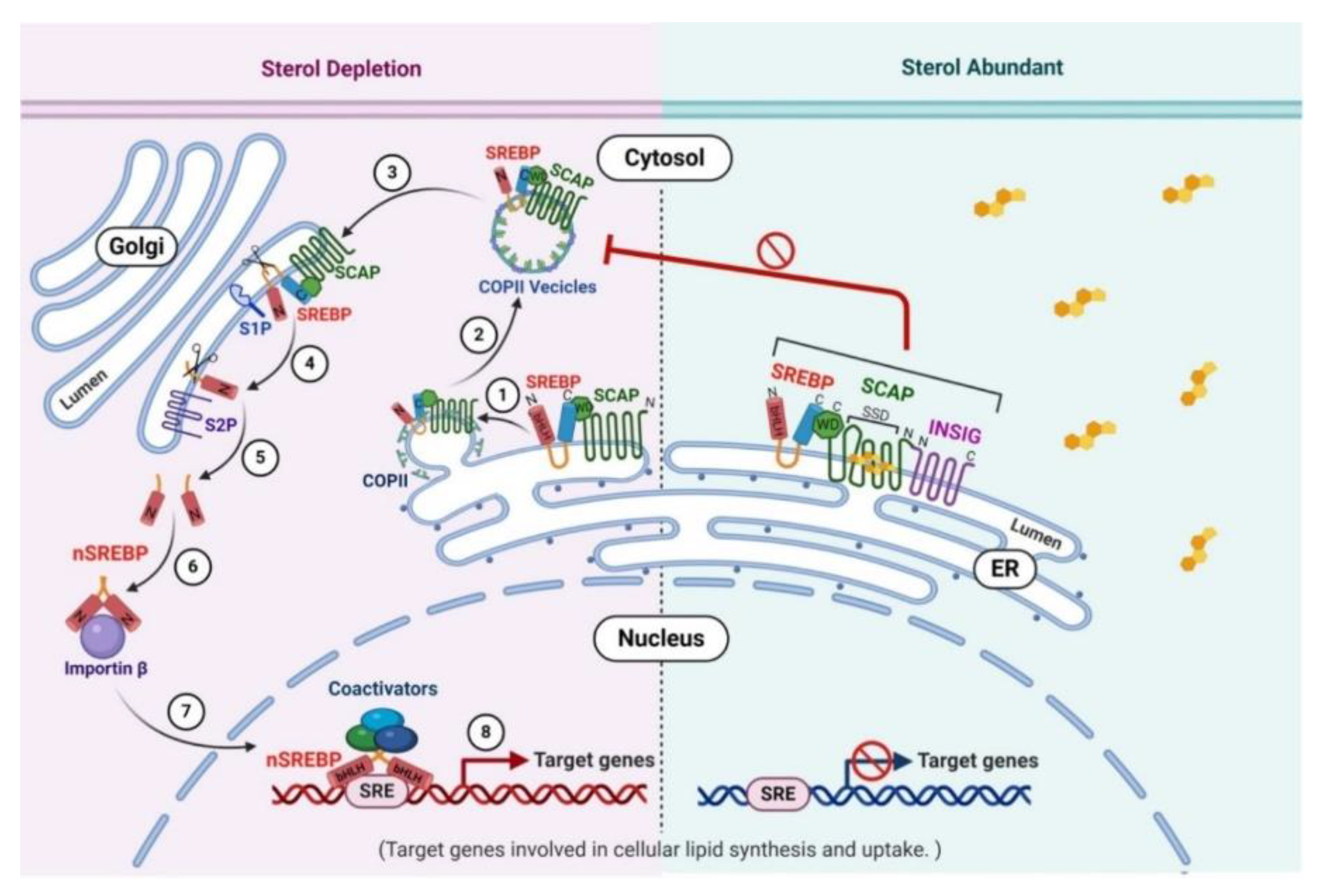
4.1. SREBP Pathway
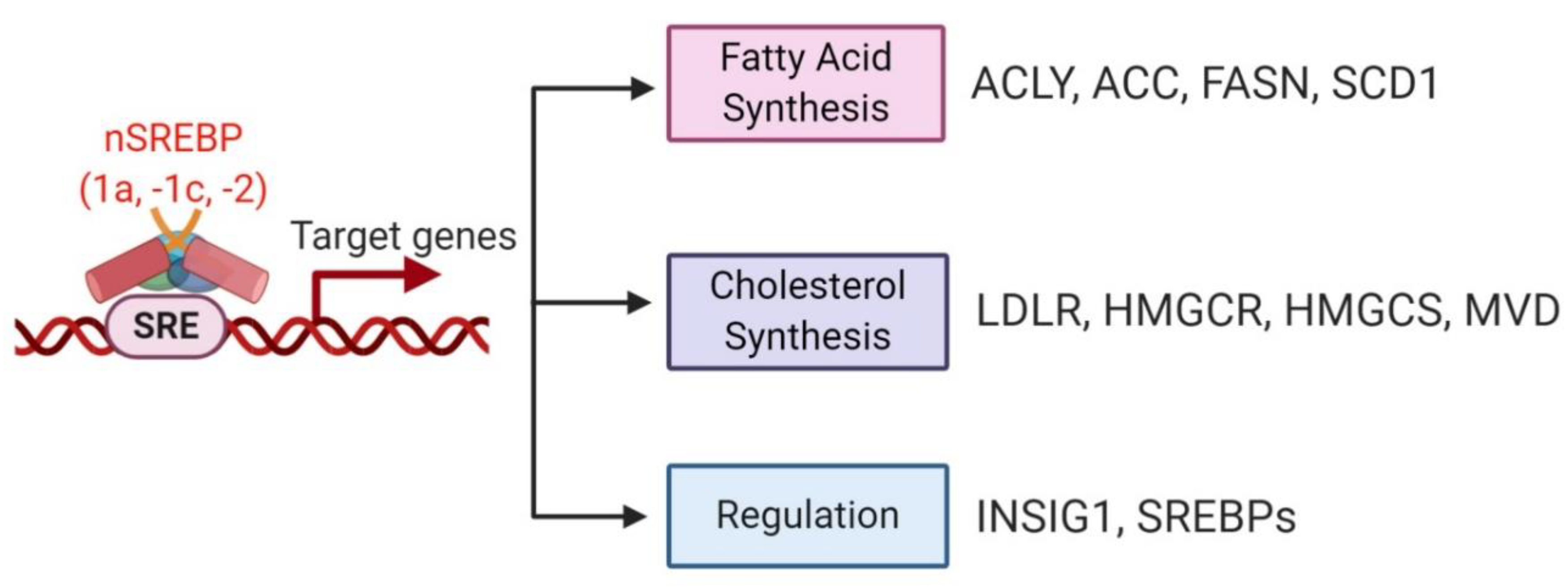
4.2. Regulation of de novo Lipogenesis by SREBP in Cancer
4.3. Regulation of SREBP by Oncogenic and Tumor Suppressor Signaling Pathways
4.4. SREBP1 and Ovarian Cancer
5. Targeting SREBP1-Mediated Lipid Metabolism Pathways in Cancer Treatment
5.1. Targeting SREBP1 with Small Molecule Inhibitors
5.1.1. Fatostatin
5.1.2. Betulin
5.1.3. PF-429242
5.1.4. BF175
5.2. Targeting SREBP-Pathway Genes
5.2.1. FASN
5.2.2. ACLY
5.2.3. ACC
5.2.4. SCD
5.2.5. HMGCR
5.2.6. CPT1
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef]
- Flegal, K.M.; Kruszon-Moran, D.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Trends in Obesity Among Adults in the United States, 2005 to 2014. JAMA 2016, 315, 2284–2291. [Google Scholar] [CrossRef] [PubMed]
- Flegal, K.M.; Carroll, M.D.; Kit, B.K.; Ogden, C.L. Prevalence of Obesity and Trends in the Distribution of Body Mass Index Among US Adults, 1999–2010. JAMA 2012, 307, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of Obesity and Severe Obesity Among Adults: United States, 2017–2018. In NCHS Data Brief, no 360; National Center for Health Statistics: Hyattsville, MD, USA, 2020. [Google Scholar]
- Stefan, N.; Haring, H.U.; Hu, F.B.; Schulze, M.B. Metabolically healthy obesity: Epidemiology, mechanisms, and clinical implications. Lancet Diabetes Endocrinol. 2013, 1, 152–162. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Colditz, G.A.; Peterson, L.L. Obesity and Cancer: Evidence, Impact, and Future Directions. Clin. Chem. 2018, 64, 154–162. [Google Scholar] [CrossRef]
- Yang, L.; Drake, B.F.; Colditz, G.A. Obesity and Other Cancers. J. Clin. Oncol. 2016, 34, 4231–4237. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Naora, H.; Montell, D.J. Ovarian cancer metastasis: Integrating insights from disparate model organisms. Nat. Rev. Cancer 2005, 5, 355–366. [Google Scholar] [CrossRef]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.L.; Leung, C.S.; Yip, K.P.; Au Yeung, C.L.; Wong, S.T.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [PubMed]
- Motohara, T.; Masuda, K.; Morotti, M.; Zheng, Y.; El-Sahhar, S.; Chong, K.Y.; Wietek, N.; Alsaadi, A.; Karaminejadranjbar, M.; Hu, Z.; et al. An evolving story of the metastatic voyage of ovarian cancer cells: Cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene 2019, 38, 2885–2898. [Google Scholar] [CrossRef] [PubMed]
- Burleson, K.M.; Casey, R.C.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R., Jr.; Skubitz, A.P. Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol. Oncol. 2004, 93, 170–181. [Google Scholar] [CrossRef]
- Schild, T.; Low, V.; Blenis, J.; Gomes, A.P. Unique Metabolic Adaptations Dictate Distal Organ-Specific Metastatic Colonization. Cancer Cell 2018, 33, 347–354. [Google Scholar] [CrossRef]
- Hilliard, T.S. The Impact of Mesothelin in the Ovarian Cancer Tumor Microenvironment. Cancers 2018, 10, 277. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Clark, R.; Krishnan, V.; Schoof, M.; Rodriguez, I.; Theriault, B.; Chekmareva, M.; Rinker-Schaeffer, C. Milky spots promote ovarian cancer metastatic colonization of peritoneal adipose in experimental models. Am. J. Pathol. 2013, 183, 576–591. [Google Scholar] [CrossRef]
- Gerber, S.A.; Rybalko, V.Y.; Bigelow, C.E.; Lugade, A.A.; Foster, T.H.; Frelinger, J.G.; Lord, E.M. Preferential attachment of peritoneal tumor metastases to omental immune aggregates and possible role of a unique vascular microenvironment in metastatic survival and growth. Am. J. Pathol. 2006, 169, 1739–1752. [Google Scholar] [CrossRef]
- Zhang, Y.; Nowicka, A.; Solley, T.N.; Wei, C.; Parikh, A.; Court, L.; Burks, J.K.; Andreeff, M.; Woodward, W.A.; Dadbin, A.; et al. Stromal Cells Derived from Visceral and Obese Adipose Tissue Promote Growth of Ovarian Cancers. PLoS ONE 2015, 10, e0136361. [Google Scholar] [CrossRef] [PubMed]
- Fortner, R.T.; Poole, E.M.; Wentzensen, N.A.; Trabert, B.; White, E.; Arslan, A.A.; Patel, A.V.; Setiawan, V.W.; Visvanathan, K.; Weiderpass, E.; et al. Ovarian cancer risk factors by tumor aggressiveness: An analysis from the Ovarian Cancer Cohort Consortium. Int. J. Cancer 2019, 145, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Nagle, C.M.; Dixon, S.C.; Jensen, A.; Kjaer, S.K.; Modugno, F.; de Fazio, A.; Fereday, S.; Hung, J.; Johnatty, S.E.; Australian Ovarian Cancer Study Group; et al. Obesity and survival among women with ovarian cancer: Results from the Ovarian Cancer Association Consortium. Br. J. Cancer 2015, 113, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.S.; Kim, H.J.; Hong, J.H.; Lee, J.K.; Lee, N.W.; Song, J.Y. Obesity and epithelial ovarian cancer survival: A systematic review and meta-analysis. J. Ovarian Res. 2014, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Protani, M.M.; Nagle, C.M.; Webb, P.M. Obesity and ovarian cancer survival: A systematic review and meta-analysis. Cancer Prev. Res. 2012, 5, 901–910. [Google Scholar] [CrossRef]
- Yang, H.S.; Yoon, C.; Myung, S.K.; Park, S.M. Effect of obesity on survival of women with epithelial ovarian cancer: A systematic review and meta-analysis of observational studies. Int. J. Gynecol. Cancer 2011, 21, 1525–1532. [Google Scholar] [CrossRef]
- Liu, Y.; Metzinger, M.N.; Lewellen, K.A.; Cripps, S.N.; Carey, K.D.; Harper, E.I.; Shi, Z.; Tarwater, L.; Grisoli, A.; Lee, E.; et al. Obesity Contributes to Ovarian Cancer Metastatic Success through Increased Lipogenesis, Enhanced Vascularity, and Decreased Infiltration of M1 Macrophages. Cancer Res. 2015, 75, 5046–5057. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef]
