Cytoskeletal Control and Wnt Signaling—APC’s Dual Contributions in Stem Cell Division and Colorectal Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. The Tumour Suppressor Apc, a Master Regulator of Gut Homeostasis
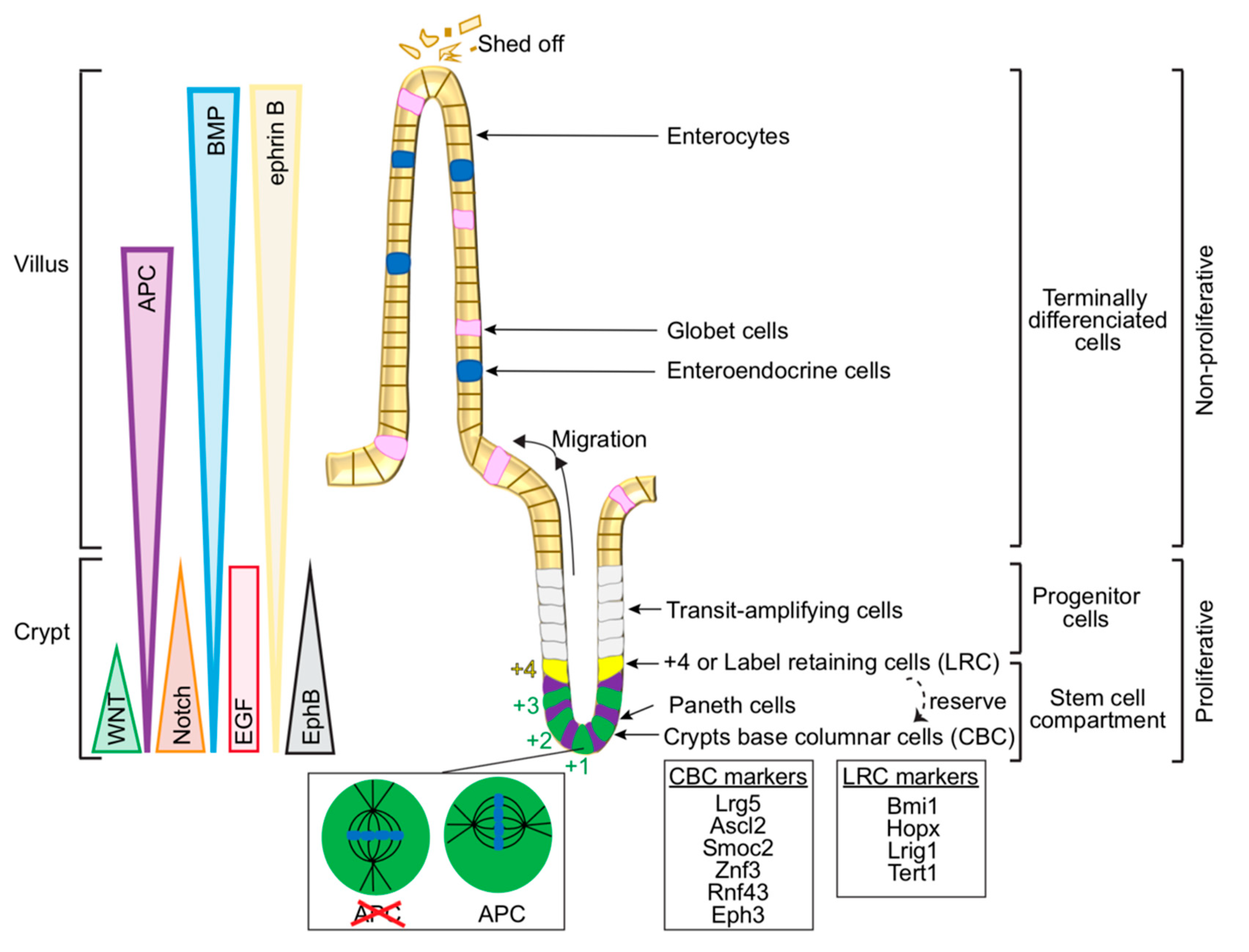
2.1. Cellular Organisation in the Gut—From Intestinal Stem Cells to Differentiated Cells
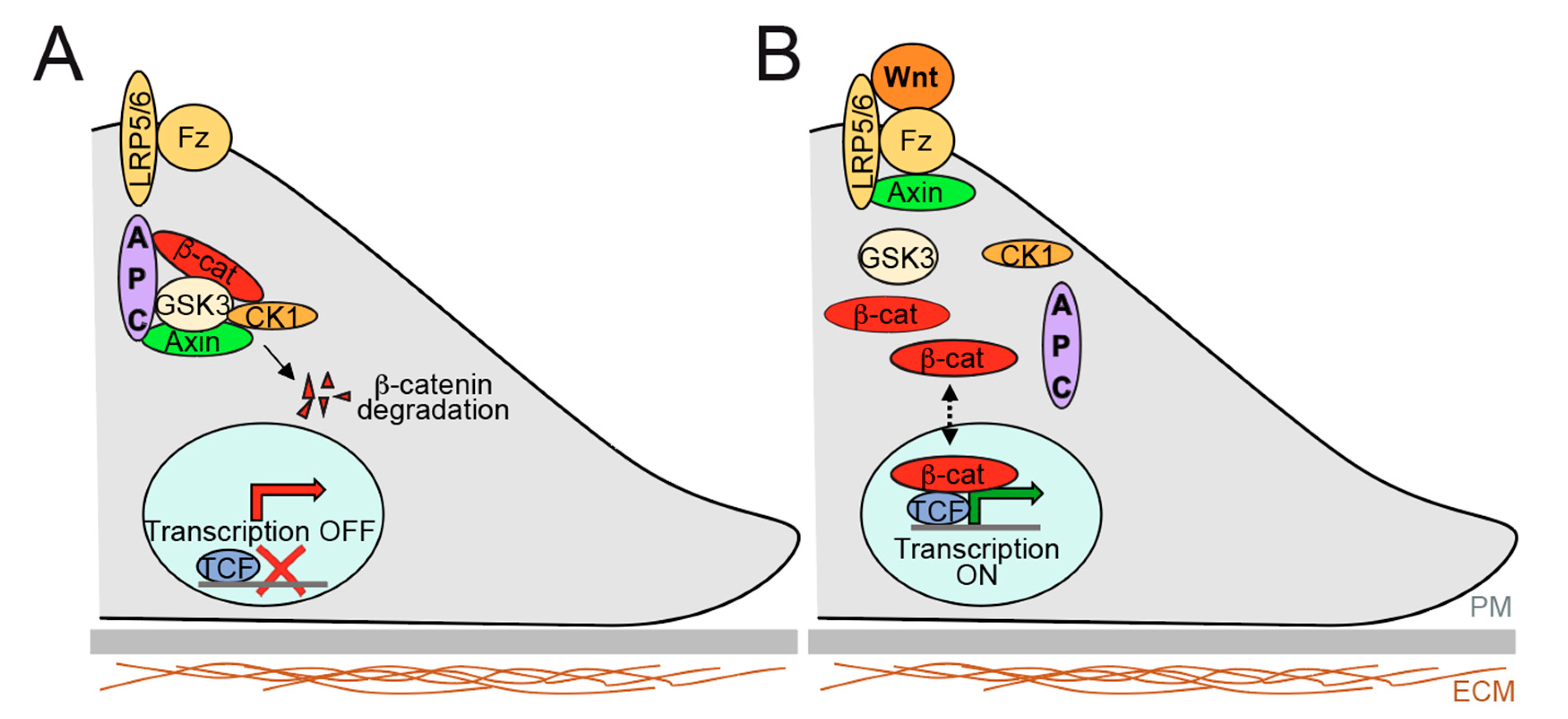
2.2. Overview of Signalling Pathways Controlling Gut Epithelium Homeostasis and Relevance to Therapeutics
2.3. Modes of Intestinal Stem Cell Division and Homeostasis
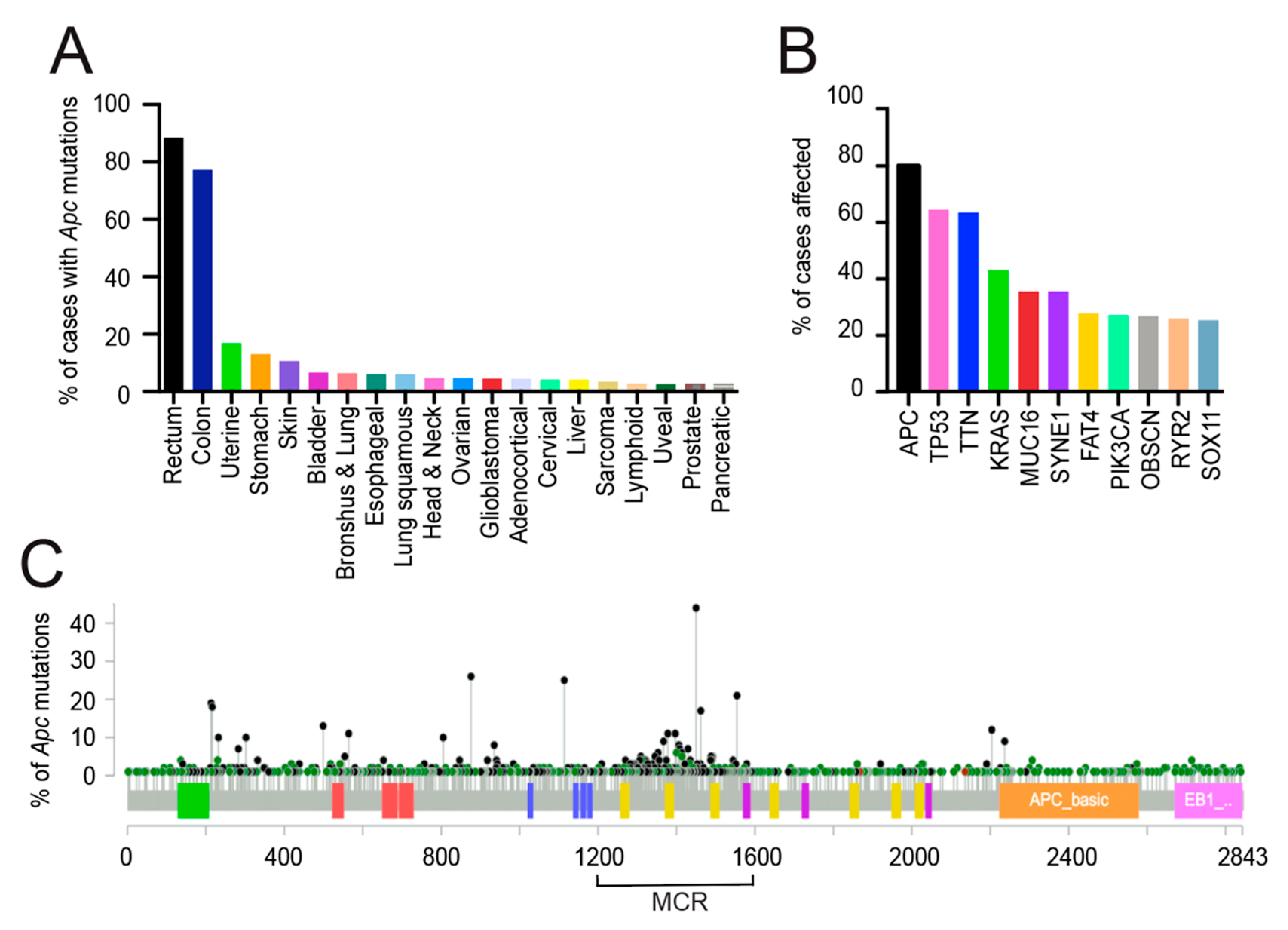
2.4. Apc as the Gatekeeper Gene in Colorectal Cancer
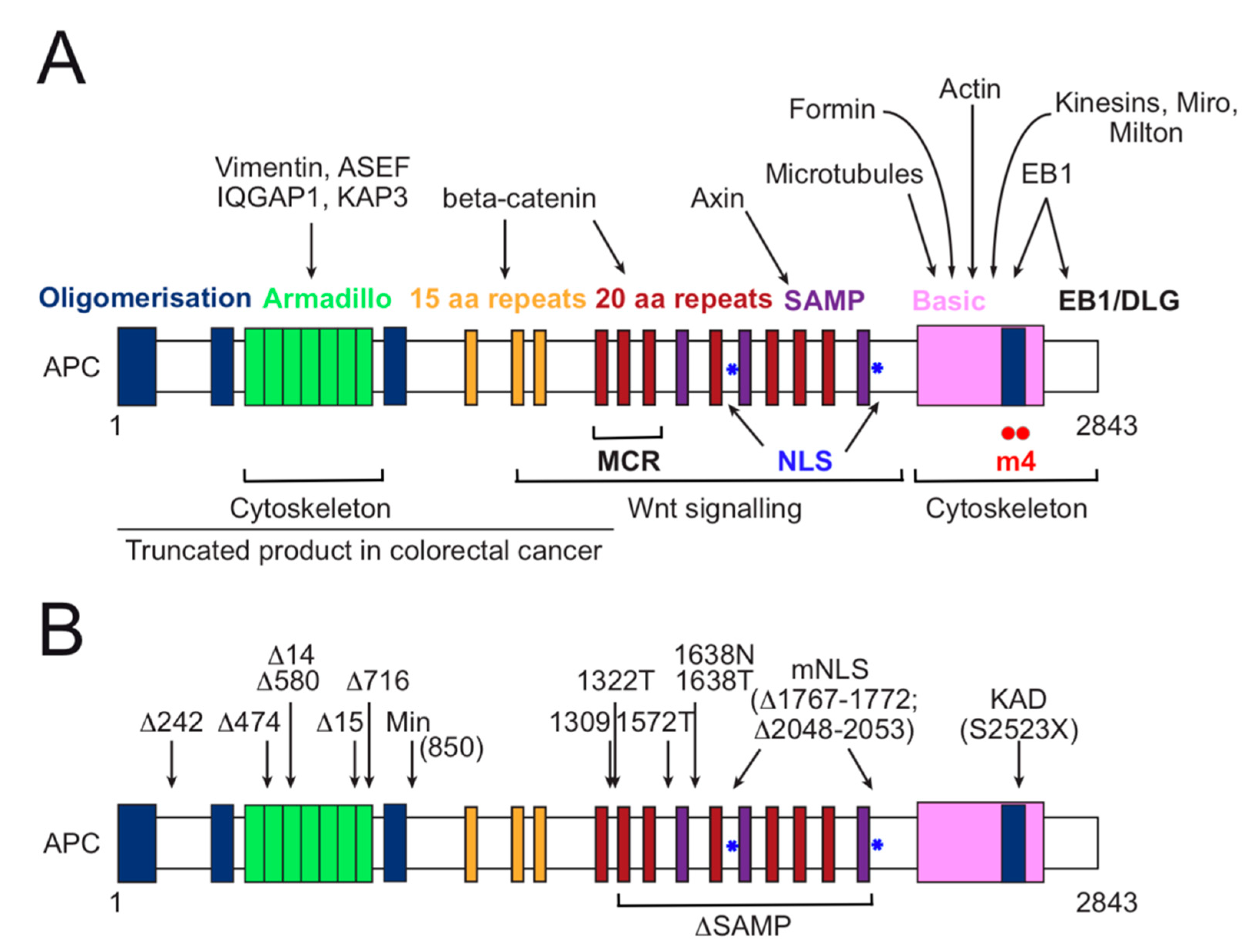
3. APC Functional Domain Analysis and Animal Models for Investigation of Colorectal Cancer
4. Control of Cell Migration beyond Wnt Signalling—APC as a Cytoskeletal Hub
4.1. Cell Migration in the Gut
4.2. APC, Cytoskeletal Dynamics and Cell Migration
5. APC Roles in Spindle Morphogenesis and Dynamics
5.1. APC at the Kinetochore-Microtubule Interface and Chromosomal Instability
5.2. Cortical APC in Spindle Orientation and Cell Migration
6. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Neumüller, R.A.; Knoblich, J.A. Dividing cellular asymmetry: Asymmetric cell division and its implications for stem cells and cancer. Genes Dev. 2009, 23, 2675–2699. [Google Scholar] [CrossRef] [PubMed]
- Knoblich, J.A. Mechanisms of asymmetric stem cell division. Cell 2008, 132, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Venkei, Z.G.; Yamashita, Y.M. Emerging mechanisms of asymmetric stem cell division. J. Cell Biol. 2018, 217, 3785–3795. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.D.; Goldstein, B. Asymmetric cell division: A new way to divide unequally. Curr. Biol. 2010, 20, R1029–R1031. [Google Scholar] [CrossRef]
- Gómez-López, S.; Lerner, R.G.; Petritsch, C. Asymmetric cell division of stem and progenitor cells during homeostasis and cancer. Cell Mol. Life Sci. 2014, 71, 575–597. [Google Scholar] [CrossRef]
- Fuchs, E.; Chen, T. A matter of life and death: Self-renewal in stem cells. EMBO Rep. 2013, 14, 39–48. [Google Scholar] [CrossRef]
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074. [Google Scholar] [CrossRef]
- Rappaport, R. Cell Division: Direct Measurement of Maximum Tension Exerted by Furrow of Echinoderm Eggs. Science 1967, 156, 1241–1243. [Google Scholar] [CrossRef]
- Campbell, E.K.M.; Werts, A.D.; Goldstein, B. A cell cycle timer for asymmetric spindle positioning. PLoS Biol. 2009, 7, e1000088. [Google Scholar]
- Siller, K.H.; Doe, C.Q. Spindle orientation during asymmetric cell division. Nat. Cell Biol. 2009, 11, 365–374. [Google Scholar] [CrossRef]
- Morin, X.; Bellaïche, Y. Mitotic spindle orientation in asymmetric and symmetric cell divisions during animal development. Dev. Cell 2011, 21, 102–119. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.S.; Johnston, C.A. Molecular pathways regulating mitotic spindle orientation in animal cells. Development 2013, 140, 1843–1856. [Google Scholar] [CrossRef] [PubMed]
- Prosser, S.L.; Pelletier, L. Mitotic spindle assembly in animal cells: A fine balancing act. Nat. Rev. Mol. Cell Biol. 2017, 18, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Elting, M.W.; Suresh, P.; Dumont, S. The Spindle: Integrating Architecture and Mechanics across Scales. Trends Cell Biol. 2018, 28, 896–910. [Google Scholar] [CrossRef]
- Tolić, I.M. Mitotic spindle: Kinetochore fibers hold on tight to interpolar bundles. Eur. Biophys J. 2018, 47, 191–203. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Medema, R.H. Mechanisms of centrosome separation and bipolar spindle assembly. Dev. Cell 2010, 19, 797–806. [Google Scholar] [CrossRef]
- Cavazza, T.; Vernos, I. The RanGTP Pathway: From Nucleo-Cytoplasmic Transport to Spindle Assembly and Beyond. Front. Cell Dev. Biol. 2015, 3, 82. [Google Scholar] [CrossRef]
- Akhmanova, A.; Steinmetz, M.O. Control of microtubule organization and dynamics: Two ends in the limelight. Nat. Rev. Mol. Cell Biol. 2015, 16, 711–726. [Google Scholar] [CrossRef]
- Obino, D.; Farina, F.; Malbec, O.; Sáez, P.J.; Maurin, M.; Gaillard, J.; Dingli, F.; Loew, D.; Gautreau, A.; Yuseff, M.-I.; et al. Actin nucleation at the centrosome controls lymphocyte polarity. Nat. Commun. 2016, 7, 10969. [Google Scholar] [CrossRef]
- Inoue, D.; Obino, D.; Pineau, J.; Farina, F.; Gaillard, J.; Guerin, C.; Blanchoin, L.; Lennon-Duménil, A.-M.; Théry, M. Actin filaments regulate microtubule growth at the centrosome. EMBO J. 2019, 38, 38. [Google Scholar] [CrossRef]
- Farina, F.; Ramkumar, N.; Brown, L.; Eweis, D.S.; Anstatt, J.; Waring, T.; Bithell, J.; Scita, G.; Thery, M.; Blanchoin, L.; et al. Local actin nucleation tunes centrosomal microtubule nucleation during passage through mitosis. EMBO J. 2019, 38, e99843. [Google Scholar] [CrossRef] [PubMed]
- Farina, F.; Gaillard, J.; Guérin, C.; Couté, Y.; Sillibourne, J.; Blanchoin, L.; Théry, M. The centrosome is an actin-organizing centre. Nat. Cell Biol. 2016, 18, 65–75. [Google Scholar] [CrossRef]
- Padgett, J.; Santos, S.D.M. From clocks to dominoes: Lessons on cell cycle remodelling from embryonic stem cells. FEBS Lett. 2020, 594, 2031–2045. [Google Scholar] [CrossRef] [PubMed]