- DeBose-Boyd, R.A.; Ye, J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem. Sci. 2018, 43, 358–368. [Google Scholar] [CrossRef]
- Jeon, T.I.; Osborne, T.F. SREBPs: Metabolic integrators in physiology and metabolism. Trends Endocrinol. Metab. 2012, 23, 65–72. [Google Scholar] [CrossRef]
- Yokoyama, C.; Wang, X.; Briggs, M.R.; Admon, A.; Wu, J.; Hua, X.; Goldstein, J.L.; Brown, M.S. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 1993, 75, 187–197. [Google Scholar] [CrossRef]
- Mukherjee, A.; Wu, J.; Barbour, S.; Fang, X. Lysophosphatidic acid activates lipogenic pathways and de novo lipid synthesis in ovarian cancer cells. J. Biol. Chem. 2012, 287, 24990–25000. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.Y.; Lu, Q.T.; Li, W.H.; Yang, N.; Dongol, S.; Zhang, X.; Jiang, J. Sterol regulatory element-binding protein 1 is required for ovarian tumor growth. Oncol. Rep. 2013, 30, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Ma, Y.; Yuan, F.; Gong, Y.; Fang, Z.; Mohamed, E.M.; Berrios, E.; Shao, H.; Fang, X. Lysophosphatidic Acid Up-Regulates Hexokinase II and Glycolysis to Promote Proliferation of Ovarian Cancer Cells. Neoplasia 2015, 17, 723–734. [Google Scholar] [CrossRef]
- Cheng, X.; Li, J.; Guo, D. SCAP/SREBPs are Central Players in Lipid Metabolism and Novel Metabolic Targets in Cancer Therapy. Curr. Top. Med. Chem. 2018, 18, 484–493. [Google Scholar] [CrossRef]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Guo, D.; Bell, E.H.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014, 20, 2619–2626. [Google Scholar] [CrossRef]
- Lim, D.; Oliva, E. Precursors and pathogenesis of ovarian carcinoma. Pathology 2013, 45, 229–242. [Google Scholar] [CrossRef]
- Zhang, S.; Dolgalev, I.; Zhang, T.; Ran, H.; Levine, D.A.; Neel, B.G. Both fallopian tube and ovarian surface epithelium are cells-of-origin for high-grade serous ovarian carcinoma. Nat. Commun. 2019, 10, 5367. [Google Scholar] [CrossRef]
- Levanon, K.; Crum, C.; Drapkin, R. New Insights Into the Pathogenesis of Serous Ovarian Cancer and Its Clinical Impact. J. Clin. Oncol. 2008, 26, 5284–5293. [Google Scholar] [CrossRef]
- Hudson, L.G.; Zeineldin, R.; Stack, M.S. Phenotypic plasticity of neoplastic ovarian epithelium: Unique cadherin profiles in tumor progression. Clin. Exp. Metastasis 2008, 25, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N.; Wong, A.S.; Choi, K.C.; Kang, S.K.; Leung, P.C. Ovarian surface epithelium: Biology, endocrinology, and pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C., Jr.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Pignata, S.; Ledermann, J.A. Latest clinical evidence and further development of PARP inhibitors in ovarian cancer. Ann. Oncol. 2018, 29, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.S. Latest clinical evidence of maintenance therapy in ovarian cancer. Curr. Opin. Obstet. Gynecol. 2020, 32, 15–21. [Google Scholar] [CrossRef]
- Schuijer, M.; Berns, E.M. TP53 and ovarian cancer. Hum. Mutat. 2003, 21, 285–291. [Google Scholar] [CrossRef]
- Berns, E.M.; Bowtell, D.D. The changing view of high-grade serous ovarian cancer. Cancer Res. 2012, 72, 2701–2704. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef]
- Nakamura, M.; Obata, T.; Daikoku, T.; Fujiwara, H. The Association and Significance of p53 in Gynecologic Cancers: The Potential of Targeted Therapy. Int. J. Mol. Sci. 2019, 20, 5482. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S.; Agarwal, R.; Kaye, S.B. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006, 7, 925–934. [Google Scholar] [CrossRef]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [PubMed]
- Barbolina, M.V.; Moss, N.M.; Westfall, S.D.; Liu, Y.; Burkhalter, R.J.; Marga, F.; Forgacs, G.; Hudson, L.G.; Stack, M.S. Microenvironmental regulation of ovarian cancer metastasis. Cancer Treat. Res. 2009, 149, 319–334. [Google Scholar] [CrossRef]
- Pradeep, S.; Kim, S.W.; Wu, S.Y.; Nishimura, M.; Chaluvally-Raghavan, P.; Miyake, T.; Pecot, C.V.; Kim, S.J.; Choi, H.J.; Bischoff, F.Z.; et al. Hematogenous metastasis of ovarian cancer: Rethinking mode of spread. Cancer Cell 2014, 26, 77–91. [Google Scholar] [CrossRef]
- Coffman, L.G.; Burgos-Ojeda, D.; Wu, R.; Cho, K.; Bai, S.; Buckanovich, R.J. New models of hematogenous ovarian cancer metastasis demonstrate preferential spread to the ovary and a requirement for the ovary for abdominal dissemination. Transl. Res. 2016, 175, 92–102.e102. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Obesity and Cancer Mechanisms: Cancer Metabolism. J. Clin. Oncol. 2016, 34, 4277–4283. [Google Scholar] [CrossRef]
- Aziz, M.; Agarwal, K.; Dasari, S.; Mitra, A.A.K. Productive Cross-Talk with the Microenvironment: A Critical Step in Ovarian Cancer Metastasis. Cancers 2019, 11, 1608. [Google Scholar] [CrossRef]
- Ghoneum, A.; Afify, H.; Salih, Z.; Kelly, M.; Said, N. Role of tumor microenvironment in the pathobiology of ovarian cancer: Insights and therapeutic opportunities. Cancer Med. 2018, 7, 5047–5056. [Google Scholar] [CrossRef]
- Ghoneum, A.; Afify, H.; Salih, Z.; Kelly, M.; Said, N. Role of tumor microenvironment in ovarian cancer pathobiology. Oncotarget 2018, 9, 22832–22849. [Google Scholar] [CrossRef] [PubMed]
- Maniati, E.; Berlato, C.; Gopinathan, G.; Heath, O.; Kotantaki, P.; Lakhani, A.; McDermott, J.; Pegrum, C.; Delaine-Smith, R.M.; Pearce, O.M.T.; et al. Mouse Ovarian Cancer Models Recapitulate the Human Tumor Microenvironment and Patient Response to Treatment. Cell Rep. 2020, 30, 525–540.e527. [Google Scholar] [CrossRef] [PubMed]
- Nwani, N.G.; Sima, L.E.; Nieves-Neira, W.; Matei, D. Targeting the Microenvironment in High Grade Serous Ovarian Cancer. Cancers 2018, 10, 266. [Google Scholar] [CrossRef] [PubMed]
- Stack, M.S.; Nephew, K.P.; Burdette, J.E.; Mitra, A.K. The Tumor Microenvironment of High Grade Serous Ovarian Cancer. Cancers 2018, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Worzfeld, T.; Pogge von Strandmann, E.; Huber, M.; Adhikary, T.; Wagner, U.; Reinartz, S.; Muller, R. The Unique Molecular and Cellular Microenvironment of Ovarian Cancer. Front. Oncol. 2017, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Luwor, R.B.; Bilandzic, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: Molecular phenotype of chemoresistant ovarian tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef]
- Preston, C.C.; Goode, E.L.; Hartmann, L.C.; Kalli, K.R.; Knutson, K.L. Immunity and immune suppression in human ovarian cancer. Immunotherapy 2011, 3, 539–556. [Google Scholar] [CrossRef]
- Takaishi, K.; Komohara, Y.; Tashiro, H.; Ohtake, H.; Nakagawa, T.; Katabuchi, H.; Takeya, M. Involvement of M2-polarized macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Sci. 2010, 101, 2128–2136. [Google Scholar] [CrossRef]
- Reinartz, S.; Schumann, T.; Finkernagel, F.; Wortmann, A.; Jansen, J.M.; Meissner, W.; Krause, M.; Schworer, A.M.; Wagner, U.; Muller-Brusselbach, S.; et al. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: Correlation of CD163 expression, cytokine levels and early relapse. Int. J. Cancer 2014, 134, 32–42. [Google Scholar] [CrossRef]
- Kulbe, H.; Chakravarty, P.; Leinster, D.A.; Charles, K.A.; Kwong, J.; Thompson, R.G.; Coward, J.I.; Schioppa, T.; Robinson, S.C.; Gallagher, W.M.; et al. A dynamic inflammatory cytokine network in the human ovarian cancer microenvironment. Cancer Res. 2012, 72, 66–75. [Google Scholar] [CrossRef]
- Peng, P.; Yan, Y.; Keng, S. Exosomes in the ascites of ovarian cancer patients: Origin and effects on anti-tumor immunity. Oncol. Rep. 2011, 25, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Huang, B.; Huang, Z.; Cai, J.; Gong, L.; Zhang, Y.; Jiang, J.; Dong, W.; Wang, Z. Tumor cellfibroblast heterotypic aggregates in malignant ascites of patients with ovarian cancer. Int. J. Mol. Med. 2019, 44, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.R.; Milner, J.J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef]
- Kolb, R.; Sutterwala, F.S.; Zhang, W. Obesity and cancer: Inflammation bridges the two. Curr. Opin. Pharmacol. 2016, 29, 77–89. [Google Scholar] [CrossRef]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, Inflammation, and Cancer. Annu. Rev. Pathol. 2016, 11, 421–449. [Google Scholar] [CrossRef]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef]
- Mukherjee, A.; Chiang, C.Y.; Daifotis, H.A.; Nieman, K.M.; Fahrmann, J.F.; Lastra, R.R.; Romero, I.L.; Fiehn, O.; Lengyel, E. Adipocyte-Induced FABP4 Expression in Ovarian Cancer Cells Promotes Metastasis and Mediates Carboplatin Resistance. Cancer Res. 2020, 80, 1748–1761. [Google Scholar] [CrossRef]
- Gharpure, K.M.; Pradeep, S.; Sans, M.; Rupaimoole, R.; Ivan, C.; Wu, S.Y.; Bayraktar, E.; Nagaraja, A.S.; Mangala, L.S.; Zhang, X.; et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun. 2018, 9, 2923. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Jarc, E.; Jusovic, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef]
- Wang, C.; Rajput, S.; Watabe, K.; Liao, D.F.; Cao, D. Acetyl-CoA carboxylase-a as a novel target for cancer therapy. Front. Biosci. 2010, 2, 515–526. [Google Scholar] [CrossRef]
- Kuhajda, F.P. Fatty-acid synthase and human cancer: New perspectives on its role in tumor biology. Nutrition 2000, 16, 202–208. [Google Scholar] [CrossRef]
- Huang, J.; Fan, X.X.; He, J.; Pan, H.; Li, R.Z.; Huang, L.; Jiang, Z.; Yao, X.J.; Liu, L.; Leung, E.L.; et al. SCD1 is associated with tumor promotion, late stage and poor survival in lung adenocarcinoma. Oncotarget 2016, 7, 39970–39979. [Google Scholar] [CrossRef] [PubMed]
- Ntambi, J.M. Regulation of stearoyl-CoA desaturase by polyunsaturated fatty acids and cholesterol. J. Lipid Res. 1999, 40, 1549–1558. [Google Scholar] [PubMed]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Bauer, D.E.; Hatzivassiliou, G.; Zhao, F.; Andreadis, C.; Thompson, C.B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 2005, 24, 6314–6322. [Google Scholar] [CrossRef]
- Yoon, S.; Lee, M.Y.; Park, S.W.; Moon, J.S.; Koh, Y.K.; Ahn, Y.H.; Park, B.W.; Kim, K.S. Up-regulation of acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J. Biol. Chem. 2007, 282, 26122–26131. [Google Scholar] [CrossRef]
- Igal, R.A. Stearoyl-CoA desaturase-1: A novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis 2010, 31, 1509–1515. [Google Scholar] [CrossRef]
- Deutsch, A.J.A.; Rinner, B.; Pichler, M.; Prochazka, K.; Pansy, K.; Bischof, M.; Fechter, K.; Hatzl, S.; Feichtinger, J.; Wenzl, K.; et al. NR4A3 Suppresses Lymphomagenesis through Induction of Proapoptotic Genes. Cancer Res. 2017, 77, 2375–2386. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Soyal, S.M.; Nofziger, C.; Dossena, S.; Paulmichl, M.; Patsch, W. Targeting SREBPs for treatment of the metabolic syndrome. Trends Pharmacol. Sci. 2015, 36, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Nohturfft, A.; DeBose-Boyd, R.A.; Scheek, S.; Goldstein, J.L.; Brown, M.S. Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proc. Natl. Acad. Sci. USA 1999, 96, 11235–11240. [Google Scholar] [CrossRef]