- Kimata, Y.; Leturcq, M.; Aradhya, R. Emerging roles of metazoan cell cycle regulators as coordinators of the cell cycle and differentiation. FEBS Lett. 2020, 594, 2061–2083. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.; Yamashita, Y.M. Fly meets yeast: Checking the correct orientation of cell division. Trends Cell Biol. 2011, 21, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Li, R. The art of choreographing asymmetric cell division. Dev. Cell 2013, 25, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Geymonat, M.; Segal, M. Intrinsic and Extrinsic Determinants Linking Spindle Pole Fate, Spindle Polarity, and Asymmetric Cell Division in the Budding Yeast S. cerevisiae. Results Probl. Cell Differ. 2017, 61, 49–82. [Google Scholar]
- Näthke, I.S. The adenomatous polyposis coli protein: The Achilles heel of the gut epithelium. Annu. Rev. Cell Dev. Biol. 2004, 20, 337–366. [Google Scholar] [CrossRef]
- Schneikert, J.; Behrens, J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut 2007, 56, 417–425. [Google Scholar] [CrossRef]
- Parker, T.W.; Neufeld, K.L. APC controls Wnt-induced β-catenin destruction complex recruitment in human colonocytes. Sci. Rep. 2020, 10, 2957. [Google Scholar] [CrossRef]
- Munemitsu, S.; Albert, I.; Souza, B.; Rubinfeld, B.; Polakis, P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc. Natl. Acad. Sci. USA 1995, 92, 3046–3050. [Google Scholar] [CrossRef]
- Rubinfeld, B.; Souza, B.; Albert, I.; Müller, O.; Chamberlain, S.H.; Masiarz, F.R.; Munemitsu, S.; Polakis, P. Association of the APC gene product with beta-catenin. Science 1993, 262, 1731–1734. [Google Scholar] [CrossRef] [PubMed]
- Pronobis, M.I.; Rusan, N.M.; Peifer, M. A novel GSK3-regulated APC: Axin interaction regulates Wnt signaling by driving a catalytic cycle of efficient βcatenin destruction. Elife 2015, 4, e08022. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.R.; Fagotto, F. The ins and outs of APC and beta-catenin nuclear transport. EMBO Rep. 2002, 3, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, K.L.; Zhang, F.; Cullen, B.R.; White, R.L. APC-mediated downregulation of beta-catenin activity involves nuclear sequestration and nuclear export. EMBO Rep. 2000, 1, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Janssen, K.-P.; Alberici, P.; Fsihi, H.; Gaspar, C.; Breukel, C.; Franken, P.; Rosty, C.; Abal, M.; El Marjou, F.; Smits, R.; et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology 2006, 131, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Fumoto, K.; Okamoto, T.; Kaibuchi, K.; Kikuchi, A. Binding of APC and dishevelled mediates Wnt5a-regulated focal adhesion dynamics in migrating cells. EMBO J. 2010, 29, 1192–1204. [Google Scholar] [CrossRef]
- Nelson, S.; Näthke, I.S. Interactions and functions of the adenomatous polyposis coli (APC) protein at a glance. J. Cell Sci. 2013, 126, 873–877. [Google Scholar] [CrossRef]
- Fodde, R. The multiple functions of tumour suppressors: It’s all in APC. Nat. Cell Biol. 2003, 5, 190–192. [Google Scholar] [CrossRef]
- Mogensen, M.M.; Tucker, J.B.; Mackie, J.B.; Prescott, A.R.; Näthke, I.S. The adenomatous polyposis coli protein unambiguously localizes to microtubule plus ends and is involved in establishing parallel arrays of microtubule bundles in highly polarized epithelial cells. J. Cell Biol. 2002, 157, 1041–1048. [Google Scholar] [CrossRef]
- Watanabe, T.; Wang, S.; Noritake, J.; Sato, K.; Fukata, M.; Takefuji, M.; Nakagawa, M.; Izumi, N.; Akiyama, T.; Kaibuchi, K. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev. Cell 2004, 7, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Mimori-Kiyosue, Y.; Shiina, N.; Tsukita, S. Adenomatous polyposis coli (APC) protein moves along microtubules and concentrates at their growing ends in epithelial cells. J. Cell Biol. 2000, 148, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Green, R.A.; Wollman, R.; Kaplan, K.B. APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol. Biol. Cell 2005, 16, 4609–4622. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, C.M.; Green, R.A.; Kaplan, K.B. APC mutations lead to cytokinetic failures in vitro and tetraploid genotypes in Min mice. J. Cell Biol. 2007, 178, 1109–1120. [Google Scholar] [CrossRef]
- Green, R.A.; Kaplan, K.B. Chromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by a dominant mutation in APC. J. Cell Biol. 2003, 163, 949–961. [Google Scholar] [CrossRef]
- Fodde, R.; Kuipers, J.; Rosenberg, C.; Smits, R.; Kielman, M.; Gaspar, C.; van Es, J.H.; Breukel, C.; Wiegant, J.; Giles, R.H.; et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat. Cell Biol. 2001, 3, 433–438. [Google Scholar] [CrossRef]
- Barth, A.I.M.; Caro-Gonzalez, H.Y.; Nelson, W.J. Role of adenomatous polyposis coli (APC) and microtubules in directional cell migration and neuronal polarization. Semin. Cell Dev. Biol. 2008, 19, 245–251. [Google Scholar] [CrossRef]
- Munemitsu, S.; Souza, B.; Müller, O.; Albert, I.; Rubinfeld, B.; Polakis, P. The APC gene product associates with microtubules in vivo and promotes their assembly in vitro. Cancer Res. 1994, 54, 3676–3681. [Google Scholar]
- Näthke, I. Relationship between the role of the adenomatous polyposis coli protein in colon cancer and its contribution to cytoskeletal regulation. Biochem. Soc. Trans. 2005, 33, 694–697. [Google Scholar] [CrossRef]
- Kroboth, K.; Newton, I.P.; Kita, K.; Dikovskaya, D.; Zumbrunn, J.; Waterman-Storer, C.M.; Näthke, I.S. Lack of adenomatous polyposis coli protein correlates with a decrease in cell migration and overall changes in microtubule stability. Mol. Biol. Cell 2007, 18, 910–918. [Google Scholar] [CrossRef]
- Okada, K.; Bartolini, F.; Deaconescu, A.M.; Moseley, J.B.; Dogic, Z.; Grigorieff, N.; Gundersen, G.G.; Goode, B.L. Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. J. Cell Biol. 2010, 189, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Moseley, J.B.; Bartolini, F.; Okada, K.; Wen, Y.; Gundersen, G.G.; Goode, B.L. Regulated binding of adenomatous polyposis coli protein to actin. J. Biol. Chem. 2007, 282, 12661–12668. [Google Scholar] [CrossRef] [PubMed]
- Juanes, M.A.; Bouguenina, H.; Eskin, J.A.; Jaiswal, R.; Badache, A.; Goode, B.L. Adenomatous polyposis coli nucleates actin assembly to drive cell migration and microtubule-induced focal adhesion turnover. J. Cell Biol. 2017, 216, 2859–2875. [Google Scholar] [CrossRef] [PubMed]
- Juanes, M.A.; Fees, C.; Hoeprich, G.J.; Jaiswal, R.; Goode, B.L. EB1 directly regulatesAPC-mediated actin nucleation. Curr. Biol. 2020, 30, 4763–4772.e8. [Google Scholar] [CrossRef]
- Breitsprecher, D.; Jaiswal, R.; Bombardier, J.P.; Gould, C.J.; Gelles, J.; Goode, B.L. Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science 2012, 336, 1164–1168. [Google Scholar] [CrossRef]
- Juanes, M.A.; Isnardon, D.; Badache, A.; Brasselet, S.; Mavrakis, M.; Goode, B.L. The role of APC-mediated actin assembly in microtubule capture and focal adhesion turnover. J. Cell Biol. 2019, 218, 3415–3435. [Google Scholar] [CrossRef] [PubMed]
- Su, L.K.; Burrell, M.; Hill, D.E.; Gyuris, J.; Brent, R.; Wiltshire, R.; Trent, J.; Vogelstein, B.; Kinzler, K.W. APC binds to the novel protein EB1. Cancer Res. 1995, 55, 2972–2977. [Google Scholar]
- Sansom, O.J.; Reed, K.R.; Hayes, A.J.; Ireland, H.; Brinkmann, H.; Newton, I.P.; Batlle, E.; Simon-Assmann, P.; Clevers, H.; Nathke, I.S.; et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004, 18, 1385–1390. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Caldwell, C.M.; Kaplan, K.B. The role of APC in mitosis and in chromosome instability. Adv. Exp. Med. Biol. 2009, 656, 51–64. [Google Scholar]
- Draviam, V.M.; Shapiro, I.; Aldridge, B.; Sorger, P.K. Misorientation and reduced stretching of aligned sister kinetochores promote chromosome missegregation in EB1- or APC-depleted cells. EMBO J. 2006, 25, 2814–2827. [Google Scholar] [CrossRef] [PubMed]
- Quyn, A.J.; Appleton, P.L.; Carey, F.A.; Steele, R.J.C.; Barker, N.; Clevers, H.; Ridgway, R.A.; Sansom, O.J.; Näthke, I.S. Spindle orientation bias in gut epithelial stem cell compartments is lost in precancerous tissue. Cell Stem Cell 2010, 6, 175–181. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, N.; Kraiczy, J.; Shivdasani, R.A. Cellular and molecular architecture of the intestinal stem cell niche. Nat. Cell Biol. 2020, 22, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The intestinal crypt, a prototype stem cell compartment. Cell 2013, 154, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Seishima, R.; Barker, N. A contemporary snapshot of intestinal stem cells and their regulation. Differentiation 2019, 108, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Habowski, A.N.; Flesher, J.L.; Bates, J.M.; Tsai, C.-F.; Martin, K.; Zhao, R.; Ganesan, A.K.; Edwards, R.A.; Shi, T.; Wiley, H.S.; et al. Transcriptomic and proteomic signatures of stemness and differentiation in the colon crypt. Commun. Biol. 2020, 3, 453. [Google Scholar] [CrossRef]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef]
- Tan, D.W.-M.; Barker, N. Intestinal stem cells and their defining niche. Curr. Top. Dev. Biol. 2014, 107, 77–107. [Google Scholar]
- Gjorevski, N.; Ordóñez-Morán, P. Intestinal Stem Cell Niche Insights Gathered from Both In Vivo and Novel In Vitro Models. Stem Cells Int. 2017, 2017, 8387297. [Google Scholar] [CrossRef]
- Karmakar, S.; Deng, L.; He, X.C.; Li, L. Intestinal epithelial regeneration: Active versus reserve stem cells and plasticity mechanisms. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G796–G802. [Google Scholar] [CrossRef]
- Yan, K.S.; Chia, L.A.; Li, X.; Ootani, A.; Su, J.; Lee, J.Y.; Su, N.; Luo, Y.; Heilshorn, S.C.; Amieva, M.R.; et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc. Natl. Acad. Sci. USA 2012, 109, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Leblond, C.P. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian Theory of the origin of the four epithelial cell types. Am J. Anat. 1974, 141, 537–561. [Google Scholar] [CrossRef]
- Barker, N.; van Oudenaarden, A.; Clevers, H. Identifying the stem cell of the intestinal crypt: Strategies and pitfalls. Cell Stem Cell 2012, 11, 452–460. [Google Scholar] [CrossRef]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 2008, 40, 915–920. [Google Scholar] [CrossRef]
- Takeda, N.; Jain, R.; LeBoeuf, M.R.; Wang, Q.; Lu, M.M.; Epstein, J.A. Interconversion between intestinal stem cell populations in distinct niches. Science 2011, 334, 1420–1424. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.; Stange, D.E.; Schepers, A.G.; van de Wetering, M.; Koo, B.-K.; Itzkovitz, S.; Volckmann, R.; Kung, K.S.; Koster, J.; Radulescu, S.; et al. The Lgr5 intestinal stem cell signature: Robust expression of proposed quiescent “+4” cell markers. EMBO J. 2012, 31, 3079–3091. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Clevers, H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010, 327, 542–545. [Google Scholar] [CrossRef]