- Rawson, R.B. The SREBP pathway—Insights from Insigs and insects. Nat. Rev. Mol. Cell Biol. 2003, 4, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Ye, J.; Rawson, R.B.; Goldstein, J.L. Regulated intramembrane proteolysis: A control mechanism conserved from bacteria to humans. Cell 2000, 100, 391–398. [Google Scholar] [CrossRef]
- Hua, X.; Yokoyama, C.; Wu, J.; Briggs, M.R.; Brown, M.S.; Goldstein, J.L.; Wang, X. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc. Natl. Acad. Sci. USA 1993, 90, 11603–11607. [Google Scholar] [CrossRef] [PubMed]
- Briggs, M.R.; Yokoyama, C.; Wang, X.; Brown, M.S.; Goldstein, J.L. Nuclear protein that binds sterol regulatory element of low density lipoprotein receptor promoter. I. Identification of the protein and delineation of its target nucleotide sequence. J. Biol. Chem. 1993, 268, 14490–14496. [Google Scholar]
- Bennett, M.K.; Lopez, J.M.; Sanchez, H.B.; Osborne, T.F. Sterol regulation of fatty acid synthase promoter. Coordinate feedback regulation of two major lipid pathways. J. Biol. Chem. 1995, 270, 25578–25583. [Google Scholar] [CrossRef]
- Hirano, Y.; Yoshida, M.; Shimizu, M.; Sato, R. Direct demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathway. J. Biol. Chem. 2001, 276, 36431–36437. [Google Scholar] [CrossRef]
- Hirano, Y.; Murata, S.; Tanaka, K.; Shimizu, M.; Sato, R. Sterol regulatory element-binding proteins are negatively regulated through SUMO-1 modification independent of the ubiquitin/26 S proteasome pathway. J. Biol. Chem. 2003, 278, 16809–16819. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Podo, F. Tumour phospholipid metabolism. NMR Biomed. 1999, 12, 413–439. [Google Scholar] [CrossRef]
- Li, J.; Ren, S.; Piao, H.L.; Wang, F.; Yin, P.; Xu, C.; Lu, X.; Ye, G.; Shao, Y.; Yan, M.; et al. Integration of lipidomics and transcriptomics unravels aberrant lipid metabolism and defines cholesteryl oleate as potential biomarker of prostate cancer. Sci. Rep. 2016, 6, 20984. [Google Scholar] [CrossRef]
- Saito, K.; Arai, E.; Maekawa, K.; Ishikawa, M.; Fujimoto, H.; Taguchi, R.; Matsumoto, K.; Kanai, Y.; Saito, Y. Lipidomic Signatures and Associated Transcriptomic Profiles of Clear Cell Renal Cell Carcinoma. Sci. Rep. 2016, 6, 28932. [Google Scholar] [CrossRef]
- Wu, Q.; Ishikawa, T.; Sirianni, R.; Tang, H.; McDonald, J.G.; Yuhanna, I.S.; Thompson, B.; Girard, L.; Mineo, C.; Brekken, R.A.; et al. 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Rep. 2013, 5, 637–645. [Google Scholar] [CrossRef]
- Nelson, E.R.; Wardell, S.E.; Jasper, J.S.; Park, S.; Suchindran, S.; Howe, M.K.; Carver, N.J.; Pillai, R.V.; Sullivan, P.M.; Sondhi, V.; et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 2013, 342, 1094–1098. [Google Scholar] [CrossRef]
- Kim, H.; Rodriguez-Navas, C.; Kollipara, R.K.; Kapur, P.; Pedrosa, I.; Brugarolas, J.; Kittler, R.; Ye, J. Unsaturated Fatty Acids Stimulate Tumor Growth through Stabilization of beta-Catenin. Cell Rep. 2015, 13, 495–503. [Google Scholar] [CrossRef]
- Khwairakpam, A.D.; Shyamananda, M.S.; Sailo, B.L.; Rathnakaram, S.R.; Padmavathi, G.; Kotoky, J.; Kunnumakkara, A.B. ATP citrate lyase (ACLY): A promising target for cancer prevention and treatment. Curr. Drug Targets 2015, 16, 156–163. [Google Scholar] [CrossRef]
- Beckers, A.; Organe, S.; Timmermans, L.; Scheys, K.; Peeters, A.; Brusselmans, K.; Verhoeven, G.; Swinnen, J.V. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007, 67, 8180–8187. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.Y.; Chang, Y.F.; Hwang, J.J. Antitumor effect of orlistat, a fatty acid synthase inhibitor, is via activation of caspase-3 on human colorectal carcinoma-bearing animal. Biomed. Pharmacother. 2011, 65, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Colomer, R.; Brunet, J.; Menendez, J.A. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin™) by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int. J. Oncol. 2007, 31, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar] [CrossRef]
- Ettinger, S.L.; Sobel, R.; Whitmore, T.G.; Akbari, M.; Bradley, D.R.; Gleave, M.E.; Nelson, C.C. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. 2004, 64, 2212–2221. [Google Scholar] [CrossRef]
- Yang, Y.; Morin, P.J.; Han, W.F.; Chen, T.; Bornman, D.M.; Gabrielson, E.W.; Pizer, E.S. Regulation of fatty acid synthase expression in breast cancer by sterol regulatory element binding protein-1c. Exp. Cell Res. 2003, 282, 132–137. [Google Scholar] [CrossRef]
- Bao, J.; Zhu, L.; Zhu, Q.; Su, J.; Liu, M.; Huang, W. SREBP-1 is an independent prognostic marker and promotes invasion and migration in breast cancer. Oncol. Lett. 2016, 12, 2409–2416. [Google Scholar] [CrossRef]
- Huang, W.C.; Li, X.; Liu, J.; Lin, J.; Chung, L.W. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol. Cancer Res. 2012, 10, 133–142. [Google Scholar] [CrossRef]
- Yin, F.; Sharen, G.; Yuan, F.; Peng, Y.; Chen, R.; Zhou, X.; Wei, H.; Li, B.; Jing, W.; Zhao, J. TIP30 regulates lipid metabolism in hepatocellular carcinoma by regulating SREBP1 through the Akt/mTOR signaling pathway. Oncogenesis 2017, 6, e347. [Google Scholar] [CrossRef]