- Mei, X.; Gu, M.; Li, M. Plasticity of Paneth cells and their ability to regulate intestinal stem cells. Stem Cell Res. Ther. 2020, 11, 349. [Google Scholar] [CrossRef]
- Stappenbeck, T.S. Paneth cell development, differentiation, and function: New molecular cues. Gastroenterology 2009, 137, 30–33. [Google Scholar] [CrossRef]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef]
- Andreu, P.; Colnot, S.; Godard, C.; Gad, S.; Chafey, P.; Niwa-Kawakita, M.; Laurent-Puig, P.; Kahn, A.; Robine, S.; Perret, C.; et al. Crypt-restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development 2005, 132, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Ritsma, L.; Ellenbroek, S.I.J.; Zomer, A.; Snippert, H.J.; de Sauvage, F.J.; Simons, B.D.; Clevers, H.; van Rheenen, J. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature 2014, 507, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Scoville, D.H.; Sato, T.; He, X.C.; Li, L. Current view: Intestinal stem cells and signaling. Gastroenterology 2008, 134, 849–864. [Google Scholar] [CrossRef]
- Beumer, J.; Clevers, H. Cell fate specification and differentiation in the adult mammalian intestine. Nat. Rev. Mol. Cell Biol. 2020, 1–15. [Google Scholar] [CrossRef]
- Takahashi, T.; Shiraishi, A. Stem cell signaling pathways in the small intestine. Int. J. Mol. Sci. 2020, 21, 2032. [Google Scholar] [CrossRef]
- Jansen, M. Marching out of the crypt. Science 2019, 365, 642–643. [Google Scholar] [CrossRef]
- Krndija, D.; El Marjou, F.; Guirao, B.; Richon, S.; Leroy, O.; Bellaiche, Y.; Hannezo, E.; Matic Vignjevic, D. Active cell migration is critical for steady-state epithelial turnover in the gut. Science 2019, 365, 705–710. [Google Scholar] [CrossRef]
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef]
- Spit, M.; Koo, B.-K.; Maurice, M.M. Tales from the crypt: Intestinal niche signals in tissue renewal, plasticity and cancer. Open Biol 2018, 8, 8. [Google Scholar] [CrossRef]
- Gehart, H.; Clevers, H. Tales from the crypt: New insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 19–34. [Google Scholar] [CrossRef]
- Kaemmerer, E.; Jeon, M.K.; Berndt, A.; Liedtke, C.; Gassler, N. Targeting wnt signaling via notch in intestinal carcinogenesis. Cancers (Basel) 2019, 11, 555. [Google Scholar] [CrossRef] [PubMed]
- van Es, J.H.; van Gijn, M.E.; Riccio, O.; van den Born, M.; Vooijs, M.; Begthel, H.; Cozijnsen, M.; Robine, S.; Winton, D.J.; Radtke, F.; et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005, 435, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Fre, S.; Huyghe, M.; Mourikis, P.; Robine, S.; Louvard, D.; Artavanis-Tsakonas, S. Notch signals control the fate of immature progenitor cells in the intestine. Nature 2005, 435, 964–968. [Google Scholar] [CrossRef] [PubMed]
- VanDussen, K.L.; Carulli, A.J.; Keeley, T.M.; Patel, S.R.; Puthoff, B.J.; Magness, S.T.; Tran, I.T.; Maillard, I.; Siebel, C.; Kolterud, Å.; et al. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development 2012, 139, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Bienz, M.; Clevers, H. Linking colorectal cancer to Wnt signaling. Cell 2000, 103, 311–320. [Google Scholar] [CrossRef]
- Giles, R.H.; van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 2003, 1653, 1–24. [Google Scholar] [CrossRef]
- Krausova, M.; Korinek, V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014, 26, 570–579. [Google Scholar] [CrossRef]
- Pinto, D.; Clevers, H. Wnt, stem cells and cancer in the intestine. Biol. Cell 2005, 97, 185–196. [Google Scholar] [CrossRef]
- Pinto, D.; Clevers, H. Wnt control of stem cells and differentiation in the intestinal epithelium. Exp. Cell Res. 2005, 306, 357–363. [Google Scholar] [CrossRef]
- van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.P.; et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [Google Scholar] [CrossRef]
- Pinto, D.; Gregorieff, A.; Begthel, H.; Clevers, H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003, 17, 1709–1713. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Virshup, D.M. Wnt signaling and drug resistance in cancer. Mol. Pharmacol. 2020, 97, 72–89. [Google Scholar] [CrossRef] [PubMed]
- Boman, B.M.; Fields, J.Z. An APC:WNT Counter-Current-Like Mechanism Regulates Cell Division along the Human Colonic Crypt Axis: A Mechanism that Explains How APC Mutations Induce Proliferative Abnormalities that Drive Colon Cancer Development. Front. Oncol. 2013, 3, 244. [Google Scholar] [CrossRef]
- Ireland, H.; Kemp, R.; Houghton, C.; Howard, L.; Clarke, A.R.; Sansom, O.J.; Winton, D.J. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: Effect of loss of beta-catenin. Gastroenterology 2004, 126, 1236–1246. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Moerer, P.; van Donselaar, E.; Huls, G.; Peters, P.J.; Clevers, H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet. 1998, 19, 379–383. [Google Scholar] [CrossRef]
- Nishikawa, S.-I.; Osawa, M. Generating quiescent stem cells. Pigment Cell Res. 2007, 20, 263–270. [Google Scholar] [CrossRef]
- Kim, M.J.; Huang, Y.; Park, J.-I. Targeting wnt signaling for gastrointestinal cancer therapy: Present and evolving views. Cancers (Basel) 2020, 12, 3638. [Google Scholar] [CrossRef]
- Fischer, M.M.; Cancilla, B.; Yeung, V.P.; Cattaruzza, F.; Chartier, C.; Murriel, C.L.; Cain, J.; Tam, R.; Cheng, C.-Y.; Evans, J.W.; et al. WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci. Adv. 2017, 3, e1700090. [Google Scholar] [CrossRef]
- Zhang, L.; Shay, J.W. Multiple roles of APC and its therapeutic implications in colorectal cancer. J. Natl. Cancer Inst. 2017, 109, 109. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Molecular genetics and targeted therapy of WNT-related human diseases (Review). Int. J. Mol. Med. 2017, 40, 587–606. [Google Scholar] [CrossRef] [PubMed]
- Makena, M.R.; Gatla, H.; Verlekar, D.; Sukhavasi, S.; Pandey, M.K.; Pramanik, K.C. Wnt/β-Catenin Signaling: The Culprit in Pancreatic Carcinogenesis and Therapeutic Resistance. Int. J. Mol. Sci. 2019, 20, 4242. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Dannappel, M.; Wan, C.; Firestein, R. Transcriptional Regulation of Wnt/β-Catenin Pathway in Colorectal Cancer. Cells 2020, 9, 2125. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Mashima, T.; Mizutani, A.; Sato, A.; Aoyama, A.; Gong, B.; Yoshida, H.; Muramatsu, Y.; Nakata, K.; Matsuura, M.; et al. APC mutations as a potential biomarker for sensitivity to tankyrase inhibitors in colorectal cancer. Mol. Cancer Ther. 2017, 16, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Schatoff, E.M.; Goswami, S.; Zafra, M.P.; Foronda, M.; Shusterman, M.; Leach, B.I.; Katti, A.; Diaz, B.J.; Dow, L.E. Distinct Colorectal Cancer-Associated APC Mutations Dictate Response to Tankyrase Inhibition. Cancer Discov. 2019, 9, 1358–1371. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Riyaz, S.; Srivastava, A.K.; Rahim, A. Tankyrase inhibitors as therapeutic targets for cancer. Curr. Top. Med. Chem. 2014, 14, 1967–1976. [Google Scholar] [CrossRef]
- Haider, K.; Rahaman, S.; Yar, M.S.; Kamal, A. Tubulin inhibitors as novel anticancer agents: An overview on patents (2013–2018). Expert Opin. Ther. Pat. 2019, 29, 623–641. [Google Scholar] [CrossRef]
- Astarita, E.M.; Hoover, C.A.; Maloney, S.M.; Nair, T.M.; Prosperi, J.R. Adenomatous polyposis coli loss controls cell cycle regulators and response to paclitaxel. BioRxiv 2020. [Google Scholar] [CrossRef]
- Maloney, S.M.; Hoover, C.A.; Morejon-Lasso, L.V.; Prosperi, J.R. Mechanisms of taxane resistance. Cancers (Basel) 2020, 12, 3323. [Google Scholar] [CrossRef]
- Moore, K.N.; Gunderson, C.C.; Sabbatini, P.; McMeekin, D.S.; Mantia-Smaldone, G.; Burger, R.A.; Morgan, M.A.; Kapoun, A.M.; Brachmann, R.K.; Stagg, R.; et al. A phase 1b dose escalation study of ipafricept (OMP54F28) in combination with paclitaxel and carboplatin in patients with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 2019, 154, 294–301. [Google Scholar] [CrossRef]
- Diamond, J.R.; Becerra, C.; Richards, D.; Mita, A.; Osborne, C.; O’Shaughnessy, J.; Zhang, C.; Henner, R.; Kapoun, A.M.; Xu, L.; et al. Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res. Treat. 2020, 184, 53–62. [Google Scholar] [CrossRef]
- Davis, S.L.; Cardin, D.B.; Shahda, S.; Lenz, H.-J.; Dotan, E.; O’Neil, B.H.; Kapoun, A.M.; Stagg, R.J.; Berlin, J.; Messersmith, W.A.; et al. A phase 1b dose escalation study of Wnt pathway inhibitor vantictumab in combination with nab-paclitaxel and gemcitabine in patients with previously untreated metastatic pancreatic cancer. Invest. New Drugs 2020, 38, 821–830. [Google Scholar] [CrossRef]
- He, S.; Nakada, D.; Morrison, S.J. Mechanisms of stem cell self-renewal. Annu. Rev. Cell Dev. Biol. 2009, 25, 377–406. [Google Scholar]
- Carulli, A.J.; Samuelson, L.C.; Schnell, S. Unraveling intestinal stem cell behavior with models of crypt dynamics. Integr. Biol. (Camb) 2014, 6, 243–257. [Google Scholar] [CrossRef]
- Joly, A.; Rousset, R. Tissue adaptation to environmental cues by symmetric and asymmetric division modes of intestinal stem cells. Int. J. Mol. Sci. 2020, 21, 6362. [Google Scholar] [CrossRef]
- Nakajima, Y.-I. Mitotic spindle orientation in epithelial homeostasis and plasticity. J. Biochem. 2018, 164, 277–284. [Google Scholar] [CrossRef]
- Théry, M.; Pépin, A.; Dressaire, E.; Chen, Y.; Bornens, M. Cell distribution of stress fibres in response to the geometry of the adhesive environment. Cell Motil. Cytoskelet. 2006, 63, 341–355. [Google Scholar] [CrossRef]
- Levi, B.P.; Morrison, S.J. Stem cells use distinct self-renewal programs at different ages. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 539–553. [Google Scholar] [CrossRef]
- Snippert, H.J.; van der Flier, L.G.; Sato, T.; van Es, J.H.; van den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; van Rheenen, J.; Simons, B.D.; et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef]
- Sei, Y.; Feng, J.; Chow, C.C.; Wank, S.A. Asymmetric cell division-dominant neutral drift model for normal intestinal stem cell homeostasis. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G64–G74. [Google Scholar] [CrossRef]
- Hageman, J.H.; Heinz, M.C.; Kretzschmar, K.; van der Vaart, J.; Clevers, H.; Snippert, H.J.G. Intestinal regeneration: Regulation by the microenvironment. Dev. Cell 2020, 54, 435–446. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Nilbert, M.C.; Su, L.K.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hedge, P.; McKechnie, D. Identification of FAP locus genes from chromosome 5q21. Science 1991, 253, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Kawasaki, Y. Wnt signalling and the actin cytoskeleton. Oncogene 2006, 25, 7538–7544. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.N.; Dove, W.F. APC and its modifiers in colon cancer. Adv. Exp. Med. Biol. 2009, 656, 85–106. [Google Scholar]