- Sun, Y.; He, W.; Luo, M.; Zhou, Y.; Chang, G.; Ren, W.; Wu, K.; Li, X.; Shen, J.; Zhao, X.; et al. SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumor Biol. 2015, 36, 4133–4141. [Google Scholar] [CrossRef]
- Wen, Y.A.; Xiong, X.; Zaytseva, Y.Y.; Napier, D.L.; Vallee, E.; Li, A.T.; Wang, C.; Weiss, H.L.; Evers, B.M.; Gao, T. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018, 9, 265. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.S.; Lee, Y.R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Nan, X.; Shi, X.; Mu, X.; Liu, B.; Zhu, H.; Yao, B.; Liu, X.; Yang, T.; Hu, Y.; et al. SREBP1 promotes the invasion of colorectal cancer accompanied upregulation of MMP7 expression and NF-kappaB pathway activation. BMC Cancer 2019, 19, 685. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Feng, F.; Wang, L.; Wang, X.; Li, Z.; Cao, Y. SREBP-1 inhibitor Betulin enhances the antitumor effect of Sorafenib on hepatocellular carcinoma via restricting cellular glycolytic activity. Cell Death Dis. 2019, 10, 672. [Google Scholar] [CrossRef]
- Li, J.N.; Mahmoud, M.A.; Han, W.F.; Ripple, M.; Pizer, E.S. Sterol regulatory element-binding protein-1 participates in the regulation of fatty acid synthase expression in colorectal neoplasia. Exp. Cell Res. 2000, 261, 159–165. [Google Scholar] [CrossRef]
- Li, S.; Qiu, L.; Wu, B.; Shen, H.; Zhu, J.; Zhou, L.; Gu, L.; Di, W. TOFA suppresses ovarian cancer cell growth in vitro and in vivo. Mol. Med. Rep. 2013, 8, 373–378. [Google Scholar] [CrossRef]
- Wang, C.; Xu, C.; Sun, M.; Luo, D.; Liao, D.F.; Cao, D. Acetyl-CoA carboxylase-alpha inhibitor TOFA induces human cancer cell apoptosis. Biochem. Biophys. Res. Commun. 2009, 385, 302–306. [Google Scholar] [CrossRef]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef]
- Pons-Tostivint, E.; Thibault, B.; Guillermet-Guibert, J. Targeting PI3K Signaling in Combination Cancer Therapy. Trends Cancer 2017, 3, 454–469. [Google Scholar] [CrossRef]
- Aoki, M.; Fujishita, T. Oncogenic Roles of the PI3K/AKT/mTOR Axis. Curr. Top. Microbiol. Immunol. 2017, 407, 153–189. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Kuroda, H.; Takahashi, R.; Sasano, T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol. Oncol. 2015, 137, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Ghoneum, A.; Abdulfattah, A.Y.; Said, N. Targeting the PI3K/AKT/mTOR/NFkappaB Axis in Ovarian Cancer. J. Cell. Immunol. 2020, 2, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Lewis, C.A.; Griffiths, B.; Santos, C.R.; Pende, M.; Schulze, A. Regulation of the SREBP transcription factors by mTORC1. Biochem. Soc. Trans. 2011, 39, 495–499. [Google Scholar] [CrossRef]
- Bakan, I.; Laplante, M. Connecting mTORC1 signaling to SREBP-1 activation. Curr. Opin. Lipidol. 2012, 23, 226–234. [Google Scholar] [CrossRef]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef]
- Lee, G.; Zheng, Y.; Cho, S.; Jang, C.; England, C.; Dempsey, J.M.; Yu, Y.; Liu, X.; He, L.; Cavaliere, P.M.; et al. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell 2017, 171, 1545–1558 e1518. [Google Scholar] [CrossRef]
- Kim, K.H.; Song, M.J.; Yoo, E.J.; Choe, S.S.; Park, S.D.; Kim, J.B. Regulatory role of glycogen synthase kinase 3 for transcriptional activity of ADD1/SREBP1c. J. Biol. Chem. 2004, 279, 51999–52006. [Google Scholar] [CrossRef]
- Berwick, D.C.; Hers, I.; Heesom, K.J.; Moule, S.K.; Tavare, J.M. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J. Biol. Chem. 2002, 277, 33895–33900. [Google Scholar] [CrossRef]
- Cui, Y.; Xing, P.; Wang, Y.; Liu, M.; Qiu, L.; Ying, G.; Li, B. NADPH accumulation is responsible for apoptosis in breast cancer cells induced by fatty acid synthase inhibition. Oncotarget 2017, 8, 32576–32585. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.J.; Yang, G.; Yang, P.; Fazakerley, D.J.; Stockli, J.; Yang, J.Y.; James, D.E. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013, 17, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, A.; Cornu, M.; Cybulski, N.; Polak, P.; Betz, C.; Trapani, F.; Terracciano, L.; Heim, M.H.; Ruegg, M.A.; Hall, M.N. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012, 15, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Aylon, Y.; Oren, M. The Hippo pathway, p53 and cholesterol. Cell Cycle 2016, 15, 2248–2255. [Google Scholar] [CrossRef]
- Bathaie, S.Z.; Ashrafi, M.; Azizian, M.; Tamanoi, F. Mevalonate Pathway and Human Cancers. Curr. Mol. Pharmacol. 2017, 10, 77–85. [Google Scholar] [CrossRef]
- Visser, S.; Yang, X. LATS tumor suppressor: A new governor of cellular homeostasis. Cell Cycle 2010, 9, 3892–3903. [Google Scholar] [CrossRef]
- Aylon, Y.; Gershoni, A.; Rotkopf, R.; Biton, I.E.; Porat, Z.; Koh, A.P.; Sun, X.; Lee, Y.; Fiel, M.I.; Hoshida, Y.; et al. The LATS2 tumor suppressor inhibits SREBP and suppresses hepatic cholesterol accumulation. Genes Dev. 2016, 30, 786–797. [Google Scholar] [CrossRef]
- Aylon, Y.; Michael, D.; Shmueli, A.; Yabuta, N.; Nojima, H.; Oren, M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006, 20, 2687–2700. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Silwal-Pandit, L.; Langerod, A.; Borresen-Dale, A.L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026252. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Nakagawa, Y.; Ide, T.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. p53 Activation in adipocytes of obese mice. J. Biol. Chem. 2003, 278, 25395–25400. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.T.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Xu, Y.; Gaudette, D.C.; Boynton, J.D.; Frankel, A.; Fang, X.J.; Sharma, A.; Hurteau, J.; Casey, G.; Goodbody, A.; Mellors, A.; et al. Characterization of an ovarian cancer activating factor in ascites from ovarian cancer patients. Clin. Cancer Res. 1995, 1, 1223–1232. [Google Scholar]
- Xu, Y.; Shen, Z.; Wiper, D.W.; Wu, M.; Morton, R.E.; Elson, P.; Kennedy, A.W.; Belinson, J.; Markman, M.; Casey, G. Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. JAMA 1998, 280, 719–723. [Google Scholar] [CrossRef]
- Xu, Y. Lysophospholipid Signaling in the Epithelial Ovarian Cancer Tumor Microenvironment. Cancers 2018, 10, 227. [Google Scholar] [CrossRef]
- Ren, J.; Xiao, Y.J.; Singh, L.S.; Zhao, X.; Zhao, Z.; Feng, L.; Rose, T.M.; Prestwich, G.D.; Xu, Y. Lysophosphatidic acid is constitutively produced by human peritoneal mesothelial cells and enhances adhesion, migration, and invasion of ovarian cancer cells. Cancer Res. 2006, 66, 3006–3014. [Google Scholar] [CrossRef]
- Volden, P.A.; Skor, M.N.; Johnson, M.B.; Singh, P.; Patel, F.N.; McClintock, M.K.; Brady, M.J.; Conzen, S.D. Mammary Adipose Tissue-Derived Lysophospholipids Promote Estrogen Receptor-Negative Mammary Epithelial Cell Proliferation. Cancer Prev. Res. 2016, 9, 367–378. [Google Scholar] [CrossRef]
- Bushweller, J.H. Targeting transcription factors in cancer—from undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef] [PubMed]
- Kamisuki, S.; Mao, Q.; Abu-Elheiga, L.; Gu, Z.; Kugimiya, A.; Kwon, Y.; Shinohara, T.; Kawazoe, Y.; Sato, S.; Asakura, K.; et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem. Biol. 2009, 16, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Machamer, C.E.; Espenshade, P.J. Fatostatin blocks ER exit of SCAP but inhibits cell growth in a SCAP-independent manner. J. Lipid Res. 2016, 57, 1564–1573. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Shi, Z.; Li, X.; Li, W.; Wang, Y.; Liu, Z.; Jiang, J. Fatostatin suppresses growth and enhances apoptosis by blocking SREBP-regulated metabolic pathways in endometrial carcinoma. Oncol. Rep. 2018, 39, 1919–1929. [Google Scholar] [CrossRef]
- Brovkovych, V.; Izhar, Y.; Danes, J.M.; Dubrovskyi, O.; Sakallioglu, I.T.; Morrow, L.M.; Atilla-Gokcumen, G.E.; Frasor, J. Fatostatin induces pro- and anti-apoptotic lipid accumulation in breast cancer. Oncogenesis 2018, 7, 66. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.T.; Hu, P.; Huang, W.C. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 2014, 13, 855–866. [Google Scholar] [CrossRef]
- Li, X.; Wu, J.B.; Chung, L.W.; Huang, W.C. Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations. Oncotarget 2015, 6, 41018–41032. [Google Scholar] [CrossRef]
- Siqingaowa; Sekar, S.; Gopalakrishnan, V.; Taghibiglou, C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem. Biophys. Res. Commun. 2017, 488, 136–140. [Google Scholar] [CrossRef]
- Gholkar, A.A.; Cheung, K.; Williams, K.J.; Lo, Y.C.; Hamideh, S.A.; Nnebe, C.; Khuu, C.; Bensinger, S.J.; Torres, J.Z. Fatostatin Inhibits Cancer Cell Proliferation by Affecting Mitotic Microtubule Spindle Assembly and Cell Division. J. Biol. Chem. 2016, 291, 17001–17008. [Google Scholar] [CrossRef]
- Tang, J.J.; Li, J.G.; Qi, W.; Qiu, W.W.; Li, P.S.; Li, B.L.; Song, B.L. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab. 2011, 13, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Krol, S.K.; Kielbus, M.; Rivero-Muller, A.; Stepulak, A. Comprehensive review on betulin as a potent anticancer agent. BioMed Res. Int. 2015, 2015, 584189. [Google Scholar] [CrossRef] [PubMed]
- Rzeski, W.; Stepulak, A.; Szymanski, M.; Juszczak, M.; Grabarska, A.; Sifringer, M.; Kaczor, J.; Kandefer-Szerszen, M. Betulin elicits anti-cancer effects in tumour primary cultures and cell lines in vitro. Basic Clin. Pharmacol. Toxicol. 2009, 105, 425–432. [Google Scholar] [CrossRef]
- Krycer, J.R.; Phan, L.; Brown, A.J. A key regulator of cholesterol homoeostasis, SREBP-2, can be targeted in prostate cancer cells with natural products. Biochem. J. 2012, 446, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yan, H.; Zhao, L.; Jia, W.; Yang, H.; Liu, L.; Zhou, X.; Miao, P.; Sun, X.; Song, S.; et al. Inhibition of SREBP increases gefitinib sensitivity in non-small cell lung cancer cells. Oncotarget 2016, 7, 52392–52403. [Google Scholar] [CrossRef]
- Li, N.; Zhou, Z.S.; Shen, Y.; Xu, J.; Miao, H.H.; Xiong, Y.; Xu, F.; Li, B.L.; Luo, J.; Song, B.L. Inhibition of the sterol regulatory element-binding protein pathway suppresses hepatocellular carcinoma by repressing inflammation in mice. Hepatology 2017, 65, 1936–1947. [Google Scholar] [CrossRef]
- Hawkins, J.L.; Robbins, M.D.; Warren, L.C.; Xia, D.; Petras, S.F.; Valentine, J.J.; Varghese, A.H.; Wang, I.K.; Subashi, T.A.; Shelly, L.D.; et al. Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element-binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. J. Pharmacol. Exp. Ther. 2008, 326, 801–808. [Google Scholar] [CrossRef]
- Yang, P.M.; Hong, Y.H.; Hsu, K.C.; Liu, T.P. p38alpha/S1P/SREBP2 activation by the SAM-competitive EZH2 inhibitor GSK343 limits its anticancer activity but creates a druggable vulnerability in hepatocellular carcinoma. Am. J. Cancer Res. 2019, 9, 2120–2139. [Google Scholar]
- Caruana, B.T.; Skoric, A.; Brown, A.J.; Lutze-Mann, L.H. Site-1 protease, a novel metabolic target for glioblastoma. Biochem. Biophys. Res. Commun. 2017, 490, 760–766. [Google Scholar] [CrossRef]