- Prosperi, J.R.; Khramtsov, A.I.; Khramtsova, G.F.; Goss, K.H. Apc mutation enhances PyMT-induced mammary tumorigenesis. PLoS ONE 2011, 6, e29339. [Google Scholar] [CrossRef]
- Cole, J.M.; Simmons, K.; Prosperi, J.R. Effect of adenomatous polyposis coli loss on tumorigenic potential in pancreatic ductal adenocarcinoma. Cells 2019, 8, 1084. [Google Scholar] [CrossRef]
- Prosperi, J.R.; Becher, K.R.; Willson, T.A.; Collins, M.H.; Witte, D.P.; Goss, K.H. The APC tumor suppressor is required for epithelial integrity in the mouse mammary gland. J. Cell Physiol. 2009, 220, 319–331. [Google Scholar] [CrossRef]
- Liu, C.; Gallagher, R.L.; Price, G.R.; Bolton, E.; Joy, C.; Harraway, J.; Venter, D.J.; Armes, J.E. Ovarian microcystic stromal tumor: A rare clinical manifestation of familial adenomatous polyposis. Int. J. Gynecol. Pathol. 2016, 35, 561–565. [Google Scholar] [CrossRef]
- Crobach, S.; van Wezel, T.; Vasen, H.F.; Morreau, H. Ovarian metastases of colorectal and duodenal cancer in familial adenomatous polyposis. Fam. Cancer 2012, 11, 671–673. [Google Scholar] [CrossRef]
- McCartney, B.M.; Näthke, I.S. Cell regulation by the Apc protein Apc as master regulator of epithelia. Curr. Opin. Cell Biol. 2008, 20, 186–193. [Google Scholar] [CrossRef]
- Cordero, J.; Vidal, M.; Sansom, O. APC as a master regulator of intestinal homeostasis and transformation: From flies to vertebrates. Cell Cycle 2009, 8, 2926–2931. [Google Scholar] [CrossRef]
- Gaspar, C.; Cardoso, J.; Franken, P.; Molenaar, L.; Morreau, H.; Möslein, G.; Sampson, J.; Boer, J.M.; de Menezes, R.X.; Fodde, R. Cross-Species comparison of human and mouse intestinal polyps reveals conserved mechanisms in adenomatous polyposis coli (APC)-driven tumorigenesis. Am. J. Pathol. 2008, 172, 1363–1380. [Google Scholar] [CrossRef]
- Olschwang, S.; Weiffenbach, B.; Laurent-Puig, P.; Melot, T.; Vassal, A.; Falls, K.; Salmon, R.J.; Parc, R.; Strong, L.; Nakamura, Y. Genetic characterization of the APC locus involved in familial adenomatous polyposis. Gastroenterology 1991, 101, 154–160. [Google Scholar] [CrossRef]
- Haggitt, R.C.; Reid, B.J. Hereditary gastrointestinal polyposis syndromes. Am. J. Surg. Pathol. 1986, 10, 871–887. [Google Scholar] [CrossRef]
- Nakamura, Y.; Nishisho, I.; Kinzler, K.W.; Vogelstein, B.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Nagase, H. Mutations of the adenomatous polyposis coli gene in familial polyposis coli patients and sporadic colorectal tumors. Princess Takamatsu Symp. 1991, 22, 285–292. [Google Scholar] [CrossRef]
- Valle, L.; de Voer, R.M.; Goldberg, Y.; Sjursen, W.; Försti, A.; Ruiz-Ponte, C.; Caldés, T.; Garré, P.; Olsen, M.F.; Nordling, M.; et al. Update on genetic predisposition to colorectal cancer and polyposis. Mol. Asp. Med. 2019, 69, 10–26. [Google Scholar] [CrossRef]
- Carr, S.; Kasi, A. Familial adenomatous polyposis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Nishisho, I.; Nakamura, Y.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Koyama, K.; Utsunomiya, J.; Baba, S.; Hedge, P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991, 253, 665–669. [Google Scholar] [CrossRef]
- Joslyn, G.; Carlson, M.; Thliveris, A.; Albertsen, H.; Gelbert, L.; Samowitz, W.; Groden, J.; Stevens, J.; Spirio, L.; Robertson, M. Identification of deletion mutations and three new genes at the familial polyposis locus. Cell 1991, 66, 601–613. [Google Scholar] [CrossRef]
- Sansom, O.J.; Meniel, V.; Wilkins, J.A.; Cole, A.M.; Oien, K.A.; Marsh, V.; Jamieson, T.J.; Guerra, C.; Ashton, G.H.; Barbacid, M.; et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 14122–14127. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Velasco, J.D.; Arvide, E.M.; Wei, S.H.; Vauthey, J.-N. Interactions of multiple gene alterations in colorectal liver metastases. Chin. Clin. Oncol. 2019, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Komarova, N.L.; Wang, L. Initiation of colorectal cancer: Where do the two hits hit? Cell Cycle 2004, 3, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, S.A.; Lipkin, M. Migrating colonic crypt epithelial cells: Primary targets for transformation. Carcinogenesis 2002, 23, 1777–1780. [Google Scholar] [CrossRef] [PubMed]
- Faux, M.C.; Ross, J.L.; Meeker, C.; Johns, T.; Ji, H.; Simpson, R.J.; Layton, M.J.; Burgess, A.W. Restoration of full-length adenomatous polyposis coli (APC) protein in a colon cancer cell line enhances cell adhesion. J. Cell Sci. 2004, 117, 427–439. [Google Scholar] [CrossRef]
- Dow, L.E.; O’Rourke, K.P.; Simon, J.; Tschaharganeh, D.F.; van Es, J.H.; Clevers, H.; Lowe, S.W. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 2015, 161, 1539–1552. [Google Scholar] [CrossRef]
- Kariv, R.; Caspi, M.; Fliss-Isakov, N.; Shorer, Y.; Shor, Y.; Rosner, G.; Brazowski, E.; Beer, G.; Cohen, S.; Rosin-Arbesfeld, R. Resorting the function of the colorectal cancer gatekeeper adenomatous polyposis coli. Int. J. Cancer 2020, 146, 1064–1074. [Google Scholar] [CrossRef]
- Zilberberg, A.; Lahav, L.; Rosin-Arbesfeld, R. Restoration of APC gene function in colorectal cancer cells by aminoglycoside- and macrolide-induced read-through of premature termination codons. Gut 2010, 59, 496–507. [Google Scholar] [CrossRef]