- Zhao, X.; Xiaoli; Zong, H.; Abdulla, A.; Yang, E.S.; Wang, Q.; Ji, J.Y.; Pessin, J.E.; Das, B.C.; Yang, F. Inhibition of SREBP transcriptional activity by a boron-containing compound improves lipid homeostasis in diet-induced obesity. Diabetes 2014, 63, 2464–2473. [Google Scholar] [CrossRef]
- Buckley, D.; Duke, G.; Heuer, T.S.; O’Farrell, M.; Wagman, A.S.; McCulloch, W.; Kemble, G. Fatty acid synthase—Modern tumor cell biology insights into a classical oncology target. Pharmacol. Ther. 2017, 177, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.F.; Infante, J.R. Molecular Pathways: Fatty Acid Synthase. Clin. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Pizer, E.S.; Wood, F.D.; Heine, H.S.; Romantsev, F.E.; Pasternack, G.R.; Kuhajda, F.P. Inhibition of fatty acid synthesis delays disease progression in a xenograft model of ovarian cancer. Cancer Res. 1996, 56, 1189–1193. [Google Scholar]
- Pizer, E.S.; Jackisch, C.; Wood, F.D.; Pasternack, G.R.; Davidson, N.E.; Kuhajda, F.P. Inhibition of fatty acid synthesis induces programmed cell death in human breast cancer cells. Cancer Res. 1996, 56, 2745–2747. [Google Scholar]
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004, 64, 2070–2075. [Google Scholar] [CrossRef]
- Carvalho, M.A.; Zecchin, K.G.; Seguin, F.; Bastos, D.C.; Agostini, M.; Rangel, A.L.; Veiga, S.S.; Raposo, H.F.; Oliveira, H.C.; Loda, M.; et al. Fatty acid synthase inhibition with Orlistat promotes apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. Int. J. Cancer 2008, 123, 2557–2565. [Google Scholar] [CrossRef]
- Relat, J.; Blancafort, A.; Oliveras, G.; Cufi, S.; Haro, D.; Marrero, P.F.; Puig, T. Different fatty acid metabolism effects of (-)-epigallocatechin-3-gallate and C75 in adenocarcinoma lung cancer. BMC Cancer 2012, 12, 280. [Google Scholar] [CrossRef]
- Gabrielson, E.W.; Pinn, M.L.; Testa, J.R.; Kuhajda, F.P. Increased fatty acid synthase is a therapeutic target in mesothelioma. Clin. Cancer Res. 2001, 7, 153–157. [Google Scholar]
- Zhou, W.; Han, W.F.; Landree, L.E.; Thupari, J.N.; Pinn, M.L.; Bililign, T.; Kim, E.K.; Vadlamudi, A.; Medghalchi, S.M.; El Meskini, R.; et al. Fatty acid synthase inhibition activates AMP-activated protein kinase in SKOV3 human ovarian cancer cells. Cancer Res. 2007, 67, 2964–2971. [Google Scholar] [CrossRef]
- Orita, H.; Coulter, J.; Lemmon, C.; Tully, E.; Vadlamudi, A.; Medghalchi, S.M.; Kuhajda, F.P.; Gabrielson, E. Selective inhibition of fatty acid synthase for lung cancer treatment. Clin. Cancer Res. 2007, 13, 7139–7145. [Google Scholar] [CrossRef] [PubMed]
- Hardwicke, M.A.; Rendina, A.R.; Williams, S.P.; Moore, M.L.; Wang, L.; Krueger, J.A.; Plant, R.N.; Totoritis, R.D.; Zhang, G.; Briand, J.; et al. A human fatty acid synthase inhibitor binds beta-ketoacyl reductase in the keto-substrate site. Nat. Chem. Biol. 2014, 10, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of de novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Fako, V.E.; Wu, X.; Pflug, B.; Liu, J.Y.; Zhang, J.T. Repositioning proton pump inhibitors as anticancer drugs by targeting the thioesterase domain of human fatty acid synthase. J. Med. Chem. 2015, 58, 778–784. [Google Scholar] [CrossRef]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- University of Texas Southwestern Medical Center. Phase 2 Study of TVB-2640 in KRAS Non-Small Cell Lung Carcinomas. 2019. Available online: https://ClinicalTrials.gov/show/NCT03808558 (accessed on 10 August 2020).
- Mayo Clinic; National Cancer Institute. FASN Inhibitor TVB-2640, Paclitaxel, and Trastuzumab in Treating Patients With HER2 Positive Advanced Breast Cancer. 2017. Available online: https://ClinicalTrials.gov/show/NCT03179904 (accessed on 10 August 2020).
- The University of Texas Health Science Center at San Antonio. TVB- 2640 in Combination With Bevacizumab in Patients with First Relapse of High Grade Astrocytoma. 2017. Available online: https://ClinicalTrials.gov/show/NCT03032484: (accessed on 10 August 2020).
- Evers, M.; University of Kentucky. TVB 2640 for Resectable Colon Cancer Other Resectable Cancers; a Window Trial. 2017. Available online: https://ClinicalTrials.gov/show/NCT02980029 (accessed on 10 August 2020).
- Sagimet Biosciences Inc. A Phase 1, First-In-Human Study of Escalating Doses of Oral TVB-2640 in Patients With Solid Tumors. 2013. Available online: https://ClinicalTrials.gov/show/NCT02223247 (accessed on 10 August 2020).
- Miller, K.; Indiana University. Inhibiting Fatty Acid Synthase to Improve Efficacy of Neoadjuvant Chemotherapy. 2015. Available online: https://ClinicalTrials.gov/show/NCT02595372 (accessed on 10 August 2020).
- Wake Forest University Health Sciences; National Cancer Institute. Fatty Acid Synthase Inhibition in Castration Refractory Prostate Cancer. 2020. Available online: https://ClinicalTrials.gov/show/NCT04337580 (accessed on 10 August 2020).
- Cancer Advances Inc. Effect of G17DT in Patients with Stage II/III Colorectal Cancer. 2000. Available online: https://ClinicalTrials.gov/show/NCT02518373 (accessed on 10 August 2020).
- Therapeutics, E. Evaluation of the Efficacy and Safety of Bempedoic Acid (ETC-1002) as Add-On to Ezetimibe Therapy in Patients with Elevated LDL-C (CLEAR Tranquility). 2016. Available online: https://ClinicalTrials.gov/show/NCT03001076 (accessed on 10 August 2020).
- Therapeutics, E.; Medpace, I. Evaluation of ETC-1002, Ezetimibe, and the Combination in Hypercholesterolemic Patients. 2013. Available online: https://ClinicalTrials.gov/show/NCT01941836 (accessed on 10 August 2020).
- Therapeutics, E. Evaluation of Long-Term Efficacy of Bempedoic Acid (ETC-1002) in Patients With Hyperlipidemia at High Cardiovascular Risk. 2016. Available online: https://ClinicalTrials.gov/show/NCT02991118 (accessed on 10 August 2020).
- Therapeutics, E. A Single Center Study to Evaluate the Efficacy and Safety of ETC 1002 in Subjects with Type 2 Diabetes. 2012. Available online: https://ClinicalTrials.gov/show/NCT01607294 (accessed on 10 August 2020).
- Luo, J.; Hong, Y.; Lu, Y.; Qiu, S.; Chaganty, B.K.; Zhang, L.; Wang, X.; Li, Q.; Fan, Z. Acetyl-CoA carboxylase rewires cancer metabolism to allow cancer cells to survive inhibition of the Warburg effect by cetuximab. Cancer Lett. 2017, 384, 39–49. [Google Scholar] [CrossRef]
- Jones, J.E.; Esler, W.P.; Patel, R.; Lanba, A.; Vera, N.B.; Pfefferkorn, J.A.; Vernochet, C. Inhibition of Acetyl-CoA Carboxylase 1 (ACC1) and 2 (ACC2) Reduces Proliferation and De Novo Lipogenesis of EGFRvIII Human Glioblastoma Cells. PLoS ONE 2017, 12, e0169566. [Google Scholar] [CrossRef]
- Ma, M.K.F.; Lau, E.Y.T.; Leung, D.H.W.; Lo, J.; Ho, N.P.Y.; Cheng, L.K.W.; Ma, S.; Lin, C.H.; Copland, J.A.; Ding, J.; et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J. Hepatol. 2017, 67, 979–990. [Google Scholar] [CrossRef]
- Pisanu, M.E.; Noto, A.; De Vitis, C.; Morrone, S.; Scognamiglio, G.; Botti, G.; Venuta, F.; Diso, D.; Jakopin, Z.; Padula, F.; et al. Blockade of Stearoyl-CoA-desaturase 1 activity reverts resistance to cisplatin in lung cancer stem cells. Cancer Lett. 2017, 406, 93–104. [Google Scholar] [CrossRef]
- Noto, A.; Raffa, S.; De Vitis, C.; Roscilli, G.; Malpicci, D.; Coluccia, P.; Di Napoli, A.; Ricci, A.; Giovagnoli, M.R.; Aurisicchio, L.; et al. Stearoyl-CoA desaturase-1 is a key factor for lung cancer-initiating cells. Cell Death Dis. 2013, 4, e947. [Google Scholar] [CrossRef]
- Potze, L.; di Franco, S.; Kessler, J.H.; Stassi, G.; Medema, J.P. Betulinic Acid Kills Colon Cancer Stem Cells. Curr. Stem Cell Res. Ther. 2016, 11, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Osmak, M. Statins and cancer: Current and future prospects. Cancer Lett. 2012, 324, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nayan, M.; Punjani, N.; Juurlink, D.N.; Finelli, A.; Austin, P.C.; Kulkarni, G.S.; Uleryk, E.; Hamilton, R.J. Statin use and kidney cancer survival outcomes: A systematic review and meta-analysis. Cancer Treat. Rev. 2017, 52, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Nayan, M.; Finelli, A.; Jewett, M.A.S.; Juurlink, D.N.; Austin, P.C.; Kulkarni, G.S.; Hamilton, R.J. Statin use and kidney cancer outcomes: A propensity score analysis. Urol. Oncol. 2016, 34, 487 e481–487 e486. [Google Scholar] [CrossRef]
- Pandyra, A.A.; Mullen, P.J.; Goard, C.A.; Ericson, E.; Sharma, P.; Kalkat, M.; Yu, R.; Pong, J.T.; Brown, K.R.; Hart, T.; et al. Genome-wide RNAi analysis reveals that simultaneous inhibition of specific mevalonate pathway genes potentiates tumor cell death. Oncotarget 2015, 6, 26909–26921. [Google Scholar] [CrossRef] [PubMed]
- Pandyra, A.; Penn, L.Z. Targeting tumor cell metabolism via the mevalonate pathway: Two hits are better than one. Mol. Cell. Oncol. 2014, 1, e969133. [Google Scholar] [CrossRef] [PubMed]
- Pandyra, A.; Mullen, P.J.; Kalkat, M.; Yu, R.; Pong, J.T.; Li, Z.; Trudel, S.; Lang, K.S.; Minden, M.D.; Schimmer, A.D.; et al. Immediate utility of two approved agents to target both the metabolic mevalonate pathway and its restorative feedback loop. Cancer Res. 2014, 74, 4772–4782. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Emerging roles of lipid metabolism in cancer progression. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 254–260. [Google Scholar] [CrossRef]
- Galicia-Vazquez, G.; Aloyz, R. Ibrutinib Resistance Is Reduced by an Inhibitor of Fatty Acid Oxidation in Primary CLL Lymphocytes. Front. Oncol. 2018, 8, 411. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.M.D.; Adamoski, D.; Ornitz Oliveira Souza, R.; Rodrigues Ascencao, C.F.; Sousa de Oliveira, K.R.; Correa-da-Silva, F.; Malta de Sa Patroni, F.; Meira Dias, M.; Consonni, S.R.; Mendes de Moraes-Vieira, P.M.; et al. Dual inhibition of glutaminase and carnitine palmitoyltransferase decreases growth and migration of glutaminase inhibition-resistant triple-negative breast cancer cells. J. Biol. Chem. 2019, 294, 9342–9357. [Google Scholar] [CrossRef] [PubMed]
- Tirado-Velez, J.M.; Joumady, I.; Saez-Benito, A.; Cozar-Castellano, I.; Perdomo, G. Inhibition of fatty acid metabolism reduces human myeloma cells proliferation. PLoS ONE 2012, 7, e46484. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijon, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glode, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef]
- Wang, Y.N.; Zeng, Z.L.; Lu, J.; Wang, Y.; Liu, Z.X.; He, M.M.; Zhao, Q.; Wang, Z.X.; Li, T.; Lu, Y.X.; et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 2018, 37, 6025–6040. [Google Scholar] [CrossRef]
- Sawyer, B.T.; Qamar, L.; Yamamoto, T.M.; McMellen, A.; Watson, Z.L.; Richer, J.K.; Behbakht, K.; Schlaepfer, I.R.; Bitler, B.G. Targeting fatty acid oxidation to promote anoikis and inhibit ovarian cancer progression. Mol. Cancer Res. 2020. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Stack, M.S. Lipid Regulatory Proteins as Potential Therapeutic Targets for Ovarian Cancer in Obese Women. Cancers 2020, 12, 3469. https://doi.org/10.3390/cancers12113469
Yang J, Stack MS. Lipid Regulatory Proteins as Potential Therapeutic Targets for Ovarian Cancer in Obese Women. Cancers. 2020; 12(11):3469. https://doi.org/10.3390/cancers12113469
Chicago/Turabian StyleYang, Jing, and M. Sharon Stack. 2020. "Lipid Regulatory Proteins as Potential Therapeutic Targets for Ovarian Cancer in Obese Women" Cancers 12, no. 11: 3469. https://doi.org/10.3390/cancers12113469
APA StyleYang, J., & Stack, M. S. (2020). Lipid Regulatory Proteins as Potential Therapeutic Targets for Ovarian Cancer in Obese Women. Cancers, 12(11), 3469. https://doi.org/10.3390/cancers12113469