- Zhang, L.; Theodoropoulos, P.C.; Eskiocak, U.; Wang, W.; Moon, Y.-A.; Posner, B.; Williams, N.S.; Wright, W.E.; Kim, S.B.; Nijhawan, D.; et al. Selective targeting of mutant adenomatous polyposis coli (APC) in colorectal cancer. Sci. Transl. Med. 2016, 8, 361ra140. [Google Scholar] [CrossRef]
- Näthke, I. APC at a glance. J. Cell Sci. 2004, 117, 4873–4875. [Google Scholar] [CrossRef]
- Ruane, P.T.; Gumy, L.F.; Bola, B.; Anderson, B.; Wozniak, M.J.; Hoogenraad, C.C.; Allan, V.J. Tumour Suppressor Adenomatous Polyposis Coli (APC) localisation is regulated by both Kinesin-1 and Kinesin-2. Sci. Rep. 2016, 6, 27456. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mills, K.M.; Brocardo, M.G.; Henderson, B.R. APC binds the Miro/Milton motor complex to stimulate transport of mitochondria to the plasma membrane. Mol. Biol. Cell 2016, 27, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Askham, J.M.; Moncur, P.; Markham, A.F.; Morrison, E.E. Regulation and function of the interaction between the APC tumour suppressor protein and EB1. Oncogene 2000, 19, 1950–1958. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef]
- Zeineldin, M.; Cunningham, J.; McGuinness, W.; Alltizer, P.; Cowley, B.; Blanchat, B.; Xu, W.; Pinson, D.; Neufeld, K.L. A knock-in mouse model reveals roles for nuclear Apc in cell proliferation, Wnt signal inhibition and tumor suppression. Oncogene 2012, 31, 2423–2437. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Zaoui, K.; Benseddik, K.; Daou, P.; Salaün, D.; Badache, A. ErbB2 receptor controls microtubule capture by recruiting ACF7 to the plasma membrane of migrating cells. Proc. Natl. Acad. Sci. USA 2010, 107, 18517–18522. [Google Scholar] [CrossRef]
- Lesko, A.C.; Goss, K.H.; Yang, F.F.; Schwertner, A.; Hulur, I.; Onel, K.; Prosperi, J.R. The APC tumor suppressor is required for epithelial cell polarization and three-dimensional morphogenesis. Biochim. Biophys. Acta 2015, 1853, 711–723. [Google Scholar] [CrossRef]
- Kaplan, K.B.; Burds, A.A.; Swedlow, J.R.; Bekir, S.S.; Sorger, P.K.; Näthke, I.S. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat. Cell Biol. 2001, 3, 429–432. [Google Scholar] [CrossRef]
- Hankey, W.; Frankel, W.L.; Groden, J. Functions of the APC tumor suppressor protein dependent and independent of canonical WNT signaling: Implications for therapeutic targeting. Cancer Metastasis Rev. 2018, 37, 159–172. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Nagase, H.; Ando, H.; Horii, A.; Ichii, S.; Nakatsuru, S.; Aoki, T.; Miki, Y.; Mori, T.; Nakamura, Y. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Hum. Mol. Genet. 1992, 1, 229–233. [Google Scholar]
- Zeineldin, M.; Neufeld, K.L. More than two decades of Apc modeling in rodents. Biochim. Biophys. Acta 2013, 1836, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Washington, K.; Zemper, A.E.D. Apc-related models of intestinal neoplasia: A brief review for pathologists. Surg. Exp. Pathol. 2019, 2, 11. [Google Scholar] [CrossRef]
- Neufeld, K.; Zeineldin, M. New insights from animal models of colon cancer: Inflammation control as a new facet on the tumor suppressor APC gem. GICTT 2015, 2015, 39–52. [Google Scholar] [CrossRef]
- Stastna, M.; Janeckova, L.; Hrckulak, D.; Kriz, V.; Korinek, V. Human Colorectal Cancer from the Perspective of Mouse Models. Genes (Basel) 2019, 10, 788. [Google Scholar] [CrossRef] [PubMed]
- Moser, A.R.; Luongo, C.; Gould, K.A.; McNeley, M.K.; Shoemaker, A.R.; Dove, W.F. ApcMin: A mouse model for intestinal and mammary tumorigenesis. Eur. J. Cancer 1995, 31A, 1061–1064. [Google Scholar] [CrossRef]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef]
- Fodde, R.; Edelmann, W.; Yang, K.; van Leeuwen, C.; Carlson, C.; Renault, B.; Breukel, C.; Alt, E.; Lipkin, M.; Khan, P.M. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc. Natl. Acad. Sci. USA 1994, 91, 8969–8973. [Google Scholar] [CrossRef]
- Smits, R.; Kielman, M.F.; Breukel, C.; Zurcher, C.; Neufeld, K.; Jagmohan-Changur, S.; Hofland, N.; van Dijk, J.; White, R.; Edelmann, W.; et al. Apc1638T: A mouse model delineating critical domains of the adenomatous polyposis coli protein involved in tumorigenesis and development. Genes Dev. 1999, 13, 1309–1321. [Google Scholar] [CrossRef]
- Parker, A.; Maclaren, O.J.; Fletcher, A.G.; Muraro, D.; Kreuzaler, P.A.; Byrne, H.M.; Maini, P.K.; Watson, A.J.M.; Pin, C. Cell proliferation within small intestinal crypts is the principal driving force for cell migration on villi. FASEB J. 2017, 31, 636–649. [Google Scholar] [CrossRef]
- Kaur, P.; Potten, C.S. Cell migration velocities in the crypts of the small intestine after cytotoxic insult are not dependent on mitotic activity. Cell Tissue Kinet. 1986, 19, 601–610. [Google Scholar] [CrossRef]
- Padrick, S.B.; Rosen, M.K. Physical mechanisms of signal integration by WASP family proteins. Annu. Rev. Biochem. 2010, 79, 707–735. [Google Scholar] [CrossRef]
- Rotty, J.D.; Wu, C.; Bear, J.E. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12. [Google Scholar] [CrossRef]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef]
- Efimova, N.; Yang, C.; Chia, J.X.; Li, N.; Lengner, C.J.; Neufeld, K.L.; Svitkina, T.M. Branched actin networks are assembled on microtubules by adenomatous polyposis coli for targeted membrane protrusion. J. Cell Biol. 2020, 219, 219. [Google Scholar] [CrossRef]
- Näthke, I.S.; Adams, C.L.; Polakis, P.; Sellin, J.H.; Nelson, W.J. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J. Cell Biol. 1996, 134, 165–179. [Google Scholar] [CrossRef]
- Rosin-Arbesfeld, R.; Ihrke, G.; Bienz, M. Actin-dependent membrane association of the APC tumour suppressor in polarized mammalian epithelial cells. EMBO J. 2001, 20, 5929–5939. [Google Scholar] [CrossRef]
- Yamashita, Y.M.; Jones, D.L.; Fuller, M.T. Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science 2003, 301, 1547–1550. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Zhang, J.; Ahmad, S.; Mao, Y. BubR1 and APC/EB1 cooperate to maintain metaphase chromosome alignment. J. Cell Biol. 2007, 178, 773–784. [Google Scholar] [CrossRef]
- Trzepacz, C.; Lowy, A.M.; Kordich, J.J.; Groden, J. Phosphorylation of the tumor suppressor adenomatous polyposis coli (APC) by the cyclin-dependent kinase p34. J. Biol. Chem. 1997, 272, 21681–21684. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Dikovskaya, D.; Schiffmann, D.; Newton, I.P.; Oakley, A.; Kroboth, K.; Sansom, O.; Jamieson, T.J.; Meniel, V.; Clarke, A.; Näthke, I.S. Loss of APC induces polyploidy as a result of a combination of defects in mitosis and apoptosis. J. Cell Biol. 2007, 176, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Sanhaji, M.; Matthess, Y.; Hörlin, A.; Lorenz, I.; Dötsch, C.; Habbe, N.; Waidmann, O.; Kurunci-Csacsko, E.; Firestein, R.; et al. PLK1 has tumor-suppressive potential in APC-truncated colon cancer cells. Nat. Commun. 2018, 9, 1106. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Raab, M.; Sanhaji, M. The role of PLK1 in cancer exhibiting chromosomal instability. Mol. Cell. Oncol. 2018, 5, e1485539. [Google Scholar] [CrossRef] [PubMed]
- Brito, D.A.; Rieder, C.L. The ability to survive mitosis in the presence of microtubule poisons differs significantly between human nontransformed (RPE-1) and cancer (U2OS, HeLa) cells. Cell Motil. Cytoskelet. 2009, 66, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Zumbrunn, J.; Kinoshita, K.; Hyman, A.A.; Näthke, I.S. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3β phosphorylation. Curr. Biol. 2001, 11, 44–49. [Google Scholar] [CrossRef]
- Gupta, K.K.; Alberico, E.O.; Näthke, I.S.; Goodson, H.V. Promoting microtubule assembly: A hypothesis for the functional significance of the +TIP network. Bioessays 2014, 36, 818–826. [Google Scholar] [CrossRef]
- Kita, K.; Wittmann, T.; Näthke, I.S.; Waterman-Storer, C.M. Adenomatous polyposis coli on microtubule plus ends in cell extensions can promote microtubule net growth with or without EB1. Mol. Biol. Cell 2006, 17, 2331–2345. [Google Scholar] [CrossRef][Green Version]
- Lui, C.; Ashton, C.; Sharma, M.; Brocardo, M.G.; Henderson, B.R. APC functions at the centrosome to stimulate microtubule growth. Int. J. Biochem. Cell Biol. 2016, 70, 39–47. [Google Scholar] [CrossRef]
- Lui, C.; Mok, M.T.S.; Henderson, B.R. Characterization of adenomatous polyposis coli protein dynamics and localization at the centrosome. Cancers (Basel) 2016, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Nunes, V.; Dantas, M.; Castro, D.; Vitiello, E.; Wang, I.; Carpi, N.; Balland, M.; Piel, M.; Aguiar, P.; Maiato, H.; et al. Centrosome-nuclear axis repositioning drives the assembly of a bipolar spindle scaffold to ensure mitotic fidelity. Mol. Biol. Cell 2020, 31, 1675–1690. [Google Scholar] [CrossRef]
- Kita, A.M.; Swider, Z.T.; Erofeev, I.; Halloran, M.C.; Goryachev, A.B.; Bement, W.M. Spindle-F-actin interactions in mitotic spindles in an intact vertebrate epithelium. Mol. Biol. Cell 2019, 30, 1645–1654. [Google Scholar] [CrossRef]
- Hoopen, R.T.; Cepeda-García, C.; Fernández-Arruti, R.; Juanes, M.A.; Delgehyr, N.; Segal, M. Mechanism for astral microtubule capture by cortical Bud6p priming spindle polarity in S. cerevisiae. Curr. Biol. 2012, 22, 1075–1083. [Google Scholar] [PubMed]
- Cepeda-García, C.; Delgehyr, N.; Ortiz, M.A.J.; ten Hoopen, R.; Zhiteneva, A.; Segal, M. Actin-mediated delivery of astral microtubules instructs Kar9p asymmetric loading to the bud-ward spindle pole. Mol. Biol. Cell 2010, 21, 2685–2695. [Google Scholar] [CrossRef]
- Moseley, J.B.; Goode, B.L. Differential activities and regulation of Saccharomyces cerevisiae formin proteins Bni1 and Bnr1 by Bud6. J. Biol. Chem. 2005, 280, 28023–28033. [Google Scholar] [CrossRef] [PubMed]
- Delgehyr, N.; Lopes, C.S.J.; Moir, C.A.; Huisman, S.M.; Segal, M. Dissecting the involvement of formins in Bud6p-mediated cortical capture of microtubules in S. cerevisiae. J. Cell Sci. 2008, 121, 3803–3814. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juanes, M.A. Cytoskeletal Control and Wnt Signaling—APC’s Dual Contributions in Stem Cell Division and Colorectal Cancer. Cancers 2020, 12, 3811. https://doi.org/10.3390/cancers12123811
Juanes MA. Cytoskeletal Control and Wnt Signaling—APC’s Dual Contributions in Stem Cell Division and Colorectal Cancer. Cancers. 2020; 12(12):3811. https://doi.org/10.3390/cancers12123811
Chicago/Turabian StyleJuanes, M. Angeles. 2020. "Cytoskeletal Control and Wnt Signaling—APC’s Dual Contributions in Stem Cell Division and Colorectal Cancer" Cancers 12, no. 12: 3811. https://doi.org/10.3390/cancers12123811
APA StyleJuanes, M. A. (2020). Cytoskeletal Control and Wnt Signaling—APC’s Dual Contributions in Stem Cell Division and Colorectal Cancer. Cancers, 12(12), 3811. https://doi.org/10.3390/cancers12123